
Novel Nucleoside Analogues as Effective Antiviral Agents for Zika Virus Infections

 , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
2.1. Compound Selection
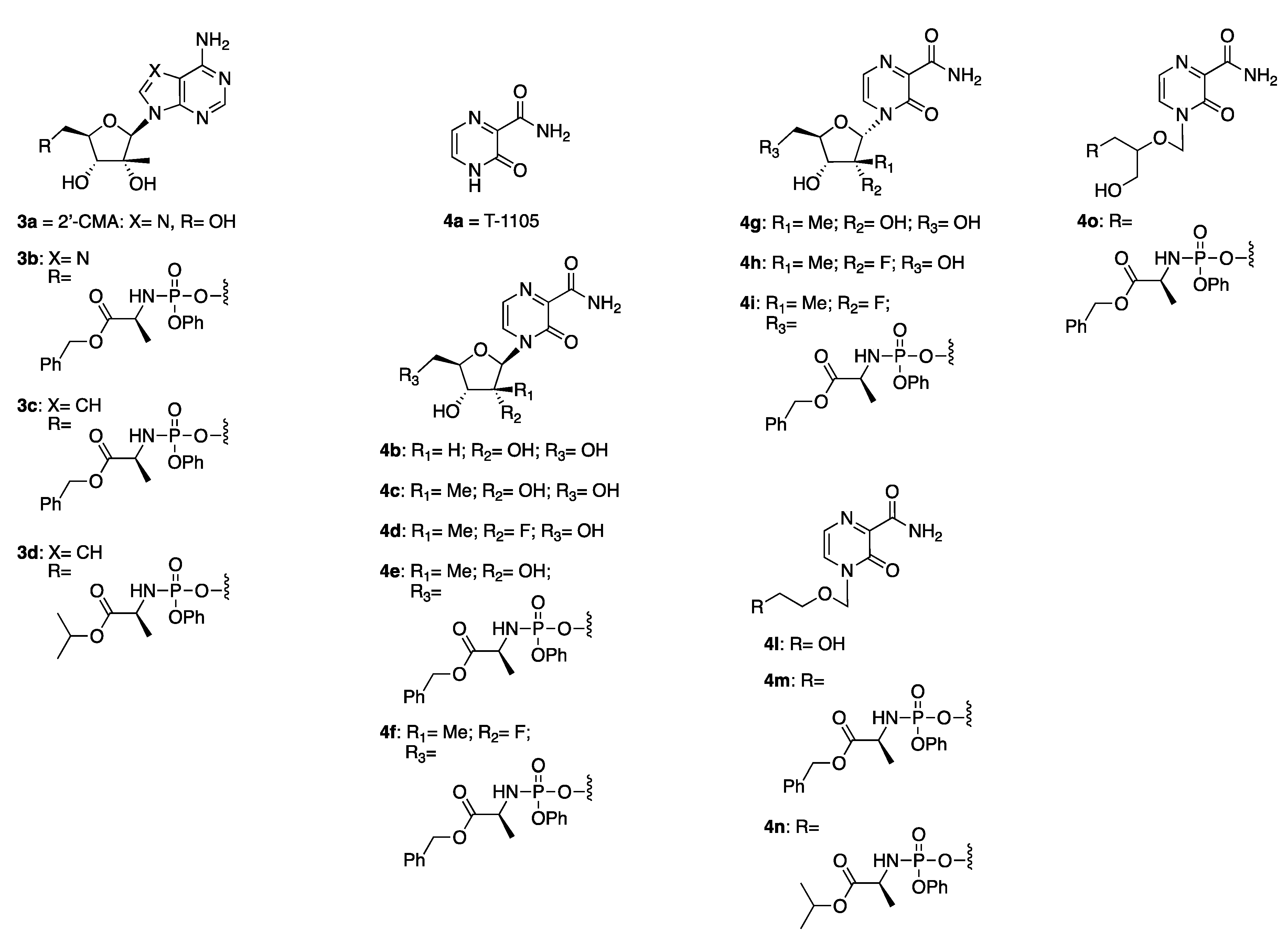
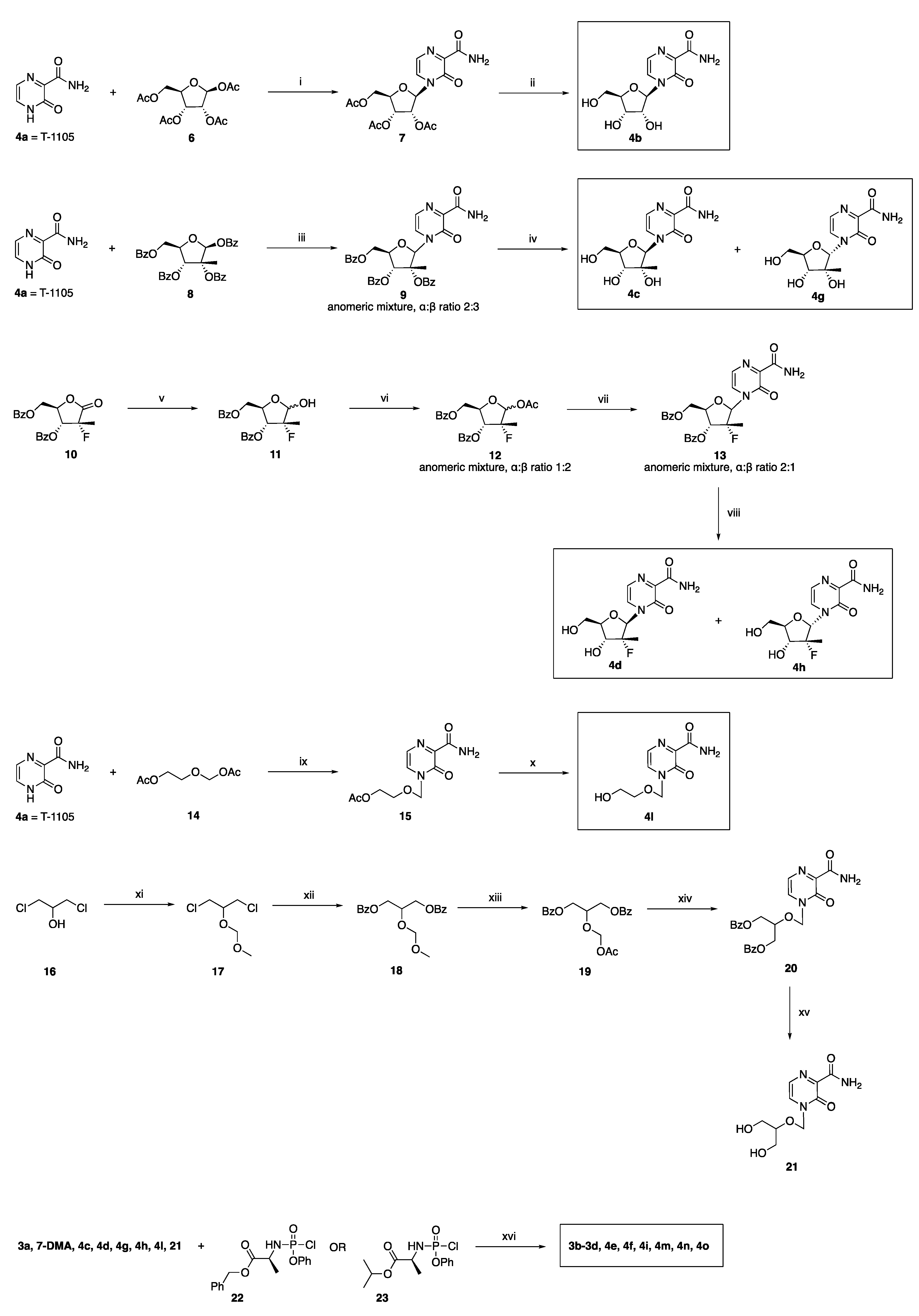
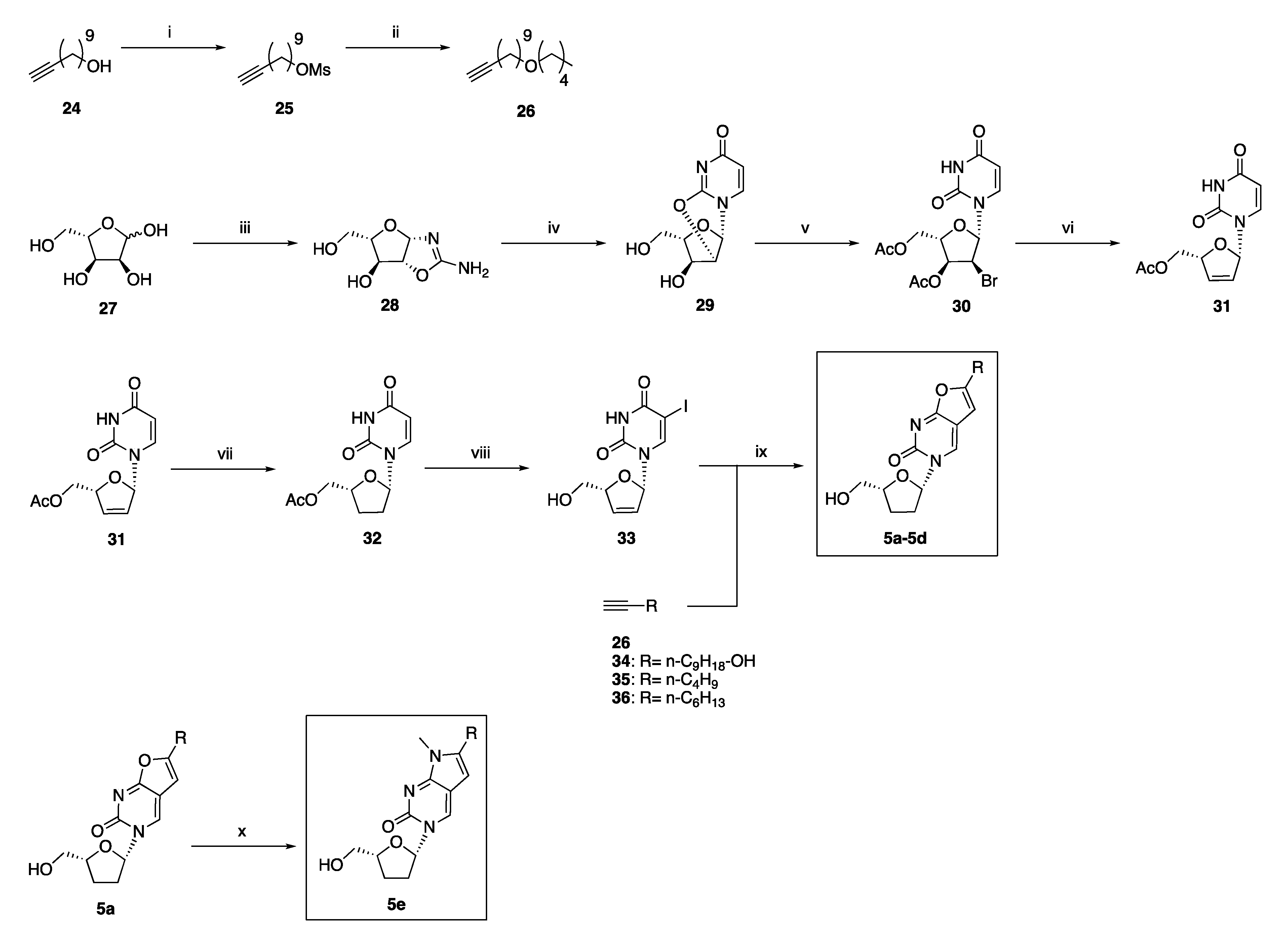
2.2. Synthetic Chemistry
2.3. Antiviral Studies
3. Materials and Methods
3.1. Synthetic Chemistry
3.1.1. General Methodologies
- -
- Linear gradient standard method (A): 90% A (0.1 min); 90–0% A (2.5 min); 0% A (0.3 min); 90% A (0.1 min); flow rate 0.5 mL/min.
- -
- Linear gradient standard method (B): 90% A (0.1 min); 90–0% A (2.1 min); 0% A (0.8 min); 90% A (0.1 min); flow rate 0.5 mL/min.
- -
- Linear gradient standard method (C): 90% A (0.1 min); 90–0% A (1.5 min); 0% A (1.4 min); 90% A (0.1 min); flow rate 0.5 mL/min.
- -
- Linear gradient standard method (D): 99% A (0.1 min); 99–80% A (2.1 min); 0% A (0.8 min); 99% A (0.1 min); flow rate 0.5 mL/min.
3.1.2. General Method for the Preparation of Novel ProTides 3b–3d, 4e, 4f, 4i, 4m–4o
2′-C-Methyladenosine 5′-O-[phenyl-(benzyloxy-l-alaninyl)] Phosphate 3b
7-Deaza-2′-C-methyladenosine 5′-O-[phenyl-(benzyloxy-l-alaninyl)] Phosphate 3c
7-Deaza-2′-C-methyladenosine 5′-O-[phenyl-(isopropoxy-l-alaninyl)] Phosphate 3d
4-(2′-C-Methyl-β-d-ribofuranosyl)-3-oxo-3,4-dihydropyrazine-2-carboxamide 5′-O-[phenyl-(benzyloxy-l-alaninyl)] Phosphate 4e
4-[(2’-R)-2’-C-Deoxy-2’-C-fluoro-2′-C-methyl-β-d-ribofuranosyl]-3-oxo-3,4-dihydropyrazine-2-carboxamide 5′-O-[phenyl-(benzyloxy-l-alaninyl)] Phosphate 4f
4-[(2’-R)-2’-C-Deoxy-2’-C-fluoro-2′-C-methyl-α -d-ribofuranosyl]-3-oxo-3,4-dihydropyrazine-2-carboxamide 5′-O-[phenyl-(benzyloxy-l-alaninyl)] Phosphate 4i
Benzyl ((2-((3-carbamoyl-2-oxopyrazin-1(2H)-yl)methoxy)ethoxy)(phenoxy)phosphoryl)-l-alaninate 4m
Isopropyl ((2-((3-carbamoyl-2-oxopyrazin-1(2H)-yl)methoxy)ethoxy)(phenoxy) phosphoryl)-l-alaninate 4n
Benzyl ((2-((3-carbamoyl-2-oxopyrazin-1(2H)-yl)methoxy)-3-hydroxypropoxy)(phenoxy) phosphoryl)-l-alaninate 4o
3.1.3. Preparation of Novel 2′-Deoxy-2′-α-fluoro-2′-β-C-methyl-ribofuranosyl Analogues 4d and 4h
4-[(2’-R)-2’-C-Deoxy-2’-C-fluoro-2′-C-methyl-β-D-ribofuranosyl]-3-oxo-3,4-dihydropyrazine-2-carboxamide 4d
4-[(2’-R)-2’-C-Deoxy-2’-C-fluoro-2′-C-methyl-α-D-ribofuranosyl]-3-oxo-3,4-dihydropyrazine-2-carboxamide 4h
