
Synthesis, In Silico and In Vitro Assessment of New Quinazolinones as Anticancer Agents via Potential AKT Inhibition




Abstract
1. Introduction
2. Results and Discussion
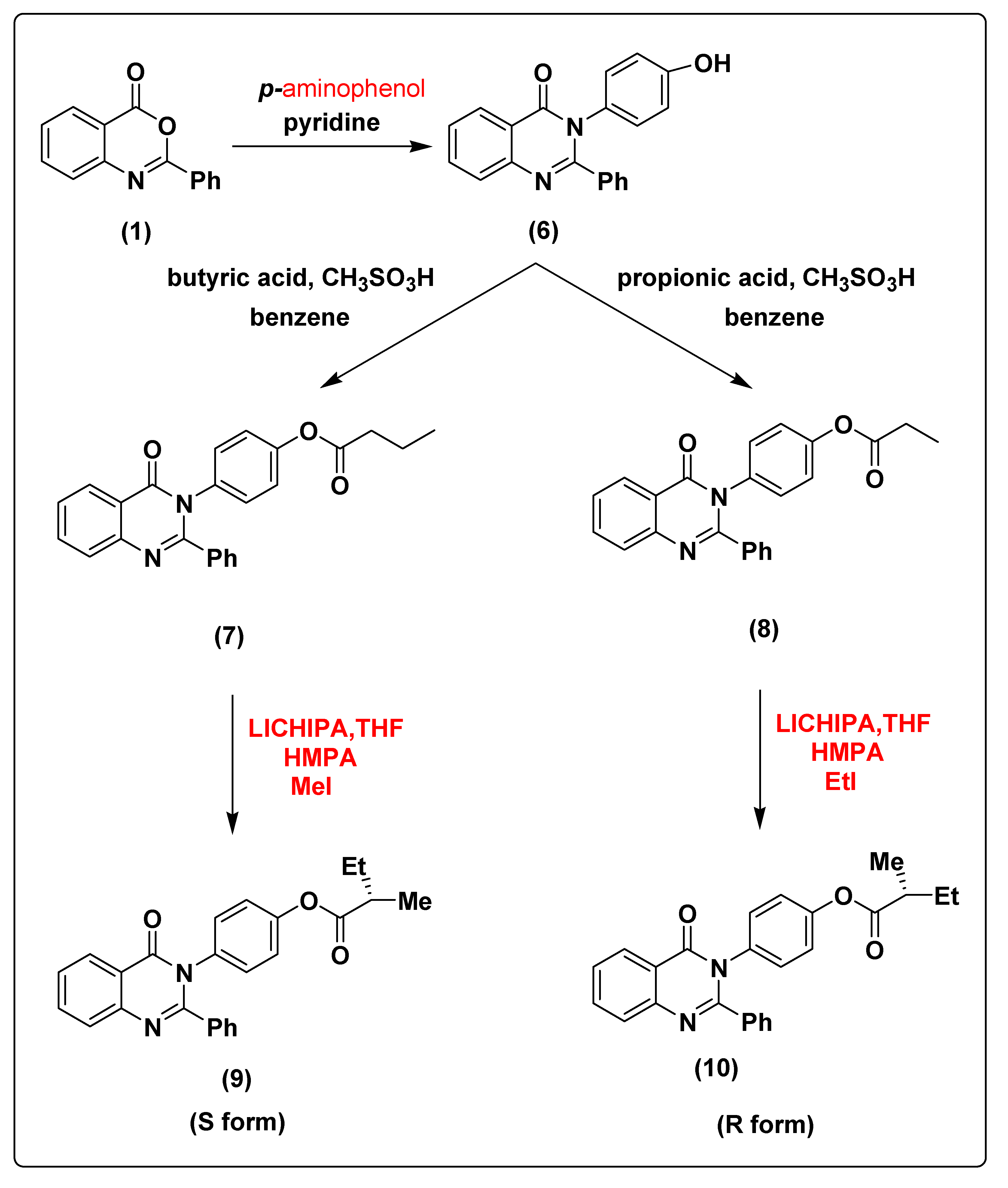
2.1. Chemistry of the Synthesized Compounds
2.2. In Silico Docking Study
2.2.1. The Crystal Structure and Active Site of the Target
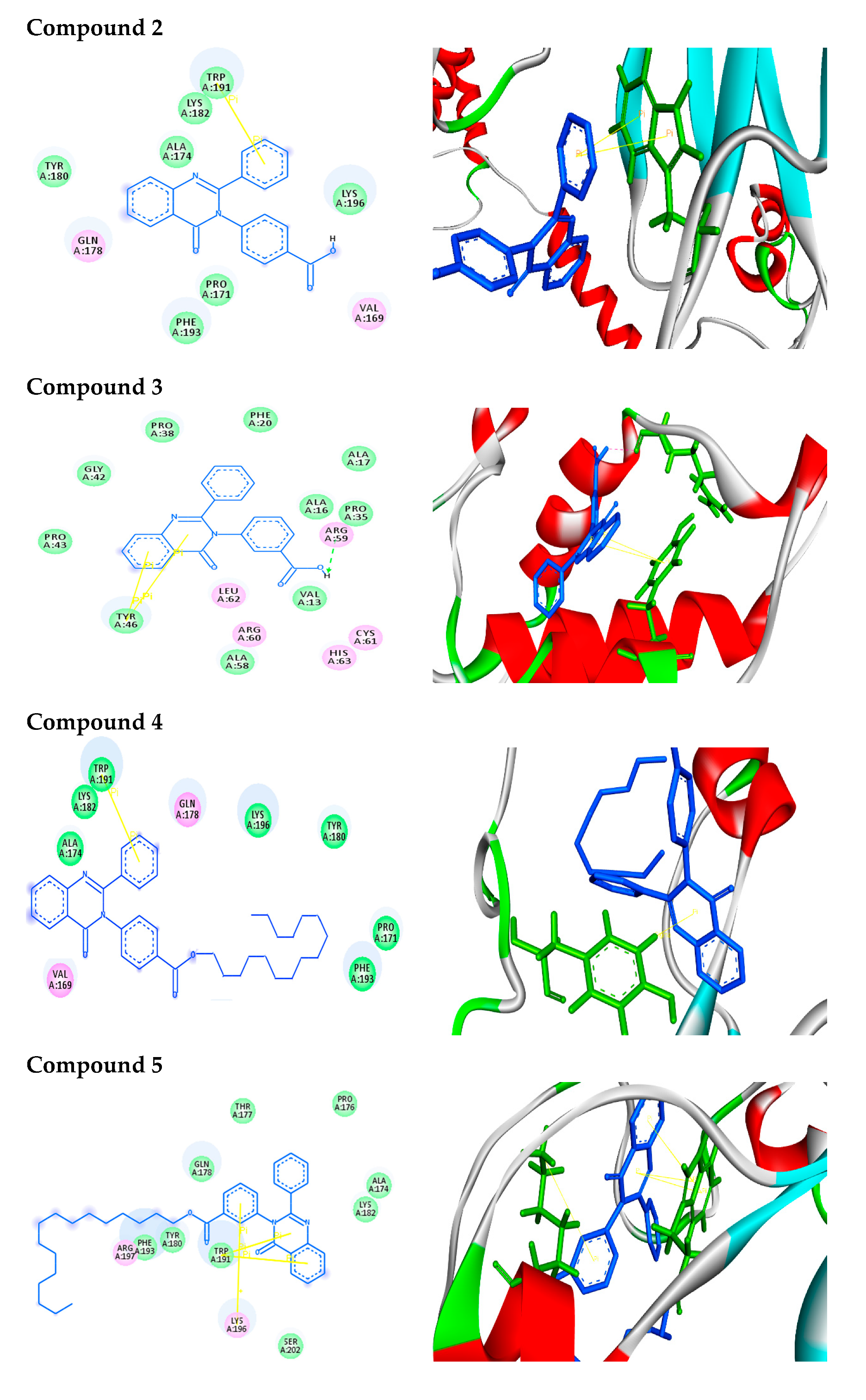
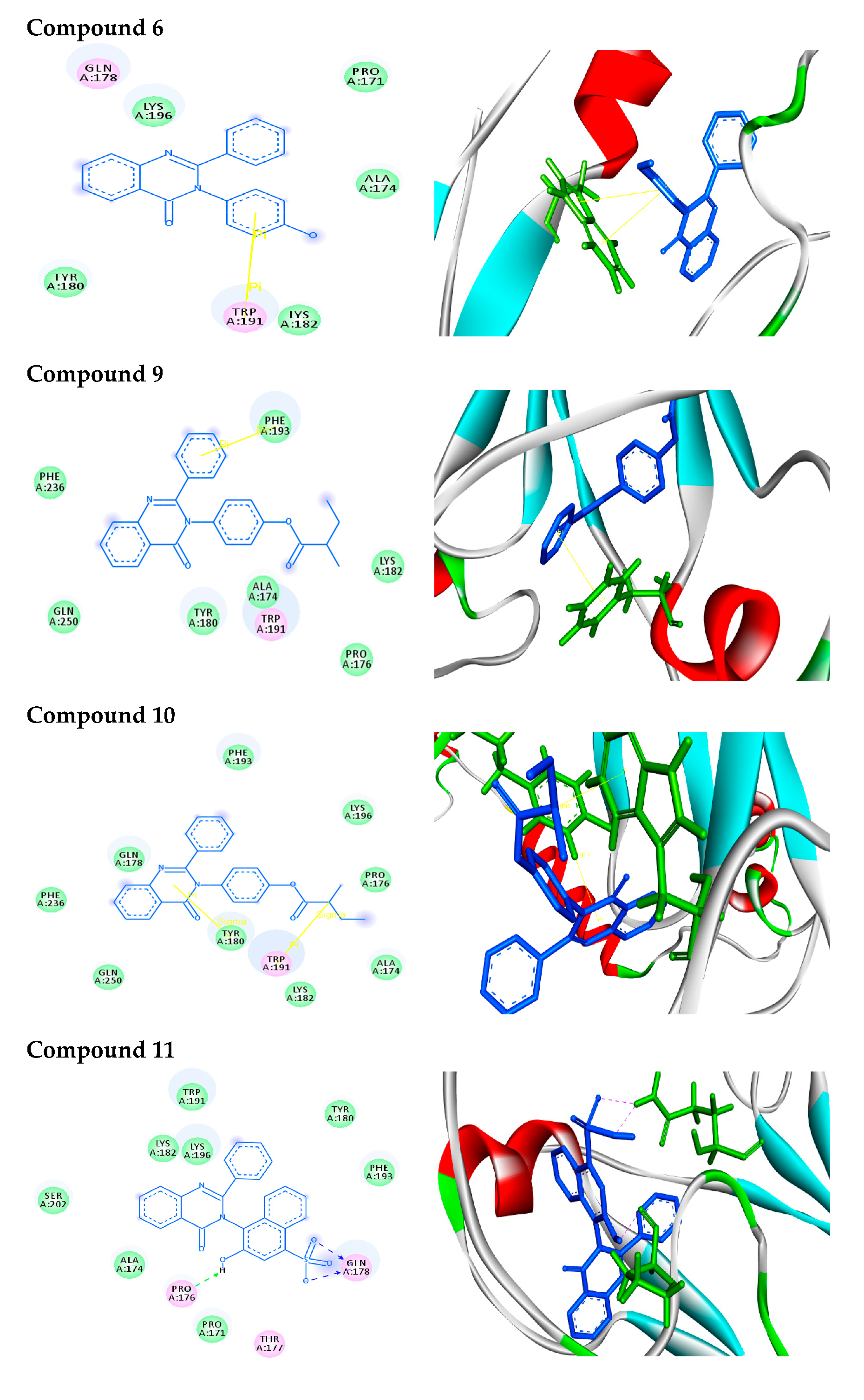
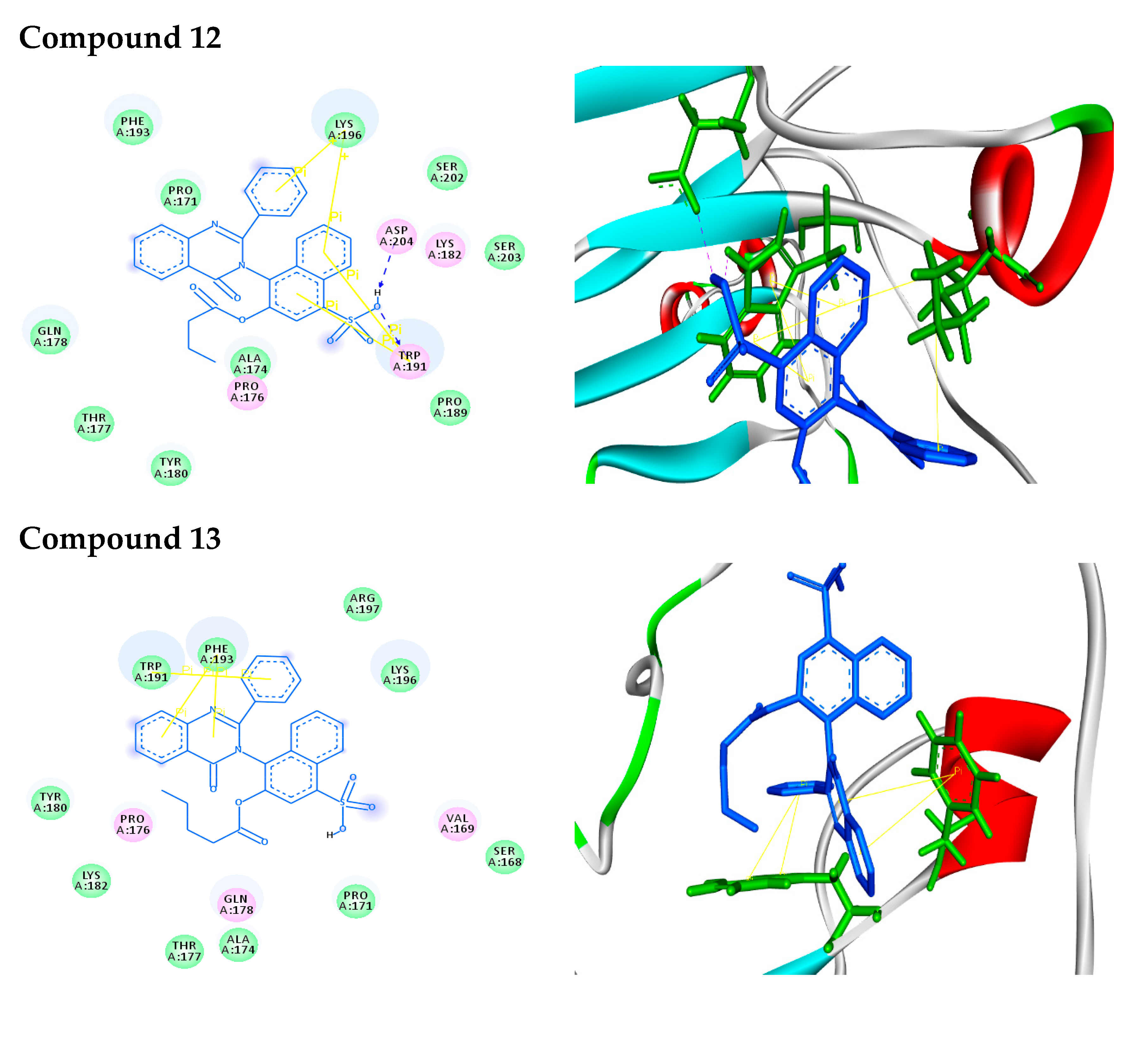
2.2.2. Molecular Docking Analysis
2.2.3. Docking Simulation
2.2.4. ADMET Property Evaluation
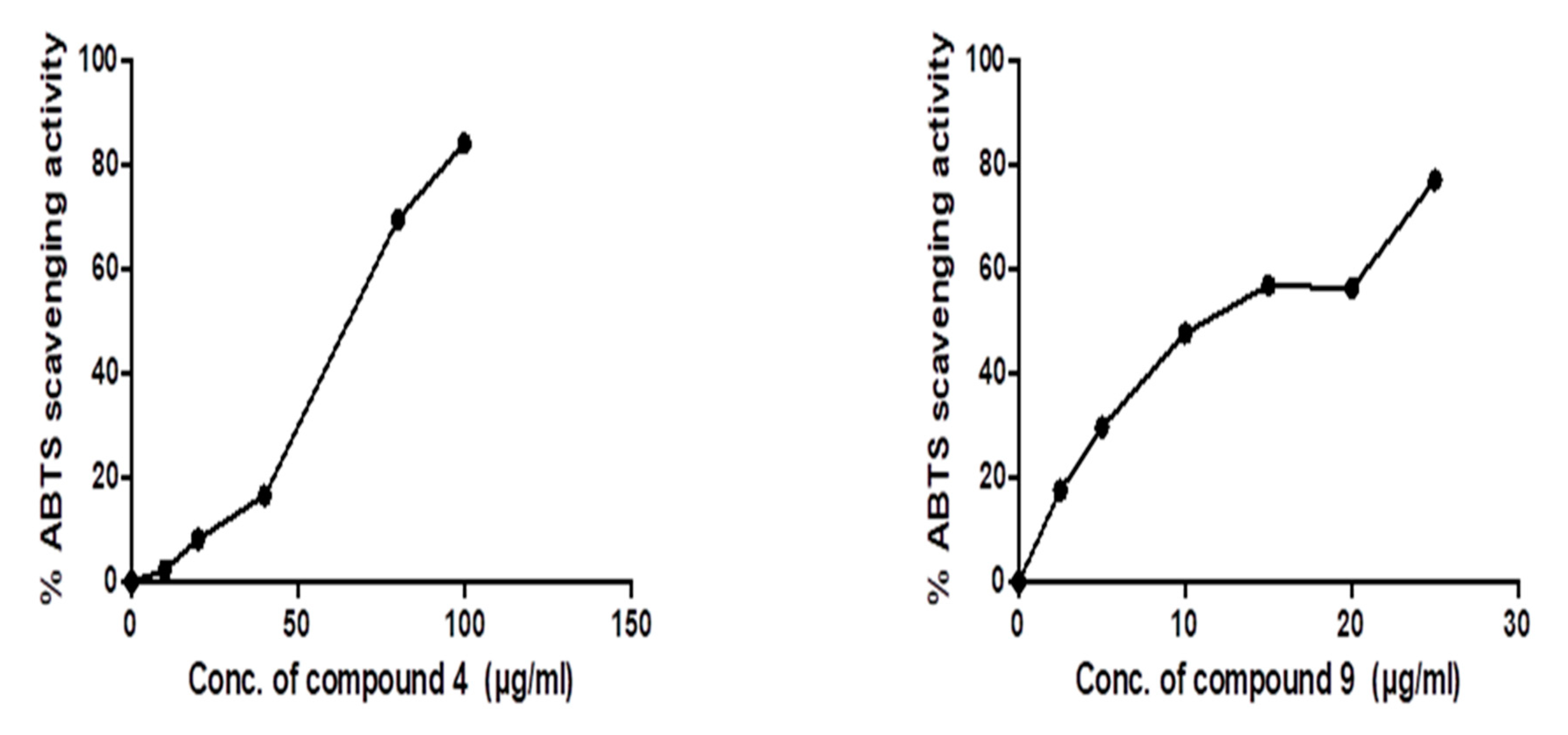
2.3. In Vitro Activity
2.4. Structure Activity Relationship (SAR)
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. Synthesis of 2-phenyl-4H-benzo[d][1,3]oxazin-4-one (1)
3.1.3. Synthesis of 4-(4-oxo-2-phenylquinazolin-3(4H)-yl)benzoic acid (2)
3.1.4. Synthesis of 3-(4-oxo-2-phenylquinazolin-3(4H)-yl)benzoic acid (3)
3.1.5. Synthesis of hexadecyl 4-(4-oxo-2-phenylquinazolin-3(4H)-yl)benzoate (4)
3.1.6. Synthesis of hexadecyl 3-(4-oxo-2-phenyl quinazolin-3(4H)-yl)benzoate (5)
3.1.7. Synthesis of 3-(4-hydroxyphenyl)-2-phenylquinazolin-4(3H)-one (6)
3.1.8. Synthesis of 4-(4-oxo-2-phenylquinazolin-3(4H)-yl)phenyl butyrate (7)
3.1.9. Synthesis of 4-(4-oxo-2-phenylquinazolin-3(4H)-yl)phenyl propionate (8)
3.1.10. Synthesis of (S)-4-(4-oxo-2-phenylquinazolin-3(4H)-yl)phenyl 2-methylbutanoate (9)
3.1.11. Synthesis of (R)-4-(4-oxo-2-phenylquinazolin-3(4H)-yl)phenyl 2-methylbutanoate (10)
3.1.12. Synthesis of 3-hydroxy-4-(4-oxo-2-phenylquinazolin-3(4H)-yl)naphthalene-1-sulfonic acid (11)
3.1.13. Synthesis of 4-(4-oxo-2-phenylquinazolin-3(4H)-yl)3-(butyryloxy)naphthalene-1-sulfonic acid (12)
3.1.14. Synthesis of 4-(4-oxo-2-phenylquinazolin-3(4H)-yl)3-(pentanoxy)naphthalene-1-sulfonic acid (13)
3.2. In Silico Study
3.3. In Vitro ABTS Radical Scavenging Antioxidant and Anticancer Activities of Predicted Compounds
3.3.1. Evaluation of ABTS Radical Scavenging Activity
3.3.2. Cell Culture
