
Antibody–Drug Conjugates for Cancer Therapy



Abstract
1. Introduction
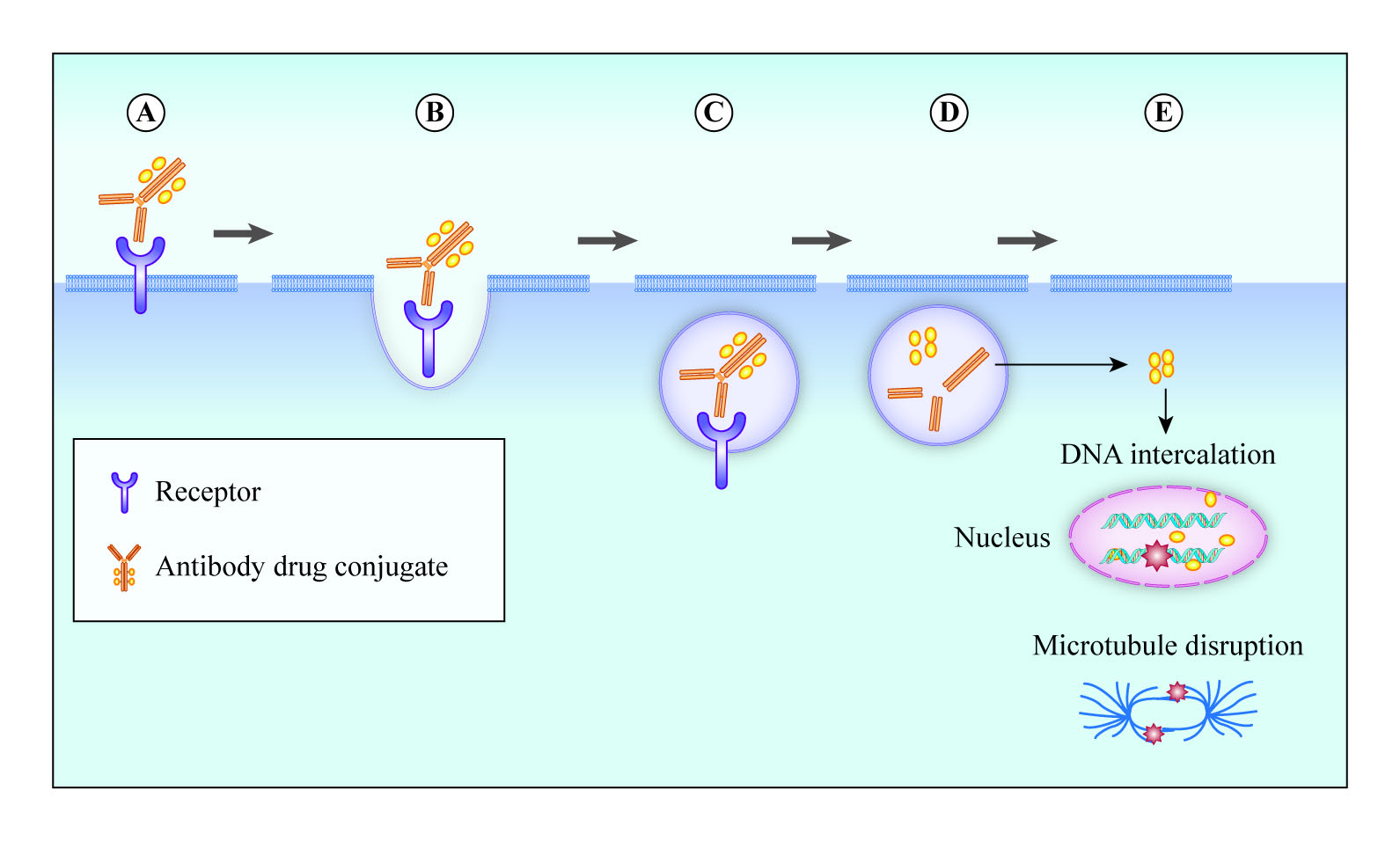
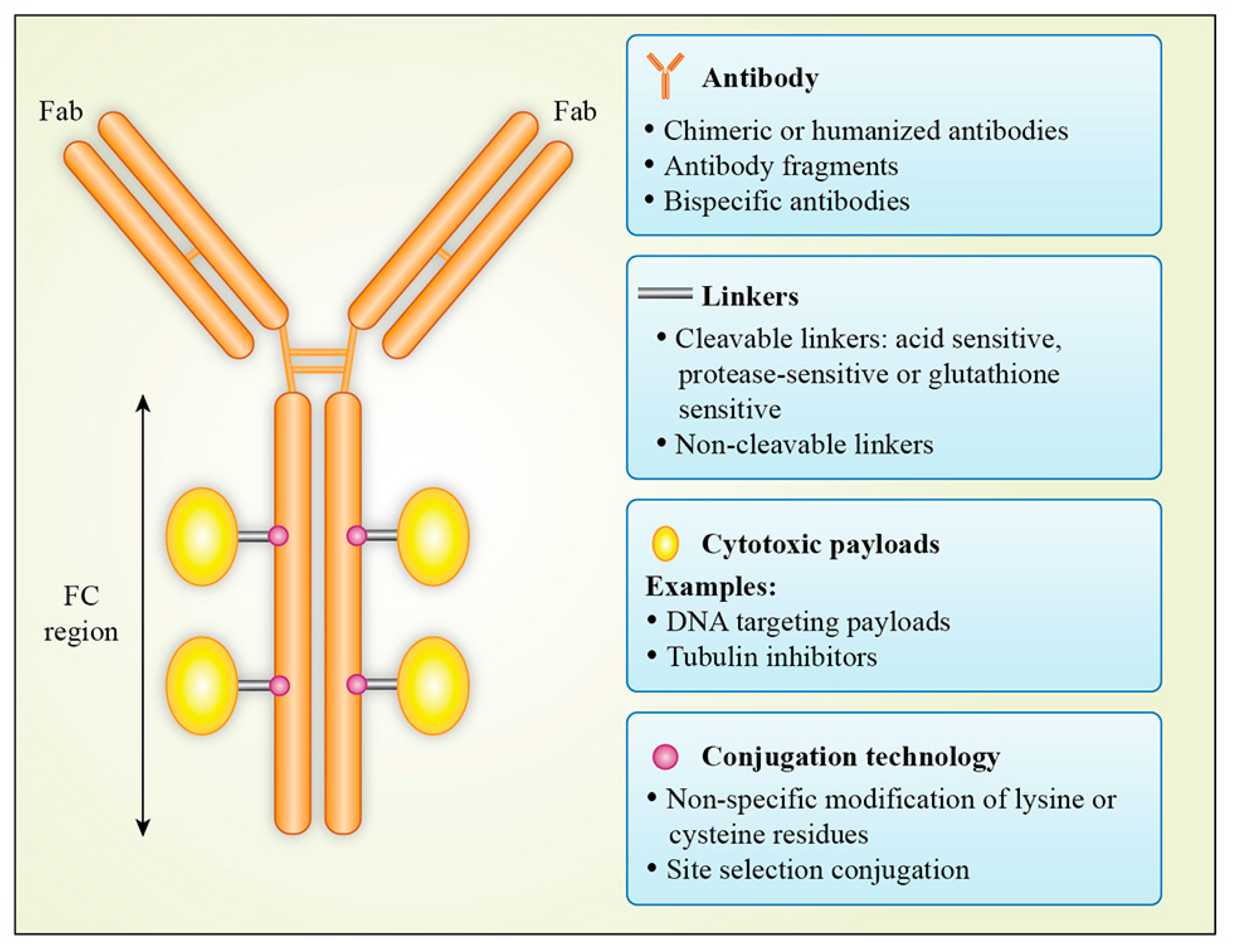
2. Design and Structure of Antibody–Drug Conjugates
2.1. Target Selection
2.2. Choice of Antibody
2.3. Cytotoxic Payload
2.4. Linkers
2.5. Antibody–Drug Conjugation
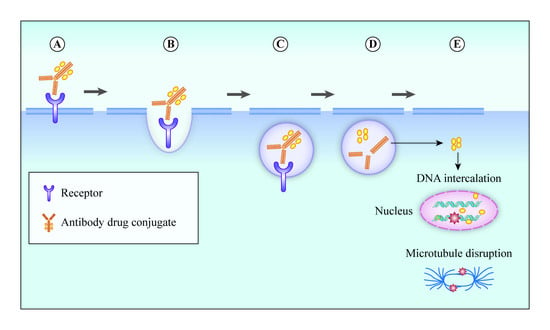
3. Mechanism of Action of ADC
4. Clinically Approved ADCs
4.1. Brentuximab Vedotin
4.2. Trastuzumab Emtansine
4.3. Inotuzumab Ozogamicin
4.4. Gemtuzumab Ozogamicin
4.5. Polatuzumab Vedotin
4.6. Trastuzumab Deruxtecan
4.7. Enfortumab Vedotin
4.8. Sacituzumab Govitecan
4.9. Belantamab Mafodotin
5. Promising ADCs in Clinical Development

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Payload | Target Antigen | Antibody–Drug Conjugates | Antibody | Linker | Lead Indication | Trial Phase | ClinicalTrials.gov Identifier | Reference |
|---|---|---|---|---|---|---|---|---|
| AGD-0182 | FLT3 | AGS62P1 (ASP1235) | Human IgG1 | Non-cleavable linker | Acute myeloid leukaemia (AML) | I | NCT02864290 | [101] |
| Amberstatin-269 | FLT3 | AGS-62P1 | Humanized IgG1 | Cleavable linker | AML | I | NCT02864290 | |
| Auristatin-0101 | ErbB2 | PF-06804103 | Humanized IgG1 | Protease-cleavable linker | Advanced solid tumors | I | NCT03284723 | [102] |
| PTK7 | Cofetuzumab pelidotin PF-7020 | Humanized IgG1 | Cleavable maleimidoca- proyl -valine-citrulline (mc-vc) PABC linker | Non-small-cell lung cancer (NSCLC), advanced solid tumors | I | NCT04189614 NCT02222922 | [103] | |
| Auristatin derivative | IGF-1R | W0101 | Humanized IgG1 | Non-cleavable maleimidoca- proyl (mc) linker | Advanced solid tumors | I/II | NCT0331638 | [104] |
| Auristatin | ErbB2 | ZW49 | Biparatopic IgG | Protease-cleavable linker | ErbB2-expressing cancers | I | NCT03821233 | [105] |