3.1.4. Preparation of 4-((2-Hydroxyethoxy)methyl)-3-oxo-3,4-dihydropyrazine-2-carboxamide 4l
3.1.5. Preparation of 4-(((1,3-Dihydroxypropan-2-yl)oxy)methyl)-3-oxo-3,4-dihydropyrazine-2-carboxamide 21
3.1.6. General Procedure for the Preparation of Novel 3-(2′,3′-Dideoxy-ribo-β-l-furanosyl)-6-substituted-furo[2,3d]pyrimidin-2-(3H)one Analogues 5b and 5c
3-(2′,3′-Dideoxy-ribo-β-l-furanosyl)-6-(1-nonanol) furo[2,3-d]pyrimidin- 2(3H)-one 5b
3-(2′,3′-Dideoxy-ribo-β-l-furanosyl)-6-(n-butil)furo[2,3-d]pyrimidin- 2(3H)-one 5c
3.2. Antiviral Studies
3.2.1. Antivirals
3.2.2. Cell Culture
3.2.3. Virus Culture
3.2.4. Viability Assay
3.2.5. Cell Adherence Assay
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dick, G.W. Zika virus. II. Pathogenicity and physical properties. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 521–534. [Google Scholar] [CrossRef]
- Ferraris, P.; Yssel, H.; Missé, D. Zika virus infection: An update. Microbes Infect. 2019, 19, 30052–30058. [Google Scholar] [CrossRef] [PubMed]
- Musso, D.; Ko, A.I.; Baud, D. Zika virus infection—After the pandemic. N. Engl. J. Med. 2019, 381, 1444–1457. [Google Scholar] [CrossRef]
- WHO. Zika: The continuing threat. Bull. World Health Organ. 2019, 97, 6–7. [Google Scholar] [CrossRef] [PubMed]
- Song, B.-H.; Yun, S.-I.; Woolley, M.; Lee, M.-L. Zika virus: History, epidemiology, transmission, and clinical presentation. J. Neuroimmunol. 2017, 380, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.J.; Carlson, C.J.; Mordecai, E.A.; Johnson, L.R. Global expansion and redistribution of Aedes-borne virus transmission risk with climate change. PLoS Negl. Trop. Dis. 2019, 13, e0007213. [Google Scholar] [CrossRef]
- Rasmussen, S.A.; Jamieson, D.J.; Honein, M.A.; Petersen, L.R. Zika virus and birth defects—Reviewing the evidence for causality. N. Engl. J. Med. 2016, 374, 1981–1987. [Google Scholar] [CrossRef]
- Krauer, F.; Riesen, M.; Reveiz, L.; Oladapo, O.T.; Martínez-Vega, R.; Porgo, T.V.; Haefliger, A.; Broutet, N.J.; Low, N. Zika virus infection as a cause of congenital brain abnormalities and Guillain-Barré syndrome: Systematic review. PLoS Med. 2017, 14, e1002203. [Google Scholar] [CrossRef]
- Broutet, N.; Krauer, F.; Riesen, M.; Khalakdina, A.; Almiron, M.; Aldighieri, S.; Espinal, M.; Low, N.; Dye, C. Zika Virus as a Cause of Neurologic Disorders. N. Engl. J. Med. 2016, 374, 1506–1509. [Google Scholar] [CrossRef]