3.3.3. Determination of Compounds Cytotoxicity on Cells (MTT Protocol)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Nepali, K.; Sharma, S.; Sharma, M.; Bedi, P.M.S.; Dhar, K.L. Rational approaches, design strategies, structure activity relationship and mechanistic insights for anticancer hybrids. Eur. J. Med. Chem. 2014, 77, 422–487. [Google Scholar] [CrossRef] [PubMed]
- Popat, K.; McQueen, K.; Feeley, T.W. The global burden of cancer. Best Pract. Res. Clin. Anaesthesiol. 2013, 27, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Nussbaumer, S.; Bonnabry, P.; Veuthey, J.-L.; Fleury-Souverain, S. Analysis of anticancer drugs: A review. Talanta 2011, 85, 2265–2289. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Foo, J.; Michor, F. Evolution of acquired resistance to anti-cancer therapy. J. Theor. Biol. 2014, 355, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.M.; Margina, D.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Saloustros, E.; Fenga, C.; Spandidos, D.A.; Libra, M.; Tsatsakis, A.M. Akt inhibitors in cancer treatment: The long journey from drug discovery to clinical use. Int. J. Oncol. 2015, 48, 869–885. [Google Scholar] [CrossRef] [PubMed]
- Brazil, D.P.; Hemmings, B.A. Ten years of protein kinase B signalling: A hard Akt to follow. Trends Biochem. Sci. 2001, 26, 657–664. [Google Scholar] [CrossRef]
- Brazil, D.P.; Park, J.; Hemmings, B.A. PKB Binding Proteins: Getting in on the Akt. Cell 2002, 111, 293–303. [Google Scholar] [CrossRef]
- Bachelder, R.E.; Ribick, M.J.; Marchetti, A.; Falcioni, R.; Soddu, S.; Davis, K.R.; Mercurio, A.M. P53 Inhibits α6β4 Integrin Survival Signaling by Promoting the Caspase 3–Dependent Cleavage of Akt/PKB. J. Cell Biol. 1999, 147, 1063–1072. [Google Scholar] [CrossRef]
- Basso, A.D.; Solit, D.B.; Chiosis, G.; Giri, B.; Tsichlis, P.; Rosen, N. Akt Forms an Intracellular Complex with Heat Shock Protein 90 (Hsp90) and Cdc37 and Is Destabilized by Inhibitors of Hsp90 Function. J. Biol. Chem. 2002, 277, 39858–39866. [Google Scholar] [CrossRef]