| Auristatin F | 5T4 | ASN-004 | Humanized scFvFc antibody | Non-cleavable linker | Advanced solid tumors | I | NCT04410224 | |
| Auristatin F-HPA (DolaLock) | SLC34A2 (NaPi2b) | XMT-1592 | Humanized IgG1 | Protease-cleavable linker | NSCLC, ovarian cancer | I/II | NCT04396340 | |
| SLC34A2 (NaPi2b) | XMT-1536 | Humanized IgG1 | Protease-cleavable linker | NSCLC, ovarian cancer | I | NCT03319628 | [106] | |
| Batansine | ErbB2 | BAT8001 | Humanized IgG1 | Non-cleavable linker | Metastatic breast cancer | I | NCT04189211 NCT04151329 | [107] |
| TROP2 | BAT8003 | IgG1 | Non-cleavable linker | Advanced epithelial cancer | I | NCT03884517 | [108] | |
| Dolastatin analogue | AG7 | ABGn-107 | Humanized IgG | Cleavable linker | Gastric, colorectal, pancreatic, or biliary cancer | I | NCT02908451 | [109] |
| DM1 | CD30 | F0002-ADC | Chimeric IgG1κ | Non-cleavable SMCC linker | Hematologic malignancies | I | NCT03894150 | [110] |
| CD37 | Naratuximab emtansine (IMGN-529, DEBIO 1562) | Humanized IgG1 | Non-cleavable SMCC linker | Non-Hodgkin’s lymphoma (NHL) | I | NCT01534715 | [111] | |
| CD56 | Lorvotuzumab Mertansine (LM, IMGN-901) | Humanized IgG1 | Disulfide linker | Multiple myeloma, small-cell lung cancer (SCLC), Merkel cell, ovarian cancer | I/II | NCT02452554 | [112,113,114] | |
| EGFR | AVID100 | IgG | Non-cleavable SMCC linker | Advanced epithelial carcinomas | I/II | NCT03094169 | [115] | |
| ErbB2 | B003 | Humanized IgG1 | Thioether linker | Metastatic breast cancer | I | NCT03953833 | ||
| DM4 | CD205 | MEN1309 (OBT076) | Humanized IgG1 | Cleavable SPDB linker | Solid tumors, breast cancer, NHL | I | NCT04064359 | [116] |
| CD138 | Indatuximab ravtansine (BT062) | Chimeric IgG4 | Cleavable disulfide linker | Multiple myeloma | I/IIa | NCT01638936 | [117,118] | |
| CD166 | CX-2009 | Probody | Cleavable SPDB linker | Solid tumors | I/II | NCT03149549 | [119] | |
