3.2. Methods
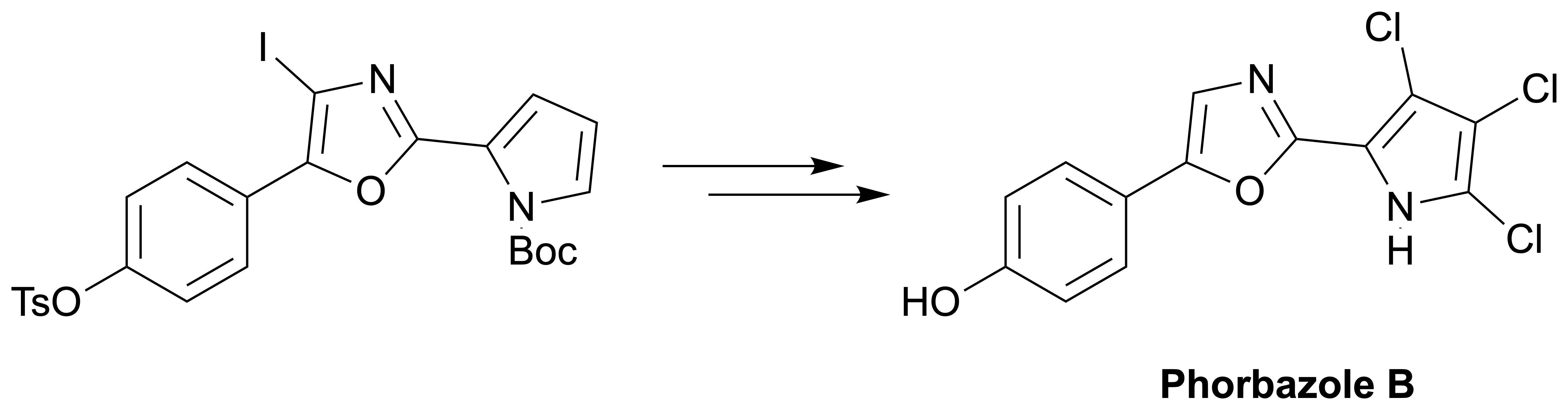
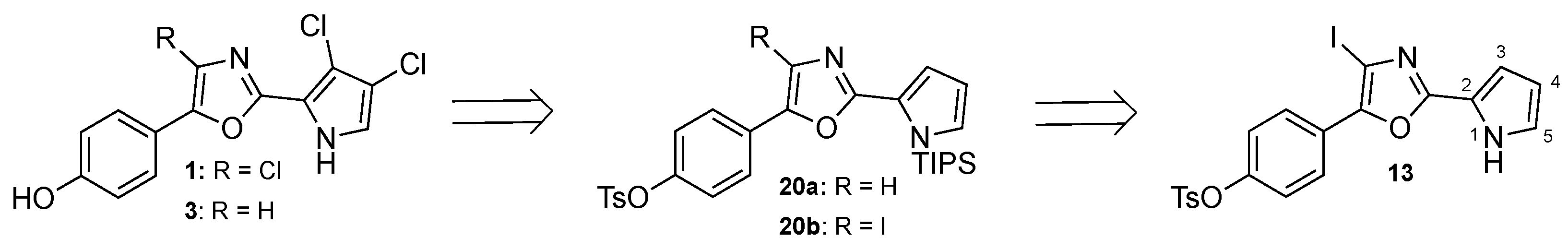
3.2.1. Synthesis of Phorbazole B (2)
The trichlorinated pyrrole 19 (7.65 mg, 12.5 µmol) was dissolved in ethanol (0.2 mL) and 6 M aqueous NaOH (50 µL) was added. The reaction mixture was heated to reflux, zinc (12 mg, 190 µmol) was added and heating was continued for 1 h. To the reaction mixture 1 mL 10% NaHCO3 was added and extracted with 3 × 1 mL CHCl3 on a Biotage phase separator with a Na2SO4 filter plug. The solution was evaporated to give the title compound.