- Xu, J.; Liu, D.; Songyang, Z. The Role of Asp-462 in Regulating Akt Activity. J. Biol. Chem. 2002, 277, 35561–35566. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Morrow, J.K.; Du-Cuny, L.; Chen, L.; Meuillet, E.J.; Mash, E.A.; Powis, G.; Zhang, S. Recent development of anticancer therapeutics targeting Akt. Recent Pat. Anti Cancer Drug Discov. 2011, 6, 146–159. [Google Scholar] [CrossRef]
- McDowell, K.A.; Riggins, G.J.; Gallia, G.L. Targeting the AKT pathway in glioblastoma. Curr. Pharm. Des. 2011, 17, 2411–2420. [Google Scholar] [CrossRef] [PubMed]
- Cassinelli, G.; Zuco, V.; Gatti, L.; Lanzi, C.; Zaffaroni, N.; Colombo, D.; Perego, P. Targeting the Akt Kinase to Modulate Survival, Invasiveness and Drug Resistance of Cancer Cells. Curr. Med. Chem. 2013, 20, 1923–1945. [Google Scholar] [CrossRef]
- Brown, J.S.; Banerji, U. Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol. Ther. 2017, 172, 101–115. [Google Scholar] [CrossRef]
- Pannipara, M.; Al-Sehemi, A.G.; Kalam, A.; Musthafa, T.N.M. Photophysics of Dihydroquinazolinone Derivatives: Experimental and Theoretical Studies. J. Fluoresc. 2017, 27, 1160–1170. [Google Scholar] [CrossRef]
- Zaghaghi, Z.; Mirjalilib, B.B.; Monfared, A. Synthesis of 2,3-Dihydroquinazolin-4(1H)-ones Promoted by Polystyrene Sulfonic Acid. Org. Chem. Res. 2019, 5, 80–86. [Google Scholar]
- Wang, H.-X.; Liu, H.-Y.; Li, W.; Zhang, S.; Wu, Z.; Li, X.; Li, C.-W.; Liu, Y.-M.; Chen, B.Q. Design, synthesis, antiproliferative and antibacterial evaluation of quinazolinone derivatives. Med. Chem. Res. 2019, 28, 203–214. [Google Scholar] [CrossRef]
- Ghaleno, M.R.; Ghaffari-Moghaddam, M.; Khajeh, M.; Oveisi, A.R.; Bohlooli, M. Iron species supported on a mesoporous zirconium metal-organic framework for visible light driven synthesis of quinazolin-4(3H)-ones through one-pot three-step tandem reaction. J. Colloid Interface Sci. 2019, 535, 214–226. [Google Scholar] [CrossRef]
- Sakr, A.; Kothayer, H.; Ibrahim, S.M.; Baraka, M.M.; Rezq, S. 1,4-Dihydroquinazolin-3(2H)-yl benzamide derivatives as anti-inflammatory and analgesic agents with an improved gastric profile: Design, synthesis, COX-1/2 inhibitory activity and molecular docking study. Bioorg. Chem. 2019, 84, 76–86. [Google Scholar] [CrossRef]