| CEACAM5 | SAR408701 | Humanized IgG1 | Cleavable linker | Solid tumors | III | NCT04154956 | [120] | |
| Folate receptor α (FRα) | Mirvetuximab soravtansine (IMGN-853) | Humanized IgG1 | Cleavable SPDB linker | NSCLC, ovarian cancer | I/II/ III | NCT01609556 NCT02631876 NCT03552471 NCT02496890 NCT03832361 | [121] | |
| Mesothelin (MSLN) | Anetumab ravtansine (BAY 94-9343) | Human IgG1 | Cleavable SPDB linker | Mesothelioma, Solid tumors | II | NCT03832361 NCT04296890 NCT03552471 NCT03023722 NCT04209855 NCT02996825 | [122,123] | |
| DUBA (Seco-duocarmycin-hydroxy-benzamide-azainodole) | ErbB2 | Trastuzumab duocarmazine (SYD985) | Humanized IgG1 | Cleavable linker | Endometrial carcinoma, metastatic breast cancer, | II/III | NCT04205630 NCT03262935 | [39] |
| B7-H3 | MGC018 | Humanized IgG1 | Cleavable valine-citrulline linker | Advanced solid tumors | I/II | NCT03729596 | [124] | |
| Duocarmycin analogue | 5T4 | SYD1875 | Humanized IgG1 | Cleavable valine-citrulline-seco linker | Advanced solid tumors | I/II | NCT04202705 | |
| Dxd (DX-8951) | B7-H3 | DS-7300a | Humanized IgG1 | Cleavable linker | Advanced solid tumors | I | NCT04145622 | |
| ErbB3 | U3-1402 | Humanized IgG1 | Cleavable linker | NSCLC, breast cancer, colorectal cancer | I/II | NCT03260491, NCT02980341, NCT04479436 | [125] | |
| GPR20 | DS-6157a | Unknown | Cleavable Gly-Gly-Phe-Gly (GGFG) linker | Advanced gastrointestinal stromal tumor | I | NCT04276415 | [126] | |
| TROP-2 | DS-1062a | Humanized IgG1 | Cleavable linker | NSCLC, Advanced solid tumors | I | NCT04526691 NCT03401385 | [127] | |
| Eribulin | FRα | MORAb-202 | Humanized IgG1 | Cleavable mal-PEG2-vc-PABC | Solid tumors | I | NCT03386942 | [128] |
| IGN (Indolino-benzodiazepine dimer) | CD123 | IMGN 632 Orphan drug designation | Humanized IgG1 | Cleavable peptide linker | AML, Acute lymphoblastic leukaemia (ALL) | I/II | NCT03386513 | [129] |