Colorless solid; yield 3.58 mg (87%);
1H-NMR (400 MHz, (CD
3)
2SO) δ = 13.57 (s, 1H), 9.86 (s, 1H), 7.63 (d,
J = 7.7, 2H), 7.60 (s, 1H),6.89 (d,
J = 7.7, 2H);
13C-NMR (101 MHz, (CD
3)
2SO) δ = 158.0, 151.6, 150.3, 125.7, 121.1, 118.3, 115.9, 115.8, 115.0, 110.0, 108.5; HRMS (ESI)
m/
z calcd. for C
13H
6O
2N
235Cl
3 [M − H]
−: 326.9500; found: 326.9496. The NMR spectra in (CD
3)
2SO (
Table S1) matched the original spectra [
1].
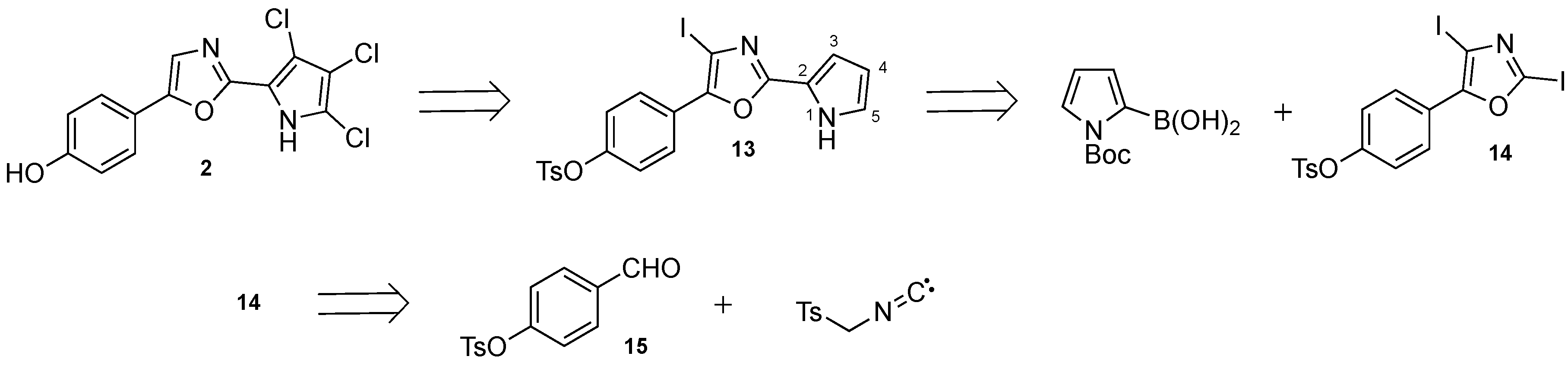
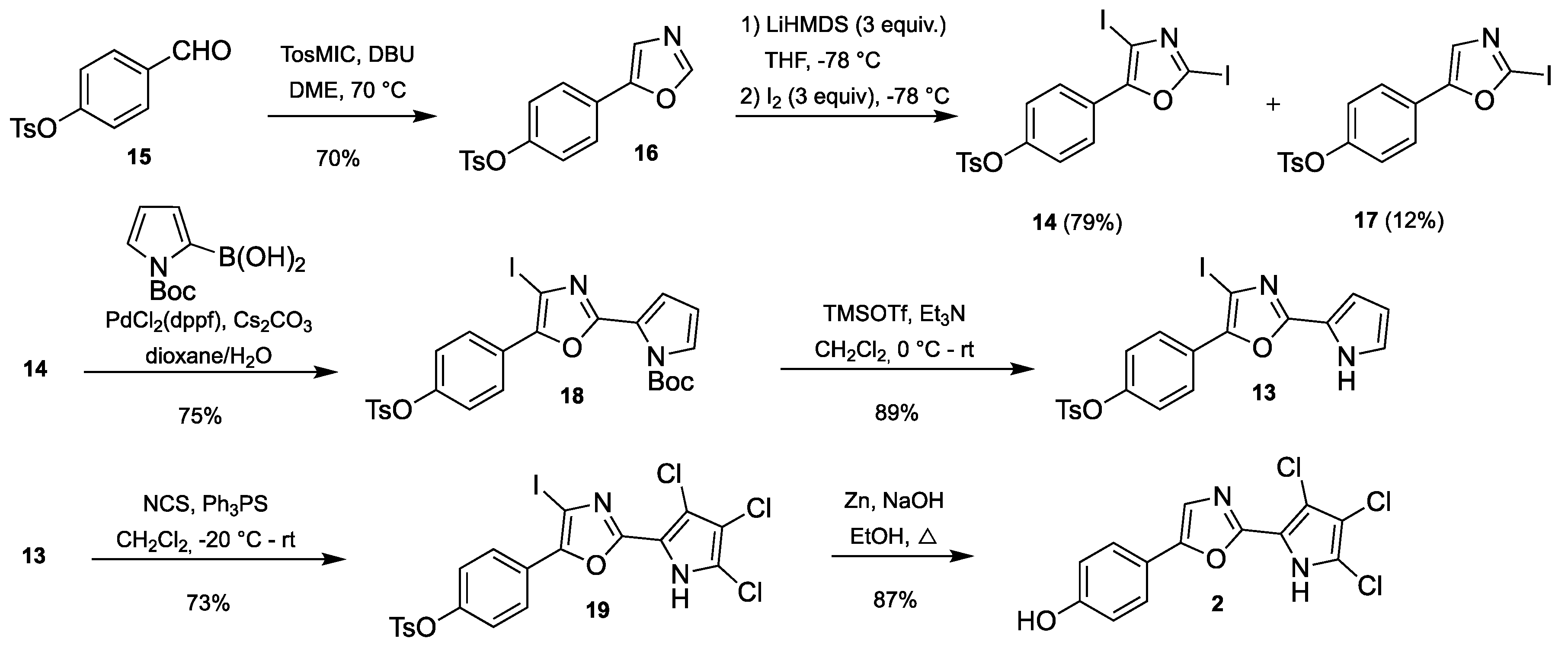
3.2.2. Synthesis of 4-(4-Iodo-2-(1H-pyrrol-2-yl)oxazol-5-yl)phenyl 4-methyl-benzenesulfonate (13)
To a solution of Boc-protected pyrrole 18 (530 mg, 0.87 mmol) in anhydrous CH2Cl2 (5 mL) at 0 °C was slowly added Et3N (1.20 mL, 8.74 mmol) and TMSOTf (1.58 mL, 8.74 mmol). The reaction mixture was stirred at room temperature for 16 h, after which no starting material could be observed by TLC analysis. The reaction mixture was diluted with ethyl acetate, quenched with cold water and extracted with ethyl acetate (3 × 20 mL). The organic phase was concentrated and dissolved in a small amount of CH2Cl2 before adsorption on a Biotage snaplet precolumn and purified by flash chromatography on a Biotage SNAP Ultra column using an eluent with 0–80% ethyl acetate in heptane to give the title compound.
