
Tri-Herbal Medicine Divya Sarva-Kalp-Kwath (Livogrit) Regulates Fatty Acid-Induced Steatosis in Human HepG2 Cells through Inhibition of Intracellular Triglycerides and Extracellular Glycerol Levels


Abstract
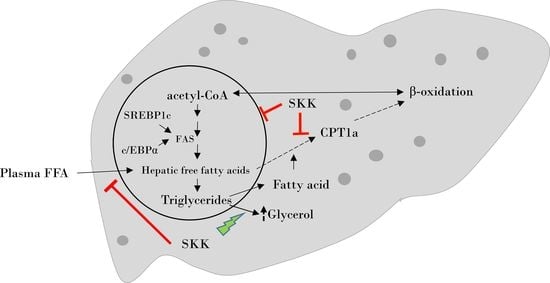
1. Introduction
2. Results
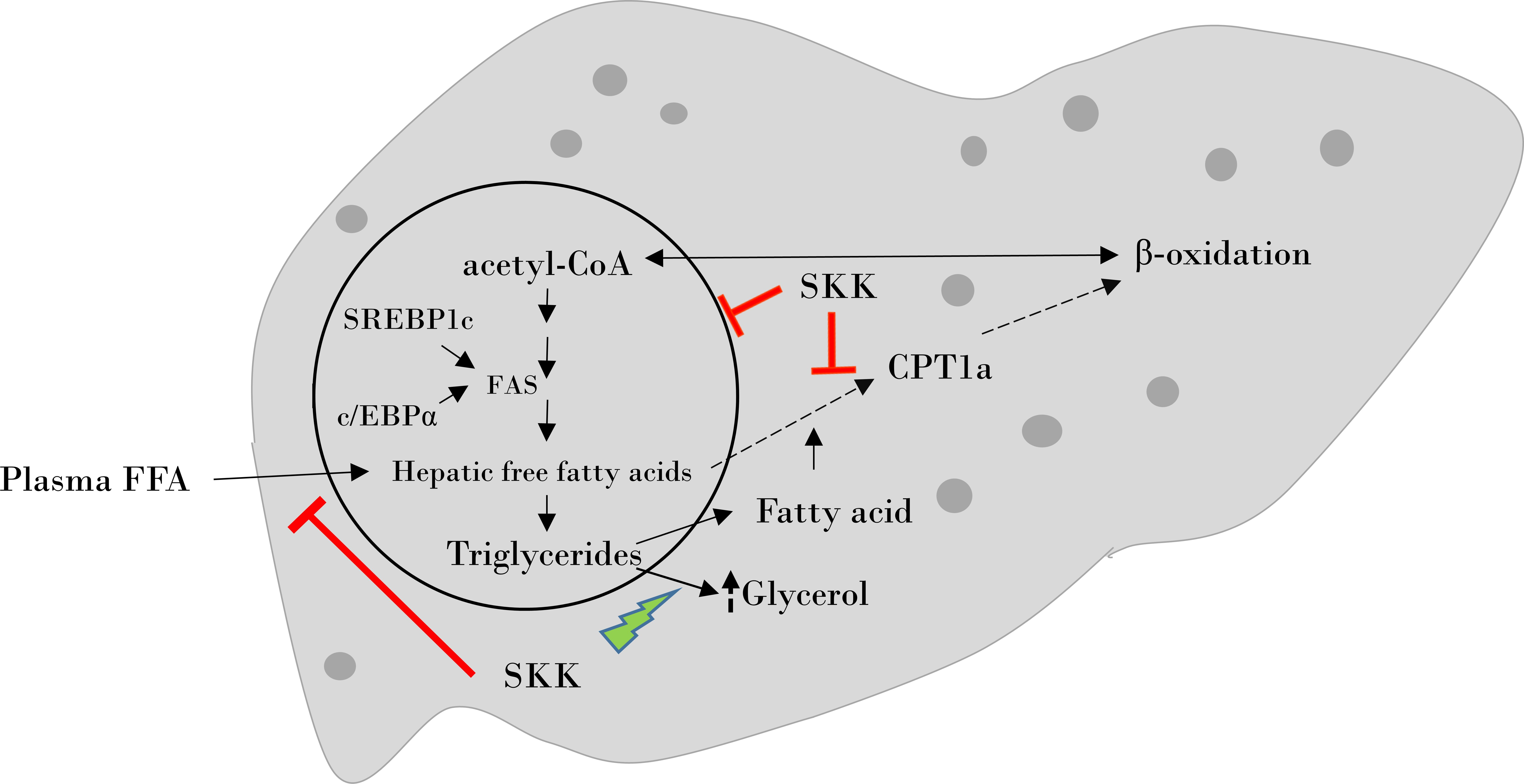
2.1. HPTLC Fingerprinting of Divya Sarva-Kalp-Kwath (SKK)
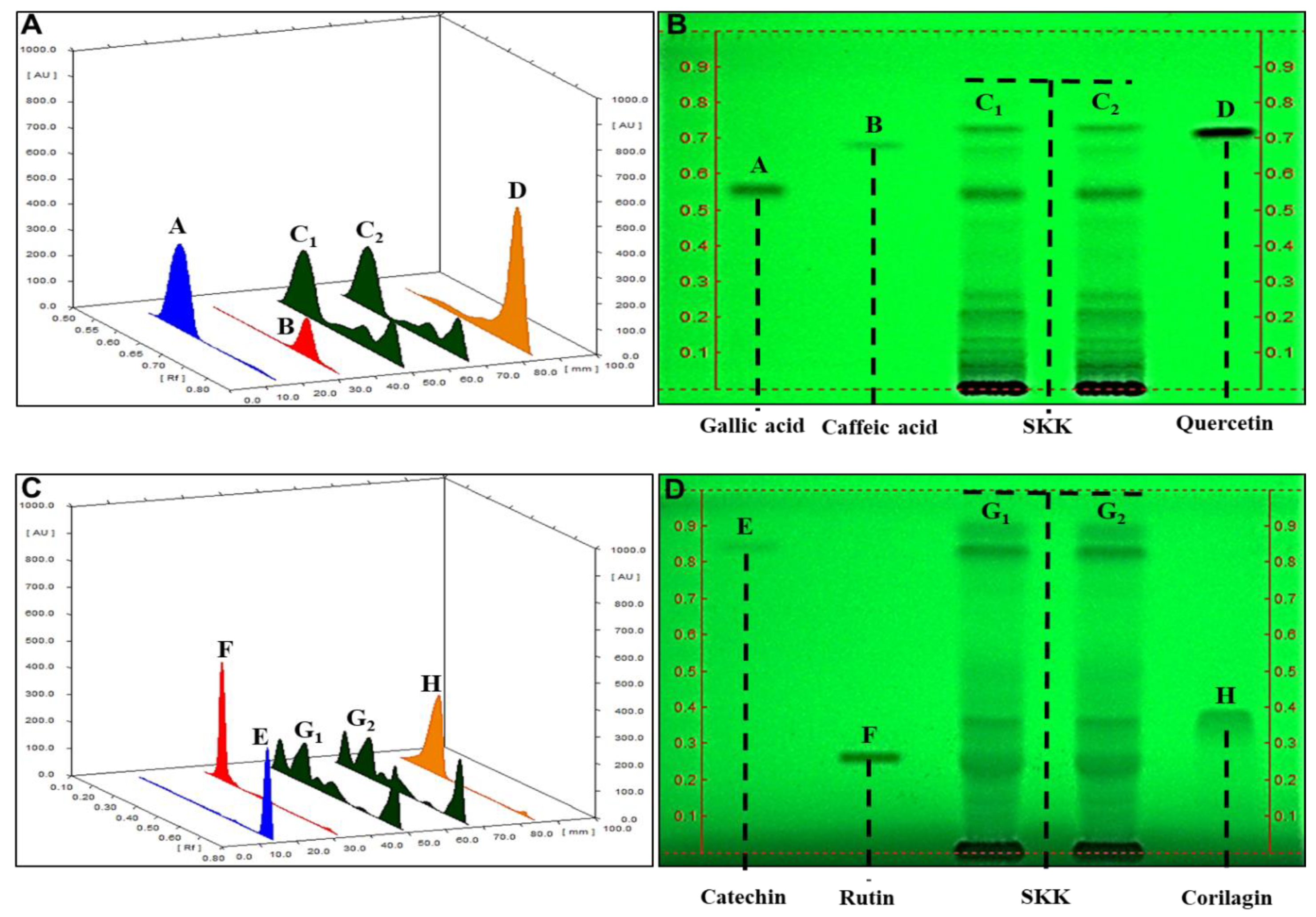
2.2. SKK Modulates HepG2 Cell Viability
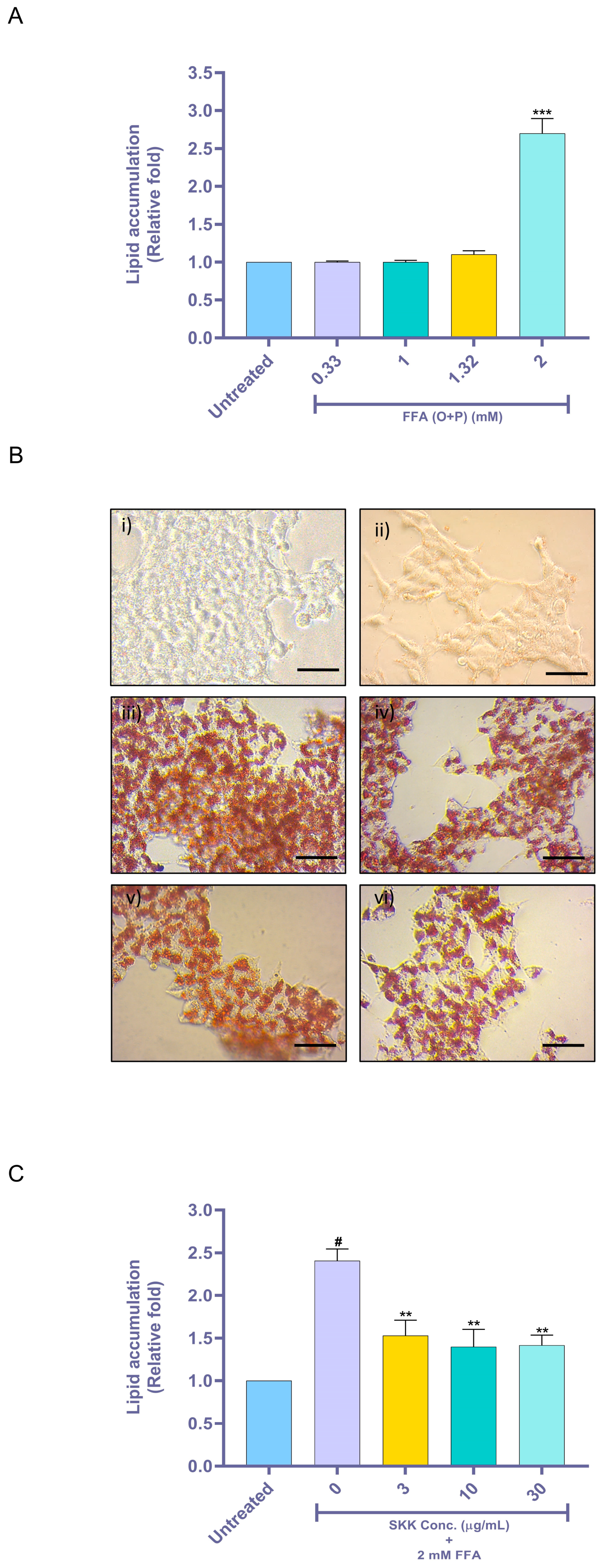
2.3. SKK Inhibits Intracellular Lipid Accumulation
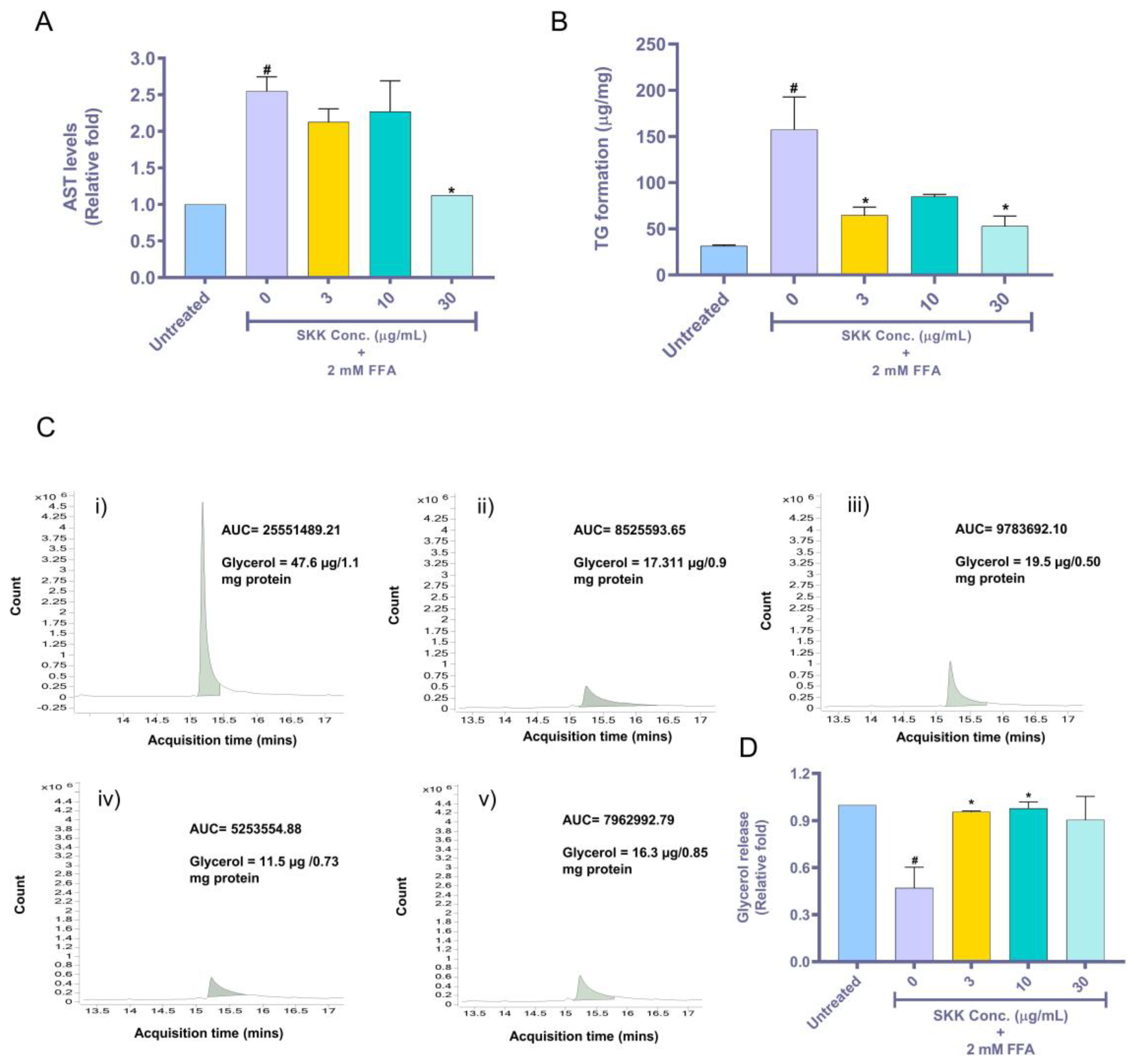
2.4. SKK Moderates Aspartate Aminotransferase (AST) Release, Intracellular Triglyceride, and Extracellular Glycerol Levels
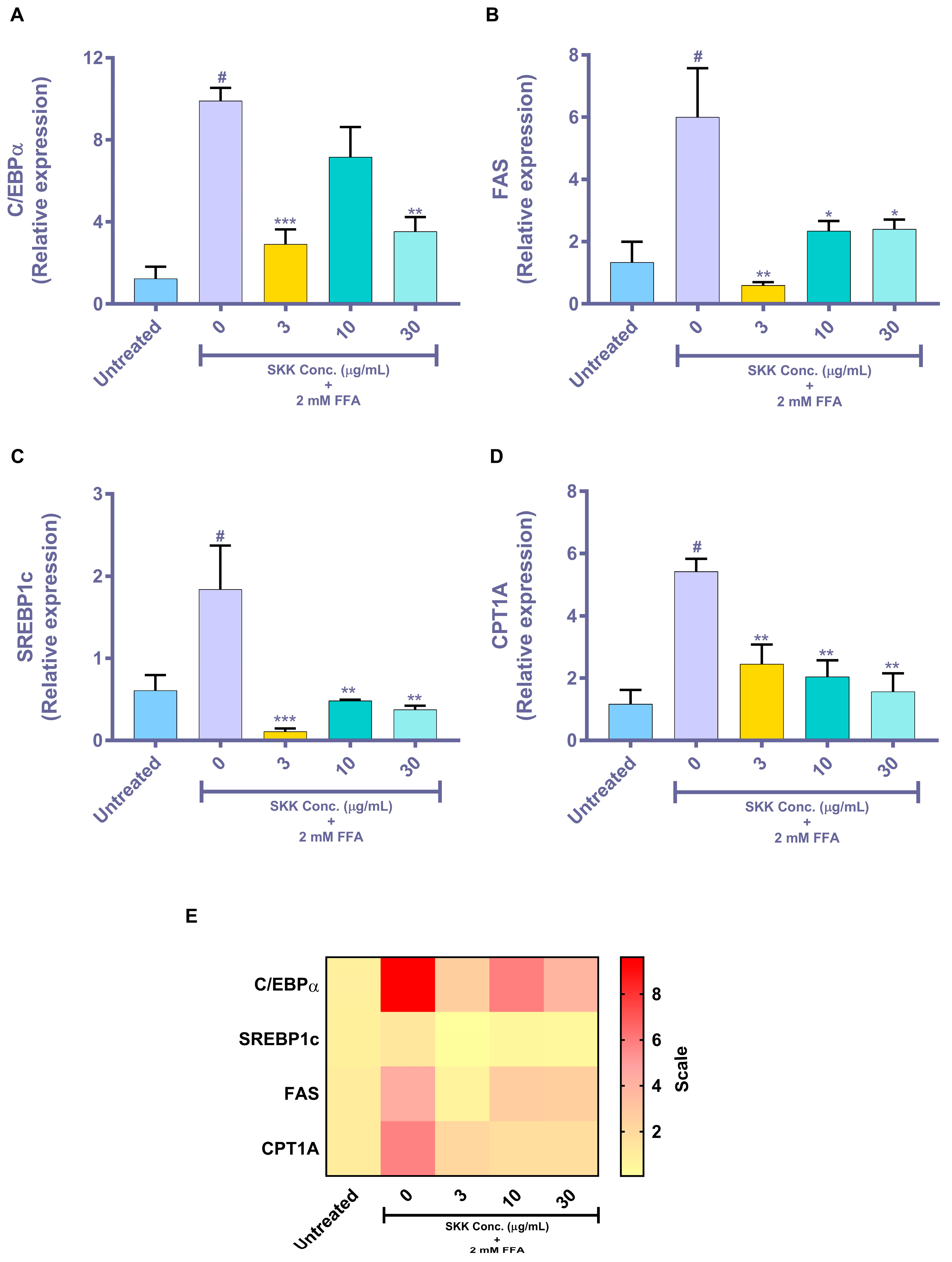
2.5. Genomic Level Changes Induced by SKK in the In Vitro Steatosis Model
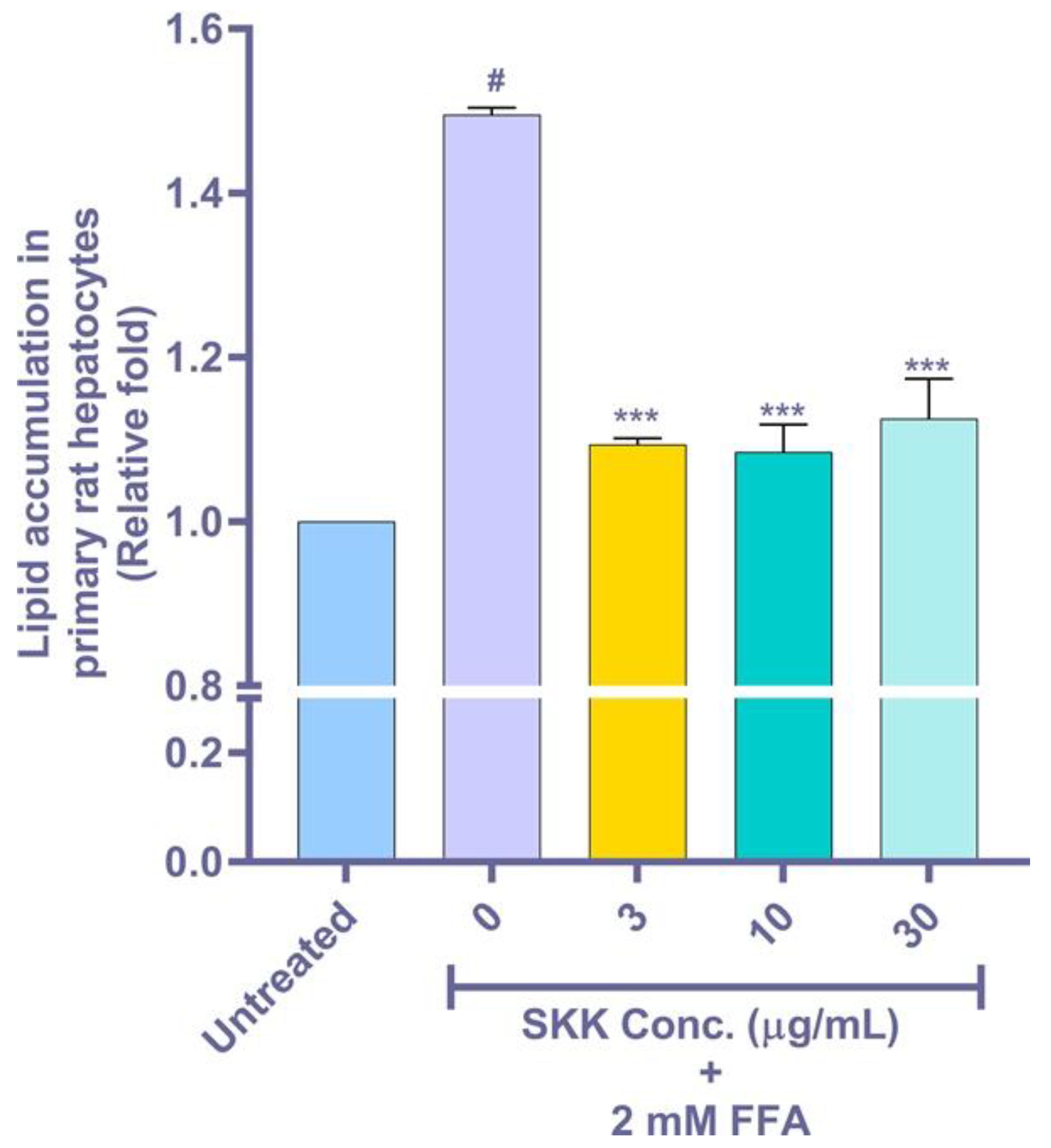
2.6. Intracellular Lipid Accumulation in the FFA Stimulated Rat Primary Hepatocytes
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Metabolite Analysis of SKK
4.3. In-Vitro Cell Culture
4.4. Cell Viability Analysis
4.5. Oil Red ‘O’ (ORO) Staining Based Lipid Accumulation Analysis
4.6. Intracellular Triglyceride Estimation
4.7. AST Estimation
4.8. Estimation of Extracellular Free Glycerol
4.9. Real-Time PCR (RT-PCR) Based Gene Expression Quantification
4.10. Primary Rat Hepatocyte Isolation and Treatment
4.11. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nassir, F.; Rector, R.S.; Hammoud, G.M.; Ibdah, J.A. Pathogenesis and Prevention of Hepatic Steatosis. Gastroenterol. Hepatol. 2015, 11, 167–175. [Google Scholar]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of Liver Diseases in the World. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Mokdad, A.H.; Forouzanfar, M.H.; Daoud, F.; Mokdad, A.; El Bcheraoui, C.; Moradi-Lakeh, M.; Kyu, H.H.; Barber, R.M.; Wagner, J.; Cercy, K.; et al. Global Burden of Diseases, Injuries, and Risk Factors for Young People’s Health During 1990–2013: A Systematic Analysis for the Global Burden of Disease Study 2013. Lancet 2016, 387, 2383–2401. [Google Scholar] [CrossRef]
- Pagadala, M.; Kasumov, T.; McCullough, A.J.; Zein, N.N.; Kirwan, J.P. Role of Ceramides in Nonalcoholic Fatty Liver Disease. Trends Endocrinol. Metab. 2012, 23, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Perumpail, B.J.; Khan, M.A.; Yoo, E.R.; Cholankeril, G.; Kim, D.; Ahmed, A. Clinical Epidemiology and Disease Burden of Nonalcoholic Fatty Liver Disease. World J. Gastroenterol. 2017, 23, 8263–8276. [Google Scholar] [CrossRef]