| MMAE | AXL | Enapotamab vedotin (HuMax-AXL-ADC) | Human IgG1 | Cleavable mc-vc linker | Solid tumors | I/II | NCT02988817 | [130] |
| AXL | BA3011 (CAB-AXL-ADC) | Humanized IgG1 | Cleavable linker | Advanced solid tumors | I/II | NCT03425279 | [131] | |
| CD71 | CX-2029 | Probody | Protease-cleavable mc-vc-PAB linker | DLBCL, Solid tumors | I/II | NCT03543813 | [132] | |
| CD228 | SGN-CD228A | Humanized IgG1 | Protease-cleavable linker | Advanced solid tumors | I | NCT04042480 | [133] | |
| C-Met | ABBV-399 Telisotuzumab vedotin | Humanized IgG1 | Cleavable mc-vc linker | Non-small-cell lung cancer | II | NCT03539536 | [134] | |
| ErbB2 | Disitimab vedotin (RC48-ADC) | Humanized IgG1 | Protease-cleavable linker | Metastatic breast cancer with low ErbB2 expression | III | NCT04400695 | [135,136] | |
| ErbB2 | ALT-P7 | Humanized IgG1 | Unknown | Breast cancer | I | NCT03281824 | [137] | |
| ErbB2 | MRG002 | IgG | Unknown | Breast cancer, gastric cancer | I/II | NCT04492488 | [138] | |
| EGFR | MRG003 | IgG | Unknown | Solid tumors | I | CTR20180310 | ||
| Globo H | OBI-999 Orphan drug approval from the FDA | Humanized IgG1 | Cleavable thiobridge linker | Advanced solid tumors | I/II | NCT04084366 | [139] | |
| Integrin beta-6 | SGN-B6A | IgG | Unknown | Advanced solid tumors | I | NCT04389632 | ||
| ROR1 | VLS-101 (UC-961ADC3) | Humanized IgG1 | Unknown | Advanced breast cancer, NSCLC, haematological malignancies | I | NCT04504916 | [140] | |
| SLC39A6 (LIV-1) | Ladiratuzumab vedotin (SGN-LIV1A) | Humanized IgG1 | Cleavable mc-vc linker | NSCLC, head and neck cancer, gastric cancer, breast cancer | I/II | NCT04032704 NCT03310957 NCT01969643 | [141] | |
| Tissue factor (TF, CD142) | Tisotumab vedotin (HuMax-TF-ADC) | Human IgG1 | Protease-cleavable mc-vc-PAB linker | Advanced ovarian cancer | II | NCT03657043 | [142] | |