Orange solid; yield 395 mg (89%); mp 53.4–54.7 °C; 1H-NMR (400 MHz, CDCl3) δ = 9.58 (bs, 1H), 7.93 (d, J = 8.8, 2H), 7.74 (d, J = 8.2, 2H), 7.34 (d, J = 8.0, 2H), 7.08 (d, J = 8.8, 2H), 7.00–6.96 (m, 1H), 6.91–6.87 (m, 1H), 6.34–6.30 (m, 1H), 2.46 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ = 157.5, 149.6, 147.4, 145.7, 132.3, 130.0, 128.7, 127.0, 126.4, 122.9, 122.6, 119.1, 111.7, 110.8, 79.7, 29.9 (grease), 21.9: HRMS (ESI) m/z calcd. for C20H16O4N2IS [M + H]+: 506.9870; found: 506.9865.
3.2.3. Synthesis of 4-(2,4-diiodooxazol-5-yl)phenyl 4-methylbenzenesulfonate (14)
Oxazole 16 (5.00 g, 15.9 mmol) was dissolved in anhydrous THF (75 mL) and cooled to −78 °C. Freshly prepared LiHMDS (1M in THF/hexanes, 47.6 mL, 47.6 mmol. Prepared by adding a 2.5 M n-BuLi solution in hexanes into HMDS in THF) was added dropwise. The resulting mixture was stirred at −78 °C for 1 h, during which the solution turned very viscous. A solution of iodine (12.10 g, 47.6 mmol) in anhydrous THF (10 mL) was added very slowly under thorough stirring. The resulting mixture was heated to room temperature for 1 h before 10% aqueous Na2S2O3 (100 mL) was added to quench the reaction and the resulting two layers were separated. The aqueous layer was extracted with ethyl acetate (2 × 50 mL) and the combined organic layers were washed with saturated NaCl (50 mL), dried over Na2SO4 and evaporated. The resulting residue was purified by flash chromatography on a Biotage SNAP Ultra column using an eluent with 5–40% ethyl acetate in heptane to give the title compound.