- Araya, J.; Rodrigo, R.; Videla, L.A.; Thielemann, L.; Orellana, M.; Pettinelli, P.; Poniachik, J. Increase in Long-Chain Polyunsaturated Fatty Acid n−6/N−3 Ratio in Relation to Hepatic Steatosis in Patients with Non-Alcoholic Fatty Liver Disease. Clin. Sci. 2004, 106, 635–643. [Google Scholar] [CrossRef]
- Joshi-Barve, S.; Barve, S.S.; Amancherla, K.; Gobejishvili, L.; Hill, D.; Cave, M.; Hote, P.; McClain, C.J. Palmitic Acid Induces Production of Proinflammatory Cytokine Interleukin-8 from Hepatocytes. Hepatology 2007, 46, 823–830. [Google Scholar] [CrossRef]
- Tang, Y.; Bian, Z.; Zhao, L.; Liu, Y.; Liang, S.; Wang, Q.; Han, X.; Peng, Y.; Chen, X.; Shen, L.; et al. Interleukin-17 Exacerbates Hepatic Steatosis and Inflammation in Non-Alcoholic Fatty Liver Disease. Clin. Exp. Immunol. 2011, 166, 281–290. [Google Scholar] [CrossRef]
- Niklas, J.; Bonin, A.; Mangin, S.; Bucher, J.; Kopacz, S.; Matz-Soja, M.; Thiel, C.; Gebhardt, R.; Hofmann, U.; Mauch, K. Central Energy Metabolism Remains Robust in Acute Steatotic Hepatocytes Challenged by a High Free Fatty Acid Load. BMB Rep. 2012, 45, 396–401. [Google Scholar] [CrossRef]
- Pan, Z.; Wang, J.-W.; Tang, H.; Li, L.; Lv, J.; Xia, L.; Han, C.; Xu, F.; He, H.; Xu, H.; et al. Effects of Palmitic Acid on Lipid Metabolism Homeostasis and Apoptosis in Goose Primary Hepatocytes. Mol. Cell. Biochem. 2010, 350, 39–46. [Google Scholar] [CrossRef]
- Gómez-Lechón, M.J.; Donato, M.T.; Martínez-Romero, A.; Jiménez, N.; Castell, J.V.; O’Connor, J.-E. A Human Hepatocellular in Vitro Model to Investigate Steatosis. Chem. Interactions 2007, 165, 106–116. [Google Scholar] [CrossRef]
- Mantzaris, M.D.; Tsianos, E.V.; Galaris, D. Interruption of Triacylglycerol Synthesis in the Endoplasmic Reticulum Is the Initiating Event for Saturated Fatty Acid-Induced Lipotoxicity in Liver Cells. FEBS J. 2010, 278, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Ricchi, M.; Odoardi, M.R.; Carulli, L.; Anzivino, C.; Ballestri, S.; Pinetti, A.; Fantoni, L.I.; Marra, F.; Bertolotti, M.; Banni, S.; et al. Differential Effect of Oleic and Palmitic Acid on Lipid Accumulation and Apoptosis in Cultured Hepatocytes. J. Gastroenterol. Hepatol. 2009, 24, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Swagell, C.D.; Henly, D.C.; Morris, C.P. Expression Analysis of a Human Hepatic Cell Line in Response to Palmitate. Biochem. Biophys. Res. Commun. 