- Masmejan, S.; Baud, D.; Musso, D.; Panchaud, A. Zika virus, vaccines, and antiviral strategies. Expert Rev. Anti-Infect. Ther. 2018, 16, 471–483. [Google Scholar] [CrossRef]
- Duarte, G.; Moron, A.; Timerman, A.; Fernandes, C.; Mariani Neto, C.; Almeida Filho, G.; Werner Junior, H.; Espírito Santo, H.; Steibel, J.; Bortoletti Filho, J.; et al. Zika virus infection in pregnant women and microcephaly. Rev. Bras. Ginecol. Obstet. RBGO Gynecol. Obstet. 2017, 39, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Chong, H.Y.; Leow, C.Y.; Abdul Majeed, A.B.; Leow, C.H. Flavivirus infection—A review of immunopathogenesis, immunological response, and immunodiagnosis. Virus Res. 2019, 274, a197770. [Google Scholar] [CrossRef] [PubMed]
- Seley-Radtke, K.L.; Yates, M.K. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antivir. Res. 2018, 154, 66–86. [Google Scholar] [CrossRef] [PubMed]
- Yates, M.K.; Seley-Radtke, K.L. The evolution of antiviral nucleoside analogues: A review for chemists and non-chemists. Part II: Complex modifications to the nucleoside scaffold. Antivir. Res. 2019, 162, 5–21. [Google Scholar] [CrossRef]
- Mehellou, Y.; Rattan, H.S.; Balzarini, J. The ProTide prodrug technology: From the concept to the clinic. J. Med. Chem. 2018, 61, 2211–2226. [Google Scholar] [CrossRef]
- Del Sarto, J.L.; Rocha, R.P.F.; Bassit, L.; Olmo, I.G.; Valiate, B.; Queiroz-Junior, C.M.; Pedrosa, C.; Ribeiro, F.M.; Guimarães, M.Z.; Rehen, S.; et al. 7-Deaza-7-fluoro-2’-C-methyladenosine inhibits Zika virus infection and viral-induced neuroinflammation. Antivir. Res. 2020, 180, 104855. [Google Scholar] [CrossRef]
- Sacramento, C.Q.; de Melo, G.R.; de Freitas, C.A.; Rocha, N.; Hoelz, L.V.B.; Miranda, M.; Fintelman-Rodrigues, N.; Marttorelli, A.; Ferreira, A.C.; Barbosa-Lima, G.; et al. The clinically approved antiviral drug sofosbuvir inhibits Zika virus replication. Sci. Rep. 2017, 7, 40920. [Google Scholar] [CrossRef]
- Bassetto, M.; Leyssen, P.; Neyts, J.; Yerukhimovich, M.M.; Frick, D.N.; Brancale, A. Computer-aided identification, synthesis and evaluation of substituted thienopyrimidines as novel inhibitors of HCV replication. Eur. J. Med. Chem. 2016, 123, 31–47. [Google Scholar] [CrossRef]