Yellow solid; yield 6.99 g (79%); mp 57.3–58.7 °C; 1H-NMR (400 MHz, CDCl3): δ = 7.86 (d, J = 8.9, 2H), 7.73 (d, J = 8.4, 2H), 7.33 (d, J = 8.0, 2H), 7.09 (d, J = 8.9, 2H), 2.46 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 156.0, 150.3, 145.8, 132.3, 130.0, 128.7, 127.4, 125.3, 123.0, 101.4, 80.00, 21.9; HRMS (ESI) m/z calcd for C16H11O4KNI2S [M + K]+: 605.8130; found: 605.8129.
3.2.4. Synthesis of 4-Formylphenyl 4-methylbenzenesulfonate (15)
4-Hydroxy-benzaldehyde (20.33 g, 166 mmol) and DMAP (68 mg, 0.61 mmol) were dissolved in a mixture of CH2Cl2 (200 mL) and Et3N (75 mL) and cooled to 0 °C. Tosyl chloride (31.7 g, 166 mmol) was dissolved in CH2Cl2 (60 mL) and added via an addition funnel for 20 min. The resulting mixture was left to heat to room temperature overnight and was then poured into water (200 mL). The phases were separated and the organic phase was washed with 1M aqueous HCl (2x100 mL), water (100 mL) and saturated NaCl (100 mL) before drying over MgSO4 and evaporation. The residue was dissolved in a small amount of ethyl acetate and heated to reflux before cooling to -20 °C overnight. The precipitated crystals were filtered off and dried to afford 25.90 g of the title compound. The mother liquid was evaporated and the crystallization was repeated to give an additional 2.81 g of the title compound. The remaining mother liquid was evaporated and crystallized from 10–15% water in ethanol to give an additional 9.10 g of the title compound.
Colorless solid; yield 37.81 g (82%); mp 72.5–73.5 °C; 1H-NMR (400 MHz, CDCl3): δ = 9.96 (s, 1H), 7.82 (d, J = 8.2, 2H), 7.71 (d, J = 7.9, 2H), 7.32 (d, J = 8.0, 2H), 7.16 (d, J = 8.2, 2H), 2.44 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ = 190.7, 154.0, 146.0, 134.9, 132.1, 131.4, 130.1, 128.6, 123.2, 21.8; HRMS (ESI) m/z calcd for C14H11O4S [M − H]−: 375.0384; found: 375.0384.
3.2.5. Synthesis of 4-(Oxazol-5-yl)phenyl 4-methylbenzenesulfonate (16)
Aldehyde 15 (2.0 g, 7.24 mmol) and TosMIC (1.48 g, 7.60 mmol) were dissolved in DME (10 mL). DBU (1.14 mL, 7.60 mmol) was added using a syringe and the resulting mixture was heated to reflux until TLC analysis showed that the starting material had been consumed (approx. 5 h). The reaction mixture was evaporated, re-dissolved in a small amount of acetone, adsorbed onto a Biotage snaplet precolumn and purified by flash chromatography on a Biotage SNAP Ultra column using an eluent with 15–100% ethyl acetate in heptane to give the title compound.
Pale yellow solid; yield 1.59 g (70%); mp 112.2–114.1 °C; 1H-NMR (400 MHz, CDCl3) δ = 7.90 (s, 1H), 7.72 (d, J = 8.3, 2H), 7.57 (d, J = 8.8, 2H), 7.32 (m, 3H), 7.05 (d, J = 8.8, 2H), 2.45 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ = 150.9, 150.5, 149.6, 145.7, 132.3, 130.0, 128.7, 126.8, 125.8, 123.2, 122.2, 21.9; HRMS (ESI) m/z calcd for C16H14NO4S+ [M + H]+: 354.0197, found 354.0201.
3.2.6. Synthesis of 4-(2-Iodooxazol-5-yl)phenyl 4-methylbenzenesulfonate (17)
The title compound was isolated as a side-product during the synthesis of 14.