2005, 328, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Yoo, J.-H.; Lee, Y.-S.; Lee, H.-J. Lonicera Caerulea Extract Attenuates Non-Alcoholic Fatty Liver Disease in Free Fatty Acid-Induced HepG2 Hepatocytes and in High Fat Diet-Fed Mice. Nutrients 2019, 11, 494. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Mera, P.; Casas, J.; Salvador, J.; Rodríguez, A.; Alonso, S.; Sebastián, D.; Soler-Vázquez, M.C.; Montironi, C.; Recalde, S.; et al. Liver CPT1A Gene Therapy Reduces diet-induced Hepatic Steatosis in Mice and Highlights Potential Lipid Biomarkers for Human NAFLD. FASEB J. 2020, 34, 11816–11837. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of Fatty Acids Stored in Liver and Secreted via Lipoproteins in Patients with Nonalcoholic Fatty Liver Disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Jian, T.; Lü, H.; Ding, X.; Wu, Y.; Zuo, Y.; Li, J.; Chen, J.; Gu, H. Polyphenol-Rich Trapa Quadrispinosa Pericarp Extract Ameliorates High-Fat Diet Induced Non-Alcoholic Fatty Liver Disease by Regulating Lipid Metabolism and Insulin Resistance in Mice. PeerJ 2019, 7, e8165. [Google Scholar] [CrossRef]
- Balkrishna, A.; Sakat, S.S.; Ranjan, R.; Joshi, K.; Shukla, S.; Joshi, K.; Verma, S.; Gupta, A.; Bhattacharya, K.; Varshney, A. Polyherbal Medicine Divya Sarva-Kalp-Kwath Ameliorates Persistent Carbon Tetrachloride Induced Biochemical and Pathological Liver Impairments in Wistar Rats and in HepG2 Cells. Front. Pharmacol. 2020, 11, 288. [Google Scholar] [CrossRef]
- Thomford, N.E.; Awortwe, C.; Dzobo, K.; Adu, F.; Chopera, D.; Wonkam, A.; Skelton, M.; Blackhurst, D.; Dandara, C. Inhibition of CYP2B6 by Medicinal Plant Extracts: Implication for Use of Efavirenz and Nevirapine-Based Highly Active Anti-Retroviral Therapy (HAART) in Resource-Limited Settings. Molecules 2016, 21, 211. [Google Scholar] [CrossRef]
- Huang, C.-Z.; Tung, Y.-T.; Hsia, S.-M.; Wu, C.-H.; Yen, G.-C. The Hepatoprotective Effect of Phyllanthus Emblica, L. Fruit on High Fat Diet-Induced Non-Alcoholic Fatty Liver Disease (NAFLD) in SD Rats. Food Funct. 2017, 8, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Tung, Y.-T.; Huang, C.-Z.; Lin, J.-H.; Yen, G.-C. Effect of Phyllanthus Emblica, L. Fruit on Methionine and Choline-Deficiency Diet-Induced Nonalcoholic Steatohepatitis. J. Food Drug Anal. 2018, 26, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, R.; Sil, P.C. Protein Isolate from the HerbPhyllanthus NiruriModulates Carbon Tetrachloride-Induced Cytotoxicity in Hepatocytes. Toxicol. Mech. Methods 2007, 17, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Harish, R.; Shivanandappa, T. Antioxidant Activity and Hepatoprotective Potential of Phyllanthus Niruri. Food Chem. 2006, 95, 180–185. [Google Scholar] [CrossRef]