- Pattnaik, A.; Palermo, N.; Sahoo, B.R.; Yuan, Z.; Hu, D.; Annamalai, A.S.; Vu, H.; Correas, I.; Prathipati, P.K.; Destache, C.J.; et al. Discovery of a non-nucleoside RNA polymerase inhibitor for blocking Zika virus replication through in silico screening. Antivir. Res. 2018, 151, 78–86. [Google Scholar] [CrossRef]
- Giancotti, G.; Rigo, I.; Pasqualetto, G.; Young, M.T.; Neyts, J.; Rocha-Pereira, J.; Brancale, A.; Ferla, S.; Bassetto, M. A new antiviral scaffold for human norovirus identified with computer-aided approaches on the viral polymerase. Sci. Rep. 2019, 9, 18413. [Google Scholar] [CrossRef]
- Eyer, L.; Nencka, R.; Huvarová, I.; Palus, M.; Joao Alves, M.; Gould, E.A.; De Clercq, E.; Růžek, D. Nucleoside inhibitors of Zika virus. J. Infect. Dis. 2016, 214, 707–711. [Google Scholar] [CrossRef]
- Zmurko, J.; Marques, R.E.; Schols, D.; Verbeken, E.; Kaptein, S.J.F.; Neyts, J. The Viral Polymerase Inhibitor 7-Deaza-2′-C-Methyladenosine Is a Potent Inhibitor of In Vitro Zika Virus Replication and Delays Disease Progression in a Robust Mouse Infection Model. PLoS Negl. Trop. Dis. 2016, 10, e0004695. [Google Scholar] [CrossRef]
- Slusarczyk, M.; Serpi, M.; Pertusati, F. Phosphoramidates and phosphonamidates (ProTides) with antiviral activity. Antivir. Chem. Chemother. 2018, 26, 2040206618775243. [Google Scholar] [CrossRef] [PubMed]
- Furuta, Y.; Gowen, B.B.; Takahashi, K.; Shiraki, K.; Smee, D.F.; Barnard, D.L. Favipiravir (T-705), a novel viral RNA polymerase inhibitor. Antivir. Res. 2013, 100, 446–454. [Google Scholar] [CrossRef]
- McGuigan, C.; Hinsinger, K.; Farleigh, L.; Pathirana, R.; Bugert, J.J. Novel Antiviral Activity of l-Dideoxy Bicyclic Nucleoside Analogues versus Vaccinia and Measles Viruses In Vitro. J. Med. Chem. 2013, 56, 1311–1322. [Google Scholar] [CrossRef]
- Suksanpaisan, L.; Susantad, T.; Smith, D.R. Characterization of dengue virus entry into HepG2 cells. J. Biomed. Sci. 2009, 16, 17. [Google Scholar] [CrossRef]
- Ke, P.-Y. The Multifaceted Roles of Autophagy in Flavivirus-Host Interactions. Int. J. Mol. Sci. 2018, 19, 3940. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, D.; Li, C.; Zheng, Z.; Fu, M.; Ni, F.; Liu, Y.; Du, T.; Wang, H.; Griffin, G.E.; et al. Characterization of Zika virus endocytic pathways in human glioblastoma cells. Front. Microbiol. 2020, 11, 242. [Google Scholar] [CrossRef]