Yellow solid; yield 837 mg (12%); mp 39.1–41.4 °C; 1H-NMR (400 MHz, CDCl3): δ = 7.71 (d, J = 8.4, 2H), 7.51 (d, J = 8.8, 2H), 7.31 (d, J = 8.0, 2H), 7.24 (s, 1H), 7.04 (d, J = 8.8, 2H), 2.46 (s, 3H); 13C-NMR (101 MHz, CDCl3): δ = 152.7, 149.9, 146.8, 145.8, 132.3, 130.0, 128.7, 126.0, 125.5, 124.0, 123.4, 29.8 (grease), 21.9; HRMS (ESI) m/z calcd for C16H13O4NIS [M + H]+: 441.9604; found: 441.9604.
3.2.7. Synthesis of t-Butyl 2-(4-iodo-5-(4-(tosyloxy)phenyl)oxazol-2-yl)-1H-pyrrole-1-carboxylate (18)
Diiodooxazol 16 (3.22 g, 5.77 mmol), Cs2CO3 (5.64 g, 17.3 mmol), (N-Boc-pyrrol-2-yl)boronic acid (1.46 g, 6.93 mmol) and PdCl2(dppf)·CH2Cl2 (235 mg, 0.289 mmol) were dissolved in a mixture of degassed dioxane (18 mL) and degassed water (6 mL). The solution was stirred at room temperature for 6 h under argon upon which TLC analysis showed some remaining starting material. Additional boronic acid (0.5 equiv.) and catalyst (5 mol%) were added and the resulting mixture was stirred for 42 h after which TLC still showed starting material left. Additional boronic acid (0.5 equiv.) and catalyst (5 mol%) were added and the mixture was stirred for 18 h, after which TLC showed no sign of starting material. The mixture was filtered through a plug of celite, diluted with ethyl acetate, washed with water (20 mL) and saturated NaCl (20 mL), dried over Na2SO4 and evaporated. The residue was purified by flash chromatography on a Biotage SNAP Ultra column using an eluent with 0–80% ethyl acetate in heptane to give the title compound.
Orange solid; yield 2.64 g (75%); mp 73.1–73.3 °C; 1H-NMR (400 MHz, CDCl3): δ = 7.94 (d, J = 8.8, 2H), 7.73 (d, J = 7.9, 2H), 7.44 (s, 1H), 7.32 (d, J = 8.1, 2H), 7.26 (s, 1H), 7.15–7.02 (m, 2H), 6.75 (s, 1H), 6.29 (d, J = 3.5, 1H), 2.45 (s, 3H), 1.44 (s, 9H); 13C-NMR (101 MHz, CDCl3): δ = 156.6, 149.8, 149.2, 148.2, 145.7, 132.4, 130.0, 128.7, 127.2, 126.4, 125.3, 122.9, 119.7, 119.7, 111.3, 85.1, 79.3, 27.8, 21.9; HRMS (ESI) m/z calcd for C25H24O6N2IS [M + H]+: 607.0394; found: 607.0394.
3.2.8. Synthesis of 4-(4-Iodo-2-(3,4,5-trichloro-1H-pyrrol-2-yl)oxazol-5-yl)phenyl 4-methylbenzenesulfonate (19)
2-(4-Iodooxazole)pyrrole 13 (101 mg, 0.20 mmol) and triphenylphosphine sulfide (12 mg, 40 µmol) were dissolved in CH2Cl2 (0.8 mL) and cooled to −20 °C. NCS (82 mg, 0.61 mmol) was added in one portion and the reaction mixture was allowed to heat to room temperature for 10 min. A small aliquot was subjected to HRMS analysis, which revealed that the trichlorination was not complete. The reaction mixture was again cooled to −20 °C and additional NCS (5 mg, 37 µmol) was added. After heating to room temperature no traces of dichlorinated compound(s) could be observed, and the reaction mixture was directly applied onto a Biotage SNAP Ultra column and purified by flash chromatography using 40–100% CH2Cl2 in heptane to give the title compound. This material was used in the next step without further purification; however, 1H-NMR analysis revealed the presence of minor amounts of residual solvent/and or grease. A small sample was purified further by normal and C18 flash chromatography for characterization by 1H- and 13C-NMR analysis.