- Jantan, I.; Haque, A.; Ilangkovan, M.; Arshad, L. An Insight Into the Modulatory Effects and Mechanisms of Action of Phyllanthus Species and Their Bioactive Metabolites on the Immune System. Front. Pharmacol. 2019, 10, 878. [Google Scholar] [CrossRef]
- Kaur, N.; Kaur, B.; Sirhindi, G. Phytochemistry and Pharmacology OfPhyllanthus niruriL: A Review. Phytotherapy Res. 2017, 31, 980–1004. [Google Scholar] [CrossRef]
- Kirtikar, K.R.; Basu, B.D. Indian Medicinal Plants, 2nd ed.; Lalit Mohan Basu Publication: Allahabad, India, 1956; p. 852. [Google Scholar]
- Chang, J.-J.; Chung, D.-J.; Lee, Y.-J.; Wen, B.-H.; Jao, H.-Y.; Wang, C.-J. Solanum NigrumPolyphenol Extracts Inhibit Hepatic Inflammation, Oxidative Stress, and Lipogenesis in High-Fat-Diet-Treated Mice. J. Agric. Food Chem. 2017, 65, 9255–9265. [Google Scholar] [CrossRef]
- Available online: https://www.ncbi.nlm.nih.gov/books/NBK482489/ (accessed on 10 September 2020).
- Fuchs, M. Non-Alcoholic Fatty Liver Disease: The Bile Acid-Activated Farnesoid X Receptor as an Emerging Treatment Target. J. Lipids 2011, 2012, 934396. [Google Scholar] [CrossRef]
- Jadeja, R.N.; Devkar, R.V.; Nammi, S. Herbal Medicines for the Treatment of Nonalcoholic Steatohepatitis: Current Scenario and Future Prospects. Evidence-Based Complement. Altern. Med. 2014, 2014, 648308. [Google Scholar] [CrossRef]
- Balkrishna, A.; Ranjan, R.; Sakat, S.S.; Sharma, V.K.; Shukla, R.; Joshi, K.; Devkar, R.; Sharma, N.; Saklani, S.; Pathak, P.; et al. Evaluation of Polyherbal Ayurvedic Formulation ‘Peedantak Vati’ for Anti-Inflammatory and Analgesic Properties. J. Ethnopharmacol. 2019, 235, 361–374. [Google Scholar] [CrossRef]
- Balkrishna, A.; Sakat, S.S.; Joshi, K.; Joshi, K.; Sharma, V.; Ranjan, R.; Bhattacharya, K.; Varshney, A. Cytokines Driven Anti-Inflammatory and Anti-Psoriasis Like Efficacies of Nutraceutical Sea Buckthorn (Hippophae Rhamnoides) Oil. Front. Pharmacol. 2019, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Balkrishna, A.; Sakat, S.S.; Joshi, K.; Paudel, S.; Joshi, D.; Joshi, K.; Ranjan, R.; Gupta, A.; Bhattacharya, K.; Varshney, A. Herbo-Mineral Formulation ‘Ashwashila’ Attenuates Rheumatoid Arthritis Symptoms in Collagen-Antibody-Induced Arthritis (CAIA) Mice Model. Sci. Rep. 2019, 9, 8025. [Google Scholar] [CrossRef]