- Huchting, J.; Winkler, M.; Nasser, H.; Meier, C. Synthesis of T-705-Ribonucleoside and T-705-Ribonucleotide and Studies of Chemical Stability. ChemMedChem 2017, 12, 652–659. [Google Scholar] [CrossRef]
- Vorbrüggen, H.; Bennua, B. Nucleoside syntheses, XXV1) A new simplified nucleoside synthesis. Chem. Ber. 1981, 114, 1279–1286. [Google Scholar] [CrossRef]
- Pierra, C.; Counor, C.; Storer, R.; Gosselin, G. Synthesis and antiviral evaluation of the 2’-C-methyl branched derivative of a nucleoside analog inhibitor of RNA viral infections, T-1106. Collect. Czech. Chem. Commun. 2011, 76, 1327–1333. [Google Scholar] [CrossRef]
- Reddy, P.G.; Chun, B.-K.; Zhang, H.-R.; Rachakonda, S.; Ross, B.S.; Sofia, M.J. Stereoselective Synthesis of PSI-352938: A β-d-2′-Deoxy-2′-α-fluoro-2′-β-C-methyl-3′,5′-cyclic Phosphate Nucleotide Prodrug for the Treatment of HCV. J. Org. Chem. 2011, 76, 3782–3790. [Google Scholar] [CrossRef]
- Puig-de-la-Bellacasa, R.; Giménez, L.; Pettersson, S.; Pascual, R.; Gonzalo, E.; Esté, J.A.; Clotet, B.; Borrell, J.I.; Teixidó, J. Diverse combinatorial design, synthesis and in vitro evaluation of new HEPT analogues as potential non-nucleoside HIV-1 reverse transcription inhibitors. Eur. J. Med. Chem. 2012, 54, 159–174. [Google Scholar] [CrossRef]
- Beauchamp, L.M.; Serling, B.L.; Kelsey, J.E.; Biron, K.K.; Collins, P.; Selway, J.; Lin, J.-C.; Schaeffer, H.J. Effect of Acyclic Pyrimidines Related to 9-[(l,3-Dihydroxy-2-propoxy)methyl]guanine on Herpesviruses. J. Med. Chem. 1988, 31, 144–149. [Google Scholar] [CrossRef]
- Serpi, M.; Madela, K.; Pertusati, F.; Slusarczyk, M. Synthesis of phosphoramidate prodrugs: ProTide approach. Curr. Protoc. Nucleic Acid. Chem. 2013, 53, 15-5. [Google Scholar] [CrossRef]
- McGuigan, C.; Bugert, J.J.; Jones, A.; Pathirana, R.; Farleigh, L.E. Dideoxyribofuranosyl Pyrimidinones as Antiviral Agents. U.S. Patent Appllication US 20130012468 A1, 10 January 2013. [Google Scholar]
- McGuigan, C.; Serpi, M.; Slusarczyk, M.; Ferrari, V.; Pertusati, F.; Meneghesso, S.; Derudas, M.; Farleigh, L.; Zanetta, P.; Bugert, J. Anti-flavivirus Activity of Different Tritylated Pyrimidine and Purine Nucleoside Analogues. ChemistryOpen 2016, 5, 227–235. [Google Scholar] [CrossRef]