Pale orange solid; yield 88 mg (73%); mp 97.7–99.4 °C; 1H-NMR (400 MHz, (CD3)2SO) δ = 8.02 (d, J = 8.9, 2H), 7.78 (d, J = 8.2, 2H), 7.50 (d, J = 8.2, 2H), 7.24 (d, J = 8.9, 2H), 2.44 (s, 3H); 13C-NMR (101 MHz, CDCl3) δ = 154.7, 149.4, 147.4, 146.5, 131.7, 130.8, 128.8, 127.3, 126.4, 123.3, 117.1, 115.0, 111.8, 109.2, 83.6, 21.7; HRMS (ESI) m/z calcd. for C20H11O4N2S35Cl237ClI [M − H]−: 608.8526; found: 608.8514.
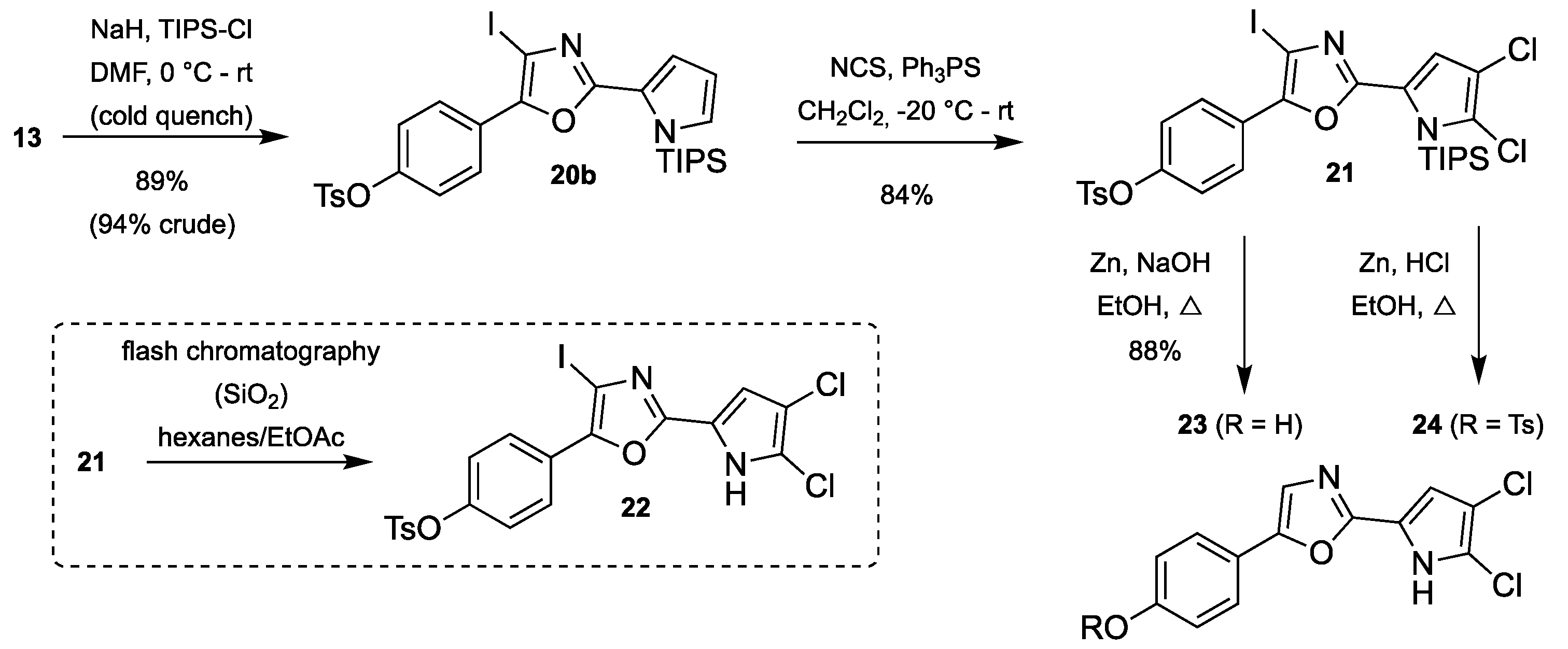
3.2.9. Synthesis of 4-(4-Iodo-2-(1-(triisopropylsilyl)-1H-pyrrol-2-yl)oxazol-5-yl)-phenyl 4-methylbenzenesulfonate (20b)
2-(4-Iodo-oxazole)pyrrole 13 (479 mg, 0.95 mmol) was dissolved in anhydrous DMF (20 mL) and the solution was cooled at 0 °C in an ice/water bath. NaH (60% in mineral oil, 92 mg, 2.28 mmol) was added slowly and the color of the solution changed from green to orange. The flask was flushed with argon and stirring was continued for 15 min before TIPS-Cl (0.25 mL, 1.14 mmol) was added dropwise upon which the reaction mixture changed to a peachy color. Stirring was continued for 15 min at 0 °C before the cooling bath was removed and stirring continued at room temperature for 2 h when TLC analysis showed complete conversion of the starting material (TLC sample quenched with ice and extracted with ethyl acetate before spotting). The reaction mixture was cooled on ice, diluted with ethyl acetate (20 mL) and quenched by slow addition of crushed ice. Phases were separated, and the aqueous phase was extracted with ethyl acetate (2 × 25 mL). The combined organic phases were washed with water (25 mL) and saturated NaCl (25 mL), dried over Na2SO4, filtered and evaporated under reduced pressure. The residue was purified by flash chromatography using a Biotage SNAP Ultra column using 0–20% ethyl acetate in heptane to give the title compound. For the most part, the crude product from this step was used in the next step without further purification.