- Gatadi, S.; Gour, J.; Shukla, M.; Kaul, G.; Das, S.; Dasgupta, A.; Madhavi, Y.V.; Chopra, S.; Nanduri, S. Synthesis and evaluation of new 4-oxoquinazolin-3(4H)-yl)benzoic acid and benzamide derivatives as potent antibacterial agents effective against multidrug resistant Staphylococcus aureus. Bioorg. Chem. 2019, 83, 569–579. [Google Scholar] [CrossRef]
- Sajjadifar, S.; Hamidi, H.; Pal, K. Revisiting of Boron Sulfonic Acid Applications in Organic Synthesis: Mini-Review. J. Chem. Rev. 2019, 1, 35–46. [Google Scholar] [CrossRef]
- Piotrowska, D.G.; Andrei, G.; Schols, D.; Snoeck, R.; Grabkowska-Drużyc, M. New Isoxazolidine-Conjugates of Quinazolinones—Synthesis, Antiviral and Cytostatic Activity. Molecules 2016, 21, 959. [Google Scholar] [CrossRef] [PubMed]
- Marzaro, G.; Castagliuolo, I.; Schirato, G.; Palu’, G.; Via, M.D.; Chilin, A.; Brun, P. Substituted quinazolinones as kinase inhibitors endowed with anti-fibrotic properties. Eur. J. Med. Chem. 2016, 115, 416–425. [Google Scholar] [CrossRef]
- Dohle, W.; Prota, A.E.; Menchon, G.; Hamel, E.; Steinmetz, M.O.; Potter, B.V.L. Tetrahydroisoquinoline Sulfamates as Potent Microtubule Disruptors: Synthesis, Antiproliferative and Antitubulin Activity of Dichlorobenzyl-Based Derivatives, and a Tubulin Cocrystal Structure. ACS Omega 2019, 4, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Saeedi, M.; Mohammadi-Khanaposhtani, M.; Pourrabia, P.; Razzaghi, N.; Ghadimi, R.; Imanparast, S.; Faramarzi, M.A.; Bandarian, F.; Esfahani, E.N.; Safavi, M.; et al. Design and synthesis of novel quinazolinone-1,2,3-triazole hybrids as new anti-diabetic agents: In vitro α-glucosidase inhibition, kinetic, and docking study. Bioorg. Chem. 2019, 83, 161–169. [Google Scholar] [CrossRef]
- Das, S.; Bhattacharjee, J.; Panda, T.K. Guanylation/cyclisation of amino acid esters using an imidazolin-2-iminato titanium initiator. Dalton Trans. 2019, 48, 7227–7235. [Google Scholar] [CrossRef]