- Sherman, K.E.; Rouster, S.D.; Kong, L.X.; Aliota, M.T.; Blackard, J.T.; Dean, G.E. Zika virus replication and cytopathic effects in liver cells. PLoS ONE 2019, 14, e0214016. [Google Scholar] [CrossRef]
- Gobillot, T.A.; Humes, D.; Sharma, A.; Kikawa, C.; Overbaugh, J. The Robust Restriction of Zika Virus by Type-I Interferon in A549 Cells Varies by Viral Lineage and Is Not Determined by IFITM3. Viruses 2020, 12, 503. [Google Scholar] [CrossRef]
- Kim, J.A.; Seong, R.K.; Kumar, M.; Shin, O.S. Favipiravir and Ribavirin Inhibit Replication of Asian and African Strains of Zika Virus in Different Cell Models. Viruses 2018, 10, 72. [Google Scholar] [CrossRef]
- Zhu, Z.; Chu, H.; Wen, L.; Yuan, S.; Chik, K.K.; Yuen, T.; Yip, C.C.; Wang, D.; Zhou, J.; Yin, F.; et al. Targeting SUMO Modification of the Non-Structural Protein 5 of Zika Virus as a Host-Targeting Antiviral Strategy. Int. J. Mol. Sci. 2019, 20, 392. [Google Scholar] [CrossRef]
- Ozdemir, A.; Ark, M. xCELLigence real time cell analysis system: A new method for cell proliferation and cytotoxicity. Niche 2013, 2, 15. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
| Cpd | R1 | R2 | X | R3 | Ar | Ar1 | Ar2 |
| 1a | C4H8 | N=C | Me | 2,5-OH-Ph | - | - | |
| 1b | C4H8 | N=C | Me | 2-OH-Ph | - | - | |
| 1c | C4H8 | N=C | Me | Pyrazine | - | - | |
| 1d | H | H | N=C | Me | 2,5-OH-Ph | - | - |
| 1e | C4H8 | N=C | Me | 2-Pyridine | - | - | |
| 1f | C3H6 | N=C | Me | 2-OH-Ph | - | - | |
| 1g | C4H8 | NHCO | - | 2,5-OH-Ph | - | - | |
| 1h | C4H8 | NHCO | - | 2,5-OH-Ph | - | - | |
| 1i | Me | Me | N=C | Me | 2-OH-Ph | - | - |
| 1l | C4H4 | N=C | Me | 2-OH-Ph | - | - | |
| 1m | C4H4 | NHCO | - | 2-Pyridine | - | - | |
| 1n | H | H | NHCO | - | 2-Pyridine | - | - |
| 1o | Me | Cl | NHCO | - | 2-Pyridine | - | - |
| 2a | - | - | C=O | - | - | 2-(3-Cl)-Benzo[b]thiophene | 2-Thiophene |
| 2b | - | - | C=O | - | - | 2-Benzo[b]thiophene | 2-Thiophene |
| 2c | - | - | C=O | - | - | 2-(3-CF3)-Benzo[b]thiophene | 2-Thiophene |
| 2d | - | - | C=O | - | - | 4-Me-Ph | 2-Thiophene |
| 2e | - | - | C=O | - | - | 2-(3-Cl)-Benzo[b]thiophene | 2-Furan |
| 2f | - | - | C=O | - | - | 2-(3-Cl)-Benzo[b]thiophene | Ph |
| 2g | - | - | C=O | - | - | Ph | Ph |
| 2h | - | - | C=O | - | - | 2-(3-Cl)-Benzo[b]thiophene | 2-Thiazole |
| 2i | - | - | C=O | - | - | 4-Me-Ph | Ph |