| MMAF | ErbB2 | ARX788 | IgG1 | pAcF linker | ErbB2-expressing cancers | I | NCT03255070 | [143] |
| ErbB2 | FS-1502 (LCB14-0110) | Humanized IgG1 | Unknown | ErbB2-positive advanced solid tumors | I | NCT03944499 | ||
| MMAF derivative (Duostatin-5) | ErbB2 | A166 | Humanized IgG1 | Unknown | ErbB2-expressing tumors | I/II | NCT03602079 | [144] |
| Maytansinoid | CD22 | TRPH-222 | Humanized IgG | Non-cleavable linker | B cell lymphoma | I | NCT03682796 | [145] |
| PBD | BCMA | MEDI2228 | Humanized IgG1 | Cleavable protease linker | Multiple myeloma | I | NCT03489525 | |
| c-MET | TR 1801-ADC (MT-8633) | Humanized IgG2 | Cleavable Val-Ala linker | c-MET-expressing tumors | I | NCT03859752 | ||
| CD19 | Loncastuximab tesirine (ADCT-402) | Humanized IgG1 | Cleavable Valine-Ala linker | NHL | III | NCT04384484 | [146] | |
| CD25 | Camidanlumab tesirine (ADCT-301) | Humanized IgG1 | Cleavable linker | AML, ALL, HL, NHL, solid tumors | I/II | NCT04052997 NCT03621982 | [147] | |
| EGFR | ABBV-321 Serclutamab talirine | IgG | Solid tumors | I | NCT03234712 | [148] | ||
| GCC | TAK-164 | Human IgG | Cleavable peptide linker | Gastrointestinal malignancies | I | NCT03449030 | [149] | |
| SC236 | CD74 | STRO-001 | Human IgG1 | Non-cleavable DBCO linker | NHL, B cell malignancies | I | NCT03424603 | [150] |
| SC209 | FR α | STRO-002 | Human IgG1 | Cleavable linker | Ovarian and endometrial cancers | I | NCT03748186 | [151] |
| SG3249 (PBD derivative) | CD22 | ADCT-602 (Epratuzumab tesirine) | Humanized IgG1 | Cleavable Val-Ala linker | B cell acute lymphoblastic leukaemia | I/II | NCT03698552 | |
| SHR152852 | c-MET | SHR-A1403 (HTI-1066) | Humanized IgG2 | Non-cleavable linker | Advanced solid tumors | I | NCT03856541 | [152] |
| PNU-159682 | ROR1 | NBE-002 | IgG | Unknown | Advanced solid tumors, triple-negative breast cancer | I | NCT04441099 | |