- Huang, W.-C.; Chen, Y.-L.; Liu, H.-C.; Wu, S.-J.; Liou, C.-J. Ginkgolide C Reduced Oleic Acid-Induced Lipid Accumulation in HepG2 Cells. Saudi Pharm. J. 2018, 26, 1178–1184. [Google Scholar] [CrossRef]
- Li, X.; Wang, R.; Zhou, N.; Wang, X.; Liu, Q.; Bai, Y.; Bai, Y.; Liu, Z.; Yang, H.; Zou, J.; et al. Quercetin Improves Insulin Resistance and Hepatic Lipid Accumulation in Vitro in a NAFLD Cell Model. Biomed. Rep. 2012, 1, 71–76. [Google Scholar] [CrossRef]
- Liu, Q.; Pan, R.; Ding, L.; Zhang, F.; Hu, L.; Ding, B.; Zhu, L.; Xia, Y.; Dou, X. Rutin Exhibits Hepatoprotective Effects in a Mouse Model of Non-Alcoholic Fatty Liver Disease by Reducing Hepatic Lipid Levels and Mitigating Lipid-Induced Oxidative Injuries. Int. Immunopharmacol. 2017, 49, 132–141. [Google Scholar] [CrossRef]
- Van De Wier, B.; Koek, G.H.; Bast, A.; Haenen, G.R.M.M. The Potential of Flavonoids in the Treatment of Non-Alcoholic Fatty Liver Disease. Crit. Rev. Food Sci. Nutr. 2015, 57, 834–855. [Google Scholar] [CrossRef]
- Yang, R.-X.; Pan, Q.; Liu, X.-L.; Zhou, D.; Xin, F.-Z.; Zhao, Z.-H.; Zhang, R.-N.; Zeng, J.; Qiao, L.; Hu, C.-X.; et al. Therapeutic Effect and Autophagy Regulation of Myriocin in Nonalcoholic Steatohepatitis. Lipids Heal. Dis. 2019, 18, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Simon, J.; Ouro, A.; Ala-Ibanibo, L.; Presa, N.; Delgado, T.C.; Martínez-Chantar, M.L. Sphingolipids in Non-Alcoholic Fatty Liver Disease and Hepatocellular Carcinoma: Ceramide Turnover. Int. J. Mol. Sci. 2019, 21, 40. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.-Q.; Li, X.-Y.; Wang, L.; Feng, Z.-L.; Li, X.-F.; Wen, Y.-F.; Han, J.-X. Palmitate Induces Fat Accumulation by Activating C/EBPβ-Mediated G0S2 Expression in HepG2 Cells. World J. Gastroenterol. 2017, 23, 7705–7715. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Aeri, V.; Gaur, P.K.; Jachak, S.M. Phytochemical, Therapeutic, and Ethnopharmacological Overview for a Traditionally Important Herb:Boerhavia DiffusaLinn. BioMed Res. Int. 2014, 2014, 808302. [Google Scholar] [CrossRef]
- Xu, Y.; Guo, W.; Zhang, C.; Chen, F.; Tan, H.Y.; Li, S.; Wang, N.; Feng, Y.-B. Herbal Medicine in the Treatment of Non-Alcoholic Fatty Liver Diseases-Efficacy, Action Mechanism, and Clinical Application. Front. Pharmacol. 2020, 11, 601. [Google Scholar] [CrossRef]