- Shagufta, S.; Ahmad, I. An insight into the therapeutic potential of quinazoline derivatives as anticancer agents. MedChemComm 2017, 8, 871–885. [Google Scholar] [CrossRef]
- Tiwary, B.K.; Zirmire, R.K.; Pradhan, K.; Nanda, A.K. Innovare Academic Sciences Preparation and Spectroscopic Characterization of Inclusion Complex of 2-Phenyl-4h-Benzo[D][1,3]Oxazin-4-One and Β-Cyclodextrin. Int. J. Pharm. Pharm. Sci. 2014, 6, 7–10. [Google Scholar]
- Henrich, S.; Feierberg, I.; Wang, T.; Blomberg, N.; Wade, R.C. Comparative binding energy analysis for binding affinity and target selectivity prediction. Proteins Struct. Funct. Bioinform. 2010, 78, 135–153. [Google Scholar] [CrossRef] [PubMed]
- El-Naggar, M.; Mohamed, M.E.; Mosallam, A.M.; Salem, W.; Rashdan, H.R.; Abdelmonsef, A.H. Synthesis, Characterization, Antibacterial Activity, and Computer-Aided Design of Novel Quinazolin-2,4-dione Derivatives as Potential Inhibitors Against Vibrio cholerae. Evol. Bioinform. 2020, 16. [Google Scholar] [CrossRef]
- Abdelmonsef, A.H.; Mosallam, A.M. Synthesis, in vitro biological evaluation and in silico docking studies of new quinazolin-2,4-dione analogues as possible anticarcinoma agents. J. Heterocycl. Chem. 2020, 1–8. [Google Scholar] [CrossRef]
- Abdelmonsef, A.H. Computer-aided identification of lung cancer inhibitors through homology modeling and virtual screening. Egypt. J. Med. Hum. Genet. 2019, 20. [Google Scholar] [CrossRef]
- Metwally, N.H.; Abdelrazek, F.M.; Eldaly, S.M. Synthesis, Molecular Docking, and Biological Evaluation of Some Novel Bis-heterocyclic Compounds Based N,N′-([1,1′-biphenyl]-4,4′-diyl)bis(2-cyanoacetamide) as Potential Anticancer Agents. J. Heterocycl. Chem. 2018, 55, 2668–2682. [Google Scholar] [CrossRef]
- Haredi Abdelmonsef, A.; Eldeeb Mohamed, M.; El-Naggar, M.; Temairk, H.; Mohamed Mosallam, A. Novel Quinazolin-2,4-Dione Hybrid Molecules as Possible Inhibitors Against Malaria: Synthesis and in silico Molecular Docking Studies. Front. Mol. Biosci. 2020, 7, 105. [Google Scholar] [CrossRef] [PubMed]
- Noser, A.; Selim, A.; Badawy, M. Asymmetric synthesis of α-alkylated acid in high enantiomeric purity using poly (Methylmethacrylate) resins. Int. J. Chem. Appl. Biol. Sci. 2014, 1, 135. [Google Scholar] [CrossRef]