| Belotecan-derived payload | TROP2 | SKB-264 | IgG | Unknown | Advanced solid tumors | I/II | NCT04152499 | |
| TLR7/TLR8 agonist | ErbB2 | BDC-1001 | Humanized IgG1 | Unknown | ErbB2-positive advanced solid tumors | I | NCT04278144 | |
| TLR8 agonist | ErbB2 | SBT6050 | IgG | Unknown | ErbB2-positive advanced solid tumors | I | NCT04460456 | |
| Unknown | ROR2 | BA 3021 (CAB-ROR2-ADC) | Humanized IgG1 | Unknown | Advanced solid tumors | I/II | NCT03504488 | |
| Unknown | CD46 | FOR-46 | Humanized IgG1 | Unknown | Multiple myeloma, prostate cancer | I | NCT03650491 NCT03575819 | |
| Unknown | Unknown | ABBV-011 | IgG | Unknown | Small-cell lung cancer | I | NCT0369194 | |
| Unknown | Unknown | ABBV-155 | IgG | Unknown | Solid tumors | I | NCT03595059 | |
| Unknown | ErbB2 | DP303c | IgG | Unknown | ErbB2-positive advanced solid tumors | I | NCT04146610 | |
| Unknown | BCMA | CC-99712 (BCMA-ADC) | IgG | Unknown | Multiple myeloma | I | NCT04036461 | |
| Unknown | ErbB2 | GQ1001 | IgG | Unknown | ErbB2-positive advanced solid tumors | I | NCT04450732 | |
| Unknown | ErbB2 | BB-1701 | IgG | Unknown | ErbB2-positive advanced solid tumors | I | NCT04257110 | |
| Unknown | ErbB2 | SHR-A1811 | IgG | Unknown | ErbB2-positive advanced solid tumors | I | NCT04513223 NCT04446260 | |
| Unknown | CCR7 | JBH492 | IgG | Unknown | CLL, NHL | I | NCT04240704 |
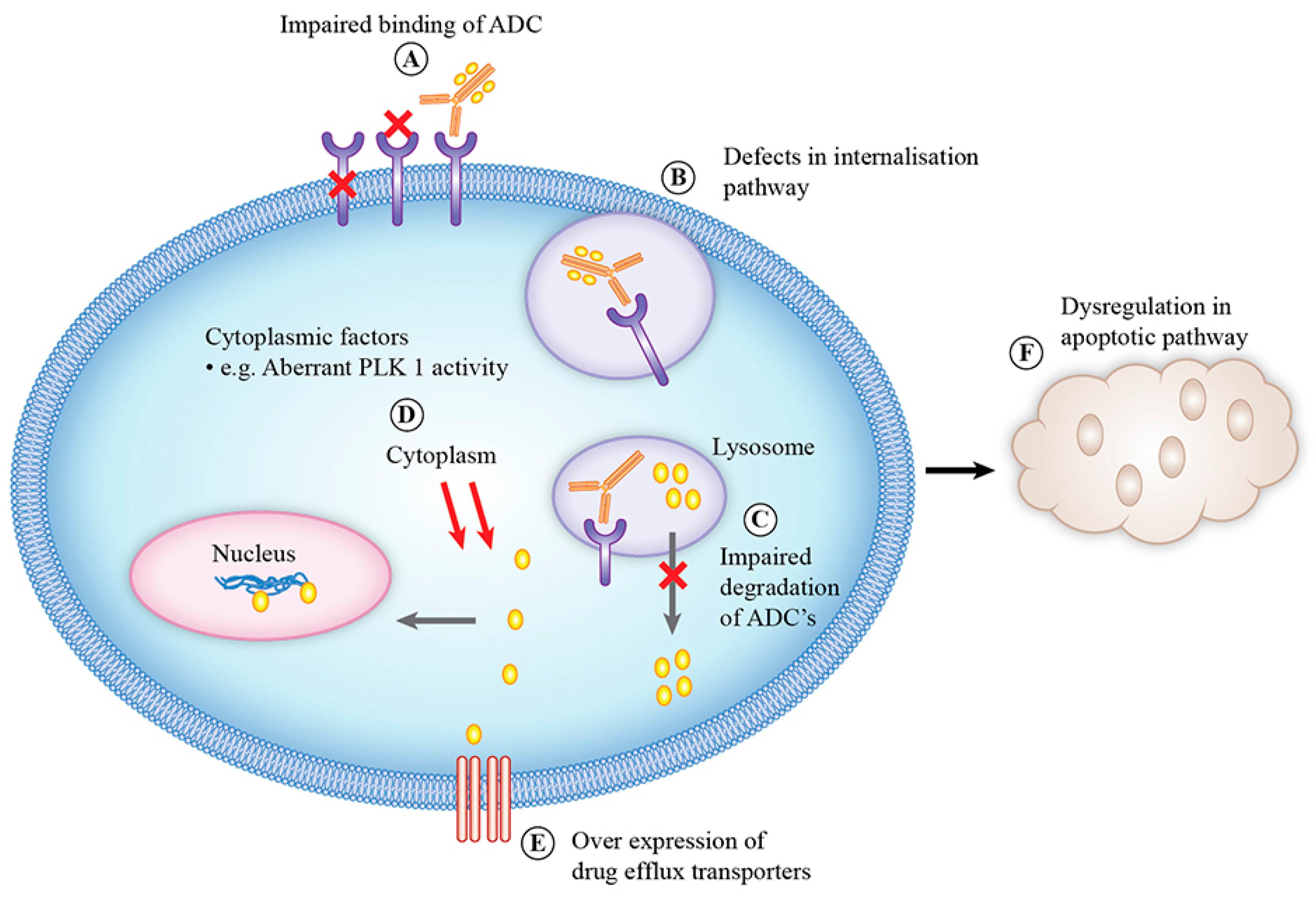
6. Mechanisms of Resistance to Antibody–Drug Conjugate Therapies
7. Role of Molecular Imaging in the Clinical Development of ADCs
7.1. Preclinical Studies
7.1.1. CD30
7.1.2. Tomoregulin (TENB2) and Six-Transmembrane Epithelial Antigen of the Prostate-1 (STEAP1)
7.1.3. Mesothelin (MSLN)
7.1.4. Leucine-Rich Repeat-Containing G Protein-Coupled Receptor 5 (LGR5)
7.2. Clinical Studies
8. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Factsheet, W. Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 29 November 2019).