| 2l | - | - | SO2 | - | - | Ph | 2-Thiophene |
| 2m | - | - | C=O | - | - | Ph | 2-Thiazole |
| Compound | % Cell Viability, ZIKV–Infected HUH7 | % Cell Viability, HUH7 (Cytotoxicity) | % Cell Viability, ZIKV–Infected Dbtrg | % Cell Viability, Dbtrg (Cytotoxicity) | % Cell Viability, ZIKV–Infected A459 | % Cell Viability, A459 (Cytotoxicity) |
|---|---|---|---|---|---|---|
| 1a | 13.5 | 23.5 | 0 | 15 | n.d. | n.d. |
| 1b | 3.4 | 14.5 | 0 | 17.3 | n.d. | n.d. |
| 1c | 8.8 | 32 | 0 | 18.4 | n.d. | n.d. |
| 1d | 8.3 | 20 | 0 | 24.9 | n.d. | n.d. |
| 1e | 0 | 11.1 | 0 | 14.1 | n.d. | n.d. |
| 1f | 4.9 | 15.3 | 0 | 19.3 | n.d. | n.d. |
| 1g | 6.1 | 46.2 | 0 | 24.3 | n.d. | n.d. |
| 1h | 5.4 | 26.1 | 0 | 21.8 | n.d. | n.d. |
| 1i | 5.7 | 16.1 | 0 | 15.5 | n.d. | n.d. |
| 1l | 12.1 | 22.0 | 0 | 18.2 | n.d. | n.d. |
| 1m | 7.9 | 25.2 | 0 | 12.6 | n.d. | n.d. |
| 1n | 5.1 | 37.9 | 0 | 14.8 | n.d. | n.d. |
| 1o | 5.2 | 30.6 | 0 | 8.9 | n.d. | n.d. |
| 2a | 30.5 ± 15.9 | 117.9 ± 42.2 | n.d. | n.d. | n.d. | n.d. |
| 2b | 25.5 ± 18.2 | 107.4 ± 35.9 | 3.1 ± 3.9 | 94.9 ± 16.2 | n.d. | n.d. |
| 2c | 0.4 ± 0.7 | 108.6 ± 55.9 | 4.1 ± 5.6 | 88.9 ± 18.5 | n.d. | n.d. |
| 2d | 4.5 ± 6.6 | 119.5 ± 47.3 | 2.8 ± 2.8 | 76.9 ± 25.7 | n.d. | n.d. |
| 2e | 11.2 ± 9.9 | 117.8 ± 40.9 | 0.25 ± 0.43 | 80.5 ± 27.2 | n.d. | n.d. |
| 2f | 3.9 ± 1.9 | 75.7 ± 54.3 | 0.8 ± 1.4 | 76 ± 27.4 | n.d. | n.d. |
| 2g | 9.5 ± 7.1 | 160.6 ± 50.6 | 0 | 109.7 ± 4.8 | n.d. | n.d. |
| 2h | 9.8 ± 6.6 | 134.9 ± 51.6 | 2.5 ± 2.2 | 114.7 ± 4.5 | n.d. | n.d. |
| 2i | 4.5 ± 3.3 | 106.4 ± 59.1 | 1.7 ± 1.7 | 96.7 ± 15.2 | n.d. | n.d. |
| 2l | 1.6 ± 3.6 | 110.1 ± 63.5 | 9.2 ± 8.5 | 91.8 ± 14.5 | n.d. | n.d. |
| 2m | 3.3 ± 2.7 | 99.9 ± 62.9 | 11 ± 7.5 | 104.2 ± 10.7 | n.d. | n.d. |
| 3a (2′-CMA) | 31.4 | 98.8 | 5.2 | 107.3 | 40 | 106.9 |
| 3b | 48.2 | 98.2 | 10.2 | 100.5 | 33.6 | 103.5 |
| 3c | 120.4 ± 80.3 | 137.6 ± 35.6 | 53.3 ± 18.9 | 87.9 ± 18.7 | n.d. | n.d. |
| 3d | 101.5 | 107.9 | 8.1 | 115.5 | 59.6 | 112.3 |
| 4a | 0.1 | 100.6 | 4.6 | 97.4 | n.d. | n.d. |
| 4b | 0.1 | 106.8 | 4.6 | 99.2 | n.d. | n.d. |
| 4c | 0.6 | 118.5 | 7.3 | 105.6 | n.d. | n.d. |
| 4d | 0 | 105.6 | 0.4 | 98.7 | n.d. | n.d. |
| 4e | 0 | 90.8 | 0 | 85.9 | n.d. | n.d. |
| 4f | 0.1 | 97.8 | 6.6 | 82.3 | n.d. | n.d. |
| 4g | 0.1 | 117.5 | 0 | 104.9 | n.d. | n.d. |