- Han, Y.; Hu, Z.; Cui, A.; Liu, Z.; Ma, F.; Xue, Y.; Liu, Y.; Zhang, F.; Zhao, Z.; Yu, Y.; et al. Post-Translational Regulation of Lipogenesis via AMPK-Dependent Phosphorylation of Insulin-Induced Gene. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Liou, C.-J.; Wei, C.-H.; Chen, Y.-L.; Cheng, C.-Y.; Wang, C.-L.; Huang, W.-C. Fisetin Protects Against Hepatic Steatosis Through Regulation of the Sirt1/AMPK and Fatty Acid β-Oxidation Signaling Pathway in High-Fat Diet-Induced Obese Mice. Cell. Physiol. Biochem. 2018, 49, 1870–1884. [Google Scholar] [CrossRef]
- Seok, J.; Jung, H.S.; Park, S.; Lee, J.O.; Kim, C.J.; Kim, G.J. Alteration of Fatty Acid Oxidation by Increased CPT1A on Replicative Senescence of Placenta-Derived Mesenchymal Stem Cells. Stem Cell Res. Ther. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Wang, Y.; Viscarra, J.; Kim, S.-J.; Sul, H.S. Transcriptional Regulation of Hepatic Lipogenesis. Nat. Rev. Mol. Cell Biol. 2015, 16, 678–689. [Google Scholar] [CrossRef]
- Tian, X.; Ru, Q.; Xiong, Q.; Wen, R.; Chen, Y. Catalpol Attenuates Hepatic Steatosis by Regulating Lipid Metabolism via AMP-Activated Protein Kinase Activation. BioMed Res. Int. 2020, 2020, 6708061. [Google Scholar] [CrossRef]
- Yuk, T.; Kim, Y.; Yang, J.; Sung, J.; Jeong, H.S.; Lee, J. Nobiletin Inhibits Hepatic Lipogenesis via Activation of AMP-Activated Protein Kinase. Evidence-Based Complement. Altern. Med. 2018, 2018, 7420265. [Google Scholar] [CrossRef]
- Zhang, Y.; Meng, T.; Zuo, L.; Bei, Y.; Zhang, Q.; Su, Z.; Huang, Y.; Pang, J.; Xiang, Q.; Yang, H. Xyloketal B Attenuates Fatty Acid-Induced Lipid Accumulation via the SREBP-1c Pathway in NAFLD Models. Mar. Drugs 2017, 15, 163. [Google Scholar] [CrossRef]
- Varshney, R.; Gupta, S.; Roy, P. Cytoprotective Effect of Kaempferol Against Palmitic Acid-Induced Pancreatic β-Cell Death through Modulation of Autophagy via AMPK/MTOR Signaling Pathway. Mol. Cell. Endocrinol. 2017, 448, 1–20. [Google Scholar] [CrossRef]
- Bauer, R.C.; Sasaki, M.; Cohen, D.M.; Cui, J.; Smith, M.A.; Yenilmez, B.O.; Steger, D.J.; Rader, D.J. Tribbles-1 Regulates Hepatic Lipogenesis through Posttranscriptional Regulation of C/EBPα. J. Clin. Investig. 2015, 125, 3809–3818. [Google Scholar] [CrossRef]
- Park, S.; Choi, Y.; Um, S.-J.; Yoon, S.K.; Park, T. Oleuropein Attenuates Hepatic Steatosis Induced by High-Fat Diet in Mice. J. Hepatol. 2011, 54, 984–993. [Google Scholar] [CrossRef]