- Selim, A.; Baren, M.; Noser, A. Enantioselective Synthesis of (S) 3-Methyl-4-Octanol as Insect Pheromone Using Polymeric Asymmetric Reagent. J. Appl. Chem. 2017, 6, 41–49. [Google Scholar]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Abdelmonsef, A.H.; Dulapalli, R.; Dasari, T.; Padmarao, L.S.; Mukkera, T.; Vuruputuri, U. Identification of Novel Antagonists for Rab38 Protein by Homology Modeling and Virtual Screening. Comb. Chem. High Throughput Screen. 2016, 19, 875–892. [Google Scholar] [CrossRef]
- Dasari, T.; Kondagari, B.; Dulapalli, R.; Abdelmonsef, A.H.; Mukkera, T.; Padmarao, L.S.; Malkhed, V.; Vuruputuri, U. Design of novel lead molecules against RhoG protein as cancer target—A computational study. J. Biomol. Struct. Dyn. 2017, 35, 3119–3139. [Google Scholar] [CrossRef] [PubMed]
- Rondla, R.; Padmarao, L.S.; Ramatenki, V.; Haredi-Abdel-Monsef, A.; Potlapally, S.R.; Vuruputuri, U. Selective ATP competitive leads of CDK4: Discovery by 3D-QSAR pharmacophore mapping and molecular docking approach. Comput. Biol. Chem. 2017, 71, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Wass, M.N.; Kelley, L.A.; Sternberg, M.J.E. 3DLigandSite: Predicting ligand-binding sites using similar structures. Nucleic Acids Res. 2010, 38, 469–473. [Google Scholar] [CrossRef] [PubMed]
- Huang, B. MetaPocket: A Meta Approach to Improve Protein Ligand Binding Site Prediction. OMICS A J. Integr. Biol. 2009, 13, 325–330. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-Molecule Library Screening by Docking with PyRx. In Chemical Biology; Springer: Berlin/Heidelberg, Germany, 2015; Volume 1263, pp. 243–250. ISBN 9780123944474. [Google Scholar]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A Comprehensive Source and Free Tool for Assessment of Chemical ADMET Properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | Binding Energy (kcal/mol) | Docked Complex (Amino Acid-Ligand) Interactions | Distance (A°) | |
|---|---|---|---|---|