Colorless oil; 558 mg (89%); 1H-NMR (400 MHz, CDCl3) δ = 7.94 (d, J = 8.8, 2H), 7.74 (d, J = 8.4, 2H), 7.33 (d, J = 8.1, 2H), 7.12–7.02 (m, 4H), 6.35 (t, J = 3.1, 1H), 2.46 (s, 3H), 1.86 (hept, J = 7.6, 3H), 1.13 (d, J = 7.6, 18H); 13C-NMR (101 MHz, CDCl3) δ = 158.0, 149.4, 147.0, 145.7, 132.4, 131.1, 130.0, 128.7, 126.9, 126.7, 124.4, 122.8, 117.6, 111.1, 79.3, 29.84 (impurity), 21.9, 18.4, 13.5; HRMS (ESI) m/z calcd. for C29H35O4N2SiSINa [M + Na]+: 685.1024; found: 685.1022.
3.2.10. Synthesis of 4-(2-(4,5-Dichloro-1-(triisopropylsilyl)-1H-pyrrol-2-yl)-4-iodo-oxazol-5-yl)phenyl 4-methylbenzenesulfonate (21)
TIPS-protected pyrrole 20b (100 mg, 0.161 mmol) and triphenylphosphine sulfide (10 mg, 32 µmol) were dissolved in CH2Cl2 (0.8 mL) and cooled to −20 °C. NCS (44 mg, 0.330 mmol) was added in one batch, and the reaction mixture was allowed to warm to room temperature for 10 min. A small aliquot was subjected to HRMS analysis, which showed that complete dichlorination had occurred. The reaction mixture was directly applied onto a Biotage SNAP Ultra column and purified using flash chromatography using 0–100% CH2Cl2 in heptane to give the title compound.
Colorless oil; yield 99 mg (84%); 1H-NMR (400 MHz, CDCl3) δ = 7.91 (d, J = 8.8, 2H), 7.74 (d, J = 8.1, 2H), 7.34 (d, J = 8.1, 2H), 7.09 (d, J = 8.8, 2H), 6.75 (s, 1H), 2.46 (s, 3H), 1.72 (hept, J = 7.5, 3H), 1.10 (d, J = 7.5, 18H); 13C-NMR (101 MHz, CDCl3) δ = 157.1, 149.9, 148.7, 145.8, 132.4, 130.0, 128.7, 127.2, 126.1, 123.8, 123.4, 123.0, 118.5, 113.8, 79.1, 29.9 (imp.), 21.9, 18.5, 13.7; HRMS (ESI) m/z calcd. for C29H33N2O4N2SiS35Cl2IK [M + K]+: 768.9984; found: 768.9987.