- Matsusue, K.; Gavrilova, O.; Lambert, G.; Brewer, H.B.; Ward, J.M.; Inoue, Y.; Leroith, D.; Gonzalez, F.J. Hepatic CCAAT/Enhancer Binding Protein α Mediates Induction of Lipogenesis and Regulation of Glucose Homeostasis in Leptin-Deficient Mice. Mol. Endocrinol. 2004, 18, 2751–2764. [Google Scholar] [CrossRef]
- Cho, S.-J.; Jung, U.J.; Choi, M.-S. Differential Effects of Low-Dose Resveratrol on Adiposity and Hepatic Steatosis in Diet-Induced Obese Mice. Br. J. Nutr. 2012, 108, 2166–2175. [Google Scholar] [CrossRef]
- Napal, L.; Marrero, P.F.; Haro, D. An Intronic Peroxisome Proliferator-Activated Receptor-Binding Sequence Mediates Fatty Acid Induction of the Human Carnitine Palmitoyltransferase 1A. J. Mol. Biol. 2005, 354, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, M.; Dessein, A.-F.; Douillard, C.; Dobbelaere, D.; Brivet, M.; Boutron, A.; Zater, M.; Mention-Mulliez, K.; Martin-Ponthieu, A.; Vianey-Saban, C.; et al. A Novel Mutation in CPT1A Resulting in Hepatic CPT Deficiency. JIMD Reports 2012, 6, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Rafiei, H.; Omidian, K.; Bandy, B. Dietary Polyphenols Protect Against Oleic Acid-Induced Steatosis in an in Vitro Model of NAFLD by Modulating Lipid Metabolism and Improving Mitochondrial Function. Nutrients 2019, 11, 541. [Google Scholar] [CrossRef]
- Parra-Vargas, M.; Sandoval-Rodríguez, A.; Rodriguez-Echevarria, R.; Dominguez-Rosales, J.A.; Santos-Garcia, A.; Armendáriz-Borunda, J. Delphinidin Ameliorates Hepatic Triglyceride Accumulation in Human HepG2 Cells, But Not in Diet-Induced Obese Mice. Nutrients 2018, 10, 1060. [Google Scholar] [CrossRef] [PubMed]
- Brown, Z.J.; Fu, Q.; Ma, C.; Kruhlak, M.; Zhang, H.; Luo, J.; Heinrich, B.; Yu, S.J.; Zhang, Q.; Wilson, A.; et al. Carnitine Palmitoyltransferase Gene Upregulation by Linoleic Acid Induces CD4+ T Cell Apoptosis Promoting HCC Development. Cell Death Dis. 2018, 9, 620. [Google Scholar] [CrossRef]
- Lin, C.-W.; Peng, Y.-J.; Lin, Y.-Y.; Mersmann, H.J.; Ding, S.T. LRRK2 Regulates CPT1A to Promote β-Oxidation in HepG2 Cells. Molecules 2020, 25, 4122. [Google Scholar] [CrossRef]
- Blainski, A.; Lopes, G.C.; De Mello, J.C.P. Application and Analysis of the Folin Ciocalteu Method for the Determination of the Total Phenolic Content from Limonium Brasiliense, L. Molecules 2013, 18, 6852–6865. [Google Scholar] [CrossRef]