| 2 |  | −7.9 | π–π interactions | |
| compound 2—TRP191 | 4.07 | |||
| compound 2—TRP191 | 3.90 | |||
| 3 |  | −9.3 | H-bonds | |
| compound 3—ARG59:O | 2.33 | |||
| π–π interactions | ||||
| compound 3—TYR46 | 4.04 | |||
| compound 3— TYR46 | 5.10 | |||
| 4 |  | −10.2 | π–σ interactions | |
| compound 4—TYR180:HE1 | 2.84 | |||
| 5 |  | −8.5 | π–π interactions | |
| compound 5—TRP191 | 3.92 | |||
| compound 5—TRP191 | 3.91 | |||
| compound 5—TRP191 | 3.99 | |||
| compound 5—TRP191 | 4.51 | |||
| π–cation interactions | ||||
| compound 5—LYS196:NZ | 6.19 | |||
| 6 |  | −8.0 | π–π interactions | |
| compound 6—TRP191 | 4.41 | |||
| compound 6—TRP191 | 3.94 | |||
| 9 |  | −9.8 | π–π interactions | |
| compound 9—PHE193 | 4.00 | |||
| 10 |  | −7.7 | π–σ interactions | |
| compound 10—TRP191 | 3.65 | |||
| compound 10—TYR180:HE1 | 2.72 | |||
| 11 |  | −8.9 | H-bonds | |
| compound 11—GLN178:HE22 | 2.10 | |||
| compound 11—GLN178:HE22 | 2.09 | |||
| compound 11—-PRO176:O | 2.07 | |||
| 12 |  | −9.2 | H-bonds | |
| compound 12—TRP191:HE1 | 2.48 | |||
| compound 12—ASP204:OD1 | 2.24 | |||
| π–π interactions | ||||
| compound 12—TRP191 | 3.90 | |||
| compound 12— TRP191 | 3.99 | |||
| compound 12— TRP191 | 3.87 | |||
| compound 12— TRP191 | 5.10 | |||
| π–cation interactions | ||||
| compound 12—LYS196:NZ | 4.67 | |||
| compound 12—LYS196:NZ | 5.24 | |||
| 13 |  | −9.1 | π–π interactions | |
| compound 13— TRP191 | 4.01 | |||
| compound 13— TRP191 | 4.41 | |||
| compound 13— PHE193 | 5.34 | |||
| compound 13— PHE193 | 4.80 |
| Molecular Weight (g/mol) | Blood–brain Barrier (BBB+) | Human Intestinal Absorption (HIA+) | Caco-2 Permeability (Caco-2) | AMES Toxicity | Carcinogenicity | |
|---|---|---|---|---|---|---|
| 2 | 342.35 | 0.958 | 0.969 | 0.619 | Nontoxic | Non carcinogenic |
| 3 | 342.35 | 0.958 | 0.969 | 0.619 | Nontoxic | Non carcinogenic |
| 4 | 566.79 | 0.979 | 0.995 | 0.531 | Nontoxic | Non carcinogenic |
| 5 | 566.79 | 0.979 | 0.995 | 0.531 | Nontoxic | Non carcinogenic |
| 6 | 314.34 | 0.975 | 0.997 | 0.548 | Nontoxic | Non carcinogenic |