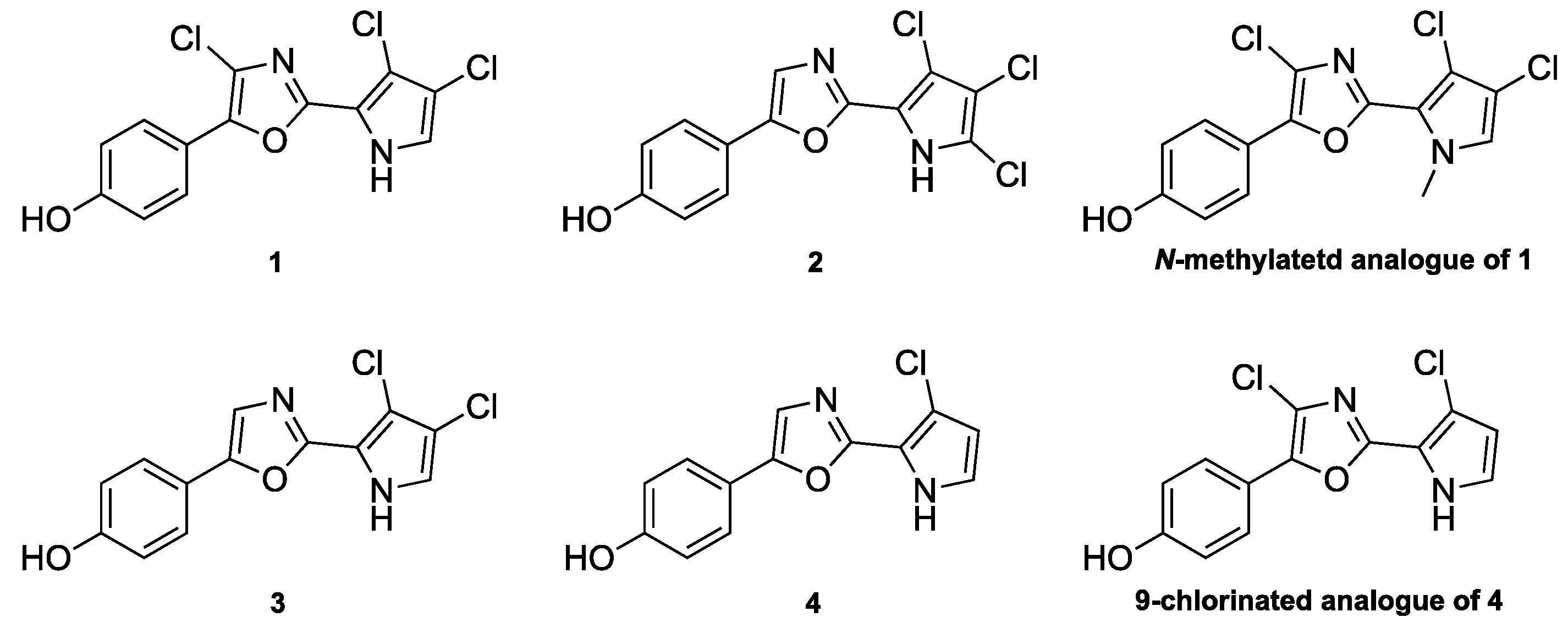
3.2.11. Synthesis of 4-(2-(4,5-Dichloro-1H-pyrrol-2-yl)oxazol-5-yl)phenol (23)
TIPS-protected dichlorinated pyrrole 21 (48 mg, 66 µmol) was dissolved in ethanol (0.7 mL) at 70 °C and 20% aqueous NaOH (0.3 mL) was added, upon which the color changed from clear to orange. The mixture was heated to reflux and, after 5 min, zinc (64 mg, 0.984 mmol) was added and heating was continued for 45 min. The reaction mixture was filtered through a pad of Celite and partitioned between ethyl acetate (10 mL) and water (10 mL). The aqueous phase was extracted with ethyl acetate (2 × 10 mL) and the combined organic phases were washed with saturated NaCl, dried over Na2SO4 and evaporated. The residue was adsorbed onto a Biotage snaplet precolumn using CH2Cl2 and purified by flash chromatography using a Biotage SNAP Ultra column using 0–100% ethyl acetate in heptane to give the title compound.
Colorless solid; yield 17 mg (88%); mp 246 °C (dec.); 1H-NMR (400 MHz, (CD3)2SO) δ = 13.12 (s, 1H), 9.82 (s, 1H), 7.62 (d, J = 8.4, 2H), 7.51 (s, 1H), 6.94–6.75 (m, 3H); 13C-NMR (101 MHz, (CD3)2SO) δ = 157.8, 153.0, 150.1, 125.6, 121.1, 118.9, 118.5, 115.8, 114.7, 109.12, 109.11; HRMS (ESI) m/z calcd. for C13H9O2N235Cl2 [M + H]+: 295.0036; found: 295.0037.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