- Davis, C.; Naci, H.; Gurpinar, E.; Poplavska, E.; Pinto, A.; Aggarwal, A. Availability of evidence of benefits on overall survival and quality of life of cancer drugs approved by European Medicines Agency: Retrospective cohort study of drug approvals 2009–13. BMJ 2017, 359, j4530. [Google Scholar] [CrossRef] [PubMed]
- Weiner, G.J. Building better monoclonal antibody-based therapeutics. Nat. Rev. Cancer 2015, 15, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Sievers, E.L.; Senter, P.D. Antibody-Drug Conjugates in Cancer Therapy. Annu. Rev. Med. 2013, 64, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V. CPhI Annual Report 2018: ADCs Growth Driven by Lack of In-House Facilities, Oncology and Integrated CDMOs. Available online: https://www.pharmoutsourcing.com/Featured-Articles/354437-CPhI-Annual-Report-2018-ADCs-Growth-Driven-by-Lack-of-In-House-Facilities-Oncology-and-Integrated-CDMOs/ (accessed on 7 July 2020).
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody–drug conjugates for cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef]
- Redman, J.M.; Hill, E.M.; AlDeghaither, D.; Weiner, L.M. Mechanisms of action of therapeutic antibodies for cancer. Mol. Immunol. 2015, 67, 28–45. [Google Scholar] [CrossRef]
- Hafeez, U.; Gan, H.K.; Scott, A.M. Monoclonal antibodies as immunomodulatory therapy against cancer and autoimmune diseases. Curr. Opin. Pharm. 2018, 41, 114–121. [Google Scholar] [CrossRef]
- Smith, L.M.; Nesterova, A.; Alley, S.C.; Torgov, M.Y.; Carter, P.J. Potent cytotoxicity of an auristatin-containing antibody-drug conjugate targeting melanoma cells expressing melanotransferrin/p97. Mol. Cancer Ther. 2006, 5, 1474. [Google Scholar] [CrossRef]
- Perez, H.L.; Cardarelli, P.; Deshpande, S.; Gangwar, S.; Schroeder, G.; Vite, G.; Borzilleri, R.M. Antibody-drug conjugates: Current status and future directions. Drug Discov. Today 2014, 19, 869–881. [Google Scholar] [CrossRef]
- Gutierrez, C.; Schiff, R. HER2: Biology, detection, and clinical implications. Arch. Pathol. Lab. Med. 2011, 135, 55–62. [Google Scholar] [CrossRef]
- Ingle, G.S.; Chan, P.; Elliott, J.M.; Chang, W.S.; Koeppen, H.; Stephan, J.P.; Scales, S.J. High CD21 expression inhibits internalization of anti-CD19 antibodies and cytotoxicity of an anti-CD19-drug conjugate. Br. J. Haematol. 2008, 140, 46–58. [Google Scholar] [CrossRef]
- Ritchie, M.; Tchistiakova, L.; Scott, N. Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. mAbs 2013, 5, 13–21. [Google Scholar] [CrossRef]
- Donaghy, H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. mAbs 2016, 8, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Sapra, P.; Stein, R.; Pickett, J.; Qu, Z.; Govindan, S.V.; Cardillo, T.M.; Hansen, H.J.; Horak, I.D.; Griffiths, G.L.; Goldenberg, D.M. Anti-CD74 antibody-doxorubicin conjugate, IMMU-110, in a human multiple myeloma xenograft and in monkeys. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 5257. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Chari, R.V. Antibody conjugate therapeutics: Challenges and potential. Clin. Cancer Res. 2011, 17, 6389–6397. [Google Scholar] [CrossRef] [PubMed]
- Hughes, B. Antibody-drug conjugates for cancer: Poised to deliver? Nat. Rev. Drug Discov. 2010, 9, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody-Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Derry, C.R.; Shreeram, A. FcRn: The neonatal Fc receptor comes of age. Nat. Rev. Immunol. 2007, 7, 715. [Google Scholar] [CrossRef]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef]
- Jochen, G.S. Isotype selection in antibody engineering. Nat. Biotechnol. 2007, 25, 1369. [Google Scholar] [CrossRef]