| 9 | 398.46 | 0.977 | 0.997 | 0.600 | Nontoxic | Non carcinogenic |
| 10 | 398.46 | 0.977 | 0.997 | 0.600 | Nontoxic | Non carcinogenic |
| 11 | 444.47 | 0.615 | 0.859 | 0.586 | Nontoxic | Non carcinogenic |
| 12 | 514.56 | 0.670 | 0.840 | 0.601 | Nontoxic | Non carcinogenic |
| 13 | 528.59 | 0.692 | 0.856 | 0.596 | Nontoxic | Non carcinogenic |
| TPSA (A2) | HBA | HBD | N rotatable | Volume (A3) | |
|---|---|---|---|---|---|
| 2 | 72.20 | 5 | 1 | 3 | 298.05 |
| 3 | 72.20 | 5 | 1 | 3 | 298.05 |
| 4 | 61.20 | 5 | 0 | 19 | 567.60 |
| 5 | 61.20 | 5 | 0 | 19 | 567.60 |
| 6 | 55.12 | 4 | 1 | 2 | 279.06 |
| 9 | 61.20 | 5 | 0 | 6 | 365.77 |
| 10 | 61.20 | 5 | 0 | 6 | 365.77 |
| 11 | 109.50 | 7 | 2 | 3 | 362.51 |
| 12 | 115.57 | 8 | 1 | 7 | 432.62 |
| 13 | 115.57 | 8 | 1 | 8 | 449.42 |
| IC50 | |||
|---|---|---|---|
| Compound | HepG2 | MCF-7 | Caco-2 |
| 4 | 53.29 ± 0.25 | 72.22 ± 0.14 | 23.31 ± 0.09 |
| 9 | 171.4 ± 0.12 | 96.58 ± 0.17 | 73.87 ± 0.13 |
| Doxorubicin | 49.38 ± 0.15 | 58.1 ± 0.07 | 5.7 ±1.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noser, A.A.; El-Naggar, M.; Donia, T.; Abdelmonsef, A.H. Synthesis, In Silico and In Vitro Assessment of New Quinazolinones as Anticancer Agents via Potential AKT Inhibition. Molecules 2020, 25, 4780. https://doi.org/10.3390/molecules25204780
Noser AA, El-Naggar M, Donia T, Abdelmonsef AH. Synthesis, In Silico and In Vitro Assessment of New Quinazolinones as Anticancer Agents via Potential AKT Inhibition. Molecules. 2020; 25(20):4780. https://doi.org/10.3390/molecules25204780
Chicago/Turabian StyleNoser, Ahmed A., Mohamed El-Naggar, Thoria Donia, and Aboubakr H. Abdelmonsef. 2020. "Synthesis, In Silico and In Vitro Assessment of New Quinazolinones as Anticancer Agents via Potential AKT Inhibition" Molecules 25, no. 20: 4780. https://doi.org/10.3390/molecules25204780
APA StyleNoser, A. A., El-Naggar, M., Donia, T., & Abdelmonsef, A. H. (2020). Synthesis, In Silico and In Vitro Assessment of New Quinazolinones as Anticancer Agents via Potential AKT Inhibition. Molecules, 25(20), 4780. https://doi.org/10.3390/molecules25204780