| 4h | 0 | 106.5 | 0.1 | 99.3 | n.d. | n.d. |
| 4i | 0.2 | 102.5 | 4.3 | 97.8 | n.d. | n.d. |
| 4l | 0 | 101.3 | 4.5 | 98.5 | n.d. | n.d. |
| 4m | 0.2 | 106.4 | 1.2 | 99.6 | n.d. | n.d. |
| 4n | 0.1 | 104.9 | 2.2 | 98 | n.d. | n.d. |
| 4o | 0.1 | 97.8 | 6.6 | 82.3 | n.d. | n.d. |
| 5a | 50.3 | 100.5 | 0 | 75.7 | 0 | 84.8 |
| 5b | 0 | 110.1 | 5.7 | 102.9 | 8.2 | 91.6 |
| 5c | 0 | 104.4 | 3.9 | 94.7 | 0 | 87.6 |
| 5d | 0.2 | 104.3 | 0.4 | 74.5 | 28.6 | 104.7 |
| 5e | 30.6 | 55.1 | 13.6 | 52 | 2.7 | 81.6 |
| Ribavirin | 61.1 ± 26.4 | 158.7 ± 16.9 | 25.1 ± 10.1 | 94.8 ± 21.6 | n.d. | n.d. |
| Control-DMSO | 100 | 100 | 100 | 100 | 100 | 100 |
| Control-ZIKV | 0 | 6 ± 1.4 | 0 | 14.4 ± 4.3 | 0 | 0 |
| Compound | EC50 a,c (μM), HUH7 | CC50 b,c (μM), HUH7 | SI d, HUH7 | EC50 a,c (μM), U251 | CC50 b,c (μM), U251 | SI d, U251 |
|---|---|---|---|---|---|---|
| 3b | 4 ± 0.2 | >20 | >5 | - | - | - |
| 3c | 11.6 ± 1.3 | 211.2 ± 4.6 | 18.2 | 8.2 ± 1.1 | 203.7 ± 16.9 | 24.8 |
| 3d | 8.3 ± 0.9 | 128.2 ± 21.1 | 15.4 | 8.6 ± 6.5 | 13.6 ± 1.7 | 1.6 |
| 5a | 10.2 ± 0.4 | >20 | >2 | - | - | - |
| Sofosbuvir | 5.8 ± 0.8 | 133.5 ± 13.6 | 23 | 7.7 ± 4.5 | 139.1 ± 26.4 | 18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bassetto, M.; Cima, C.M.; Basso, M.; Salerno, M.; Schwarze, F.; Friese, D.; Bugert, J.J.; Brancale, A. Novel Nucleoside Analogues as Effective Antiviral Agents for Zika Virus Infections. Molecules 2020, 25, 4813. https://doi.org/10.3390/molecules25204813
Bassetto M, Cima CM, Basso M, Salerno M, Schwarze F, Friese D, Bugert JJ, Brancale A. Novel Nucleoside Analogues as Effective Antiviral Agents for Zika Virus Infections. Molecules. 2020; 25(20):4813. https://doi.org/10.3390/molecules25204813
Chicago/Turabian StyleBassetto, Marcella, Cecilia M. Cima, Mattia Basso, Martina Salerno, Frank Schwarze, Daniela Friese, Joachim J. Bugert, and Andrea Brancale. 2020. "Novel Nucleoside Analogues as Effective Antiviral Agents for Zika Virus Infections" Molecules 25, no. 20: 4813. https://doi.org/10.3390/molecules25204813
APA StyleBassetto, M., Cima, C. M., Basso, M., Salerno, M., Schwarze, F., Friese, D., Bugert, J. J., & Brancale, A. (2020). Novel Nucleoside Analogues as Effective Antiviral Agents for Zika Virus Infections. Molecules, 25(20), 4813. https://doi.org/10.3390/molecules25204813