- Correia, I.R. Stability of IgG isotypes in serum. mAbs 2010, 2, 221–232. [Google Scholar] [CrossRef]
- Adlersberg, J.B. The immunoglobulin hinge (Interdomain) region. Ric. Clin. E Lab. 1976, 6, 191. [Google Scholar] [CrossRef]
- Saito, S.; Namisaki, H.; Hiraishi, K.; Takahashi, N.; Iida, S. A stable engineered human IgG3 antibody with decreased aggregation during antibody expression and low pH stress. Protein Sci. 2019, 28, 900–909. [Google Scholar] [CrossRef]
- Hoffmann, R.M.; Coumbe, B.G.T.; Josephs, D.H.; Mele, S.; Ilieva, K.M.; Cheung, A.; Tutt, A.N.; Spicer, J.F.; Thurston, D.E.; Crescioli, S.; et al. Antibody structure and engineering considerations for the design and function of Antibody Drug Conjugates (ADCs). Oncoimmunology 2017, 7, e1395127. [Google Scholar] [CrossRef]
- L’Italien, L.; Orozco, O.; Abrams, T.; Cantagallo, L.; Connor, A.; Desai, J.; Ebersbach, H.; Gelderblom, H.; Hoffmaster, K.; Lees, E.; et al. Mechanistic Insights of an Immunological Adverse Event Induced by an Anti-KIT Antibody Drug Conjugate and Mitigation Strategies. Clin. Cancer Res. 2018, 24, 3465–3474. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Li, Y.-Y.; Liu, X.-Y.; Lu, X.-L.; Cao, X.; Jiao, B.-H. Marine Antibody-Drug Conjugates: Design Strategies and Research Progress. Mar. Drugs 2017, 15, 18. [Google Scholar] [CrossRef]
- Yasunaga, M.; Manabe, S.; Tsuji, A.; Furuta, M.; Ogata, K.; Koga, Y.; Saga, T.; Matsumura, Y. Development of Antibody-Drug Conjugates Using DDS and Molecular Imaging. Bioengineering 2017, 4, 78. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Basal, J.; Pegram, M.; Do-Youn, O.; Dieras, V.; Guardino, E.; et al. Trastuzumab Emtansine for HER2-positive advanced breast cancer. (Clinical report). N. Engl. J. Med. 2012, 367, 1783. [Google Scholar] [CrossRef]
- Shen, W.C.; Ballou, B.; Ryser, H.J.; Hakala, T.R. Targeting, internalization, and cytotoxicity of methotrexate-monoclonal anti-stage-specific embryonic antigen-1 antibody conjugates in cultured F-9 teratocarcinoma cells. Cancer Res. 1986, 46, 3912–3916. [Google Scholar]
- Johnson, D.A.; Laguzza, B.C. Antitumor xenograft activity with a conjugate of a Vinca derivative and the squamous carcinoma-reactive monoclonal antibody PF1/D. Cancer Res. 1987, 47, 3118–3122. [Google Scholar] [CrossRef]
- Dillman, R.O.; Johnson, D.E.; Shawler, D.L.; Koziol, J.A. Superiority of an acid-labile daunorubicin-monoclonal antibody immunoconjugate compared to free drug. Cancer Res. 1988, 48, 6097–6102. [Google Scholar]
- Tolcher, A.W.; Sugarman, S.; Gelmon, K.A.; Cohen, R.; Saleh, M.; Isaacs, C.; Young, L.; Healey, D.; Onetto, N.; Slichenmyer, W. Randomized Phase II Study of BR96-Doxorubicin Conjugate in Patients with Metastatic Breast Cancer. J. Clin. Oncol. 1999, 17, 478. [Google Scholar] [CrossRef]
- Senter, P.D. Potent antibody drug conjugates for cancer therapy. Curr. Opin. Chem. Biol. 2009, 13, 235–244. [Google Scholar] [CrossRef]
- Wellcome Sanger Institute. Genomics of Drug Sensitivity in Cancer. Available online: https://www.cancerrxgene.org/compound/Mitomycin-C/136/overview/ic50 (accessed on 13 July 2020).
- Li, F.; Emmerton, K.K.; Jonas, M.; Zhang, X.; Miyamoto, J.B.; Setter, J.R.; Nicholas, N.D.; Okeley, N.M.; Lyon, R.P.; Benjamin, D.R.; et al. Intracellular Released Payload Influences Potency and Bystander-Killing Effects of Antibody-Drug Conjugates in Preclinical Models. Cancer Res. 2016, 76, 2710–2719. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Elgersma, R.C.; Coumans, R.G.E.; Huijbregts, T.; Menge, W.M.P.B.; Joosten, J.A.F.; Spijker, H.J.; de Groot, F.M.H.; van der Lee, M.M.C.; Ubink, R.; van den Dobbelsteen, D.J.; et al. Design, Synthesis, and Evaluation of Linker-Duocarmycin Payloads: Toward Selection of HER2-