- Fan, H.; Chen, Y.-Y.; Bei, W.-J.; Wang, L.-Y.; Chen, B.-T.; Guo, J. In VitroScreening for Antihepatic Steatosis Active Components Within Coptidis Rhizoma Alkaloids Extract Using Liver Cell Extraction With HPLC Analysis and a Free Fatty Acid-Induced Hepatic Steatosis HepG2 Cell Assay. Evid. Based Complement. Altern. Med. 2013, 2013, 459390. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Hillebrand, A.; Wang, D.Q.-H.; Liu, M. Isolation and Primary Culture of Rat Hepatic Cells. J. Vis. Exp. 2012, 10, 3791. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer Sequence |
|---|---|
| C/EBPα | F: 5′-TGGACAAGAACAGCAACGAGTA-3′ R: 5′-ATTGTCACTGGTCAGCTCCAG-3′ |
| SREBP1c | F: 5′-GCGCCTTGACAGGTGAAGTC-3′ R: 5′-GCCAGGGAAGTCACTGTCTTG-3′ |
| FAS | F: 5′-CCCCTGATGAAGAAGGATCA-3′ R: 5′-ACTCCACAGGTGGGAACAAG-3′ |
| CPT1A | F: 5′-CCTCCGTAGCTGACTCGGTA-3′ R: 5′-GGAGTGACCGTGAACTGAAA-3′ |
| PPIA | F: 5′-CCCACCGTGTTCTTCGACATT-3′ R: 5′-GGACCCGTATGCTTTAGGATGA-3′ |
Sample Availability: Samples of the compounds are not available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balkrishna, A.; Gohel, V.; Singh, R.; Joshi, M.; Varshney, Y.; Srivastava, J.; Bhattacharya, K.; Varshney, A. Tri-Herbal Medicine Divya Sarva-Kalp-Kwath (Livogrit) Regulates Fatty Acid-Induced Steatosis in Human HepG2 Cells through Inhibition of Intracellular Triglycerides and Extracellular Glycerol Levels. Molecules 2020, 25, 4849. https://doi.org/10.3390/molecules25204849
Balkrishna A, Gohel V, Singh R, Joshi M, Varshney Y, Srivastava J, Bhattacharya K, Varshney A. Tri-Herbal Medicine Divya Sarva-Kalp-Kwath (Livogrit) Regulates Fatty Acid-Induced Steatosis in Human HepG2 Cells through Inhibition of Intracellular Triglycerides and Extracellular Glycerol Levels. Molecules. 2020; 25(20):4849. https://doi.org/10.3390/molecules25204849
Chicago/Turabian StyleBalkrishna, Acharya, Vivek Gohel, Rani Singh, Monali Joshi, Yash Varshney, Jyotish Srivastava, Kunal Bhattacharya, and Anurag Varshney. 2020. "Tri-Herbal Medicine Divya Sarva-Kalp-Kwath (Livogrit) Regulates Fatty Acid-Induced Steatosis in Human HepG2 Cells through Inhibition of Intracellular Triglycerides and Extracellular Glycerol Levels" Molecules 25, no. 20: 4849. https://doi.org/10.3390/molecules25204849
APA StyleBalkrishna, A., Gohel, V., Singh, R., Joshi, M., Varshney, Y., Srivastava, J., Bhattacharya, K., & Varshney, A. (2020). Tri-Herbal Medicine Divya Sarva-Kalp-Kwath (Livogrit) Regulates Fatty Acid-Induced Steatosis in Human HepG2 Cells through Inhibition of Intracellular Triglycerides and Extracellular Glycerol Levels. Molecules, 25(20), 4849. https://doi.org/10.3390/molecules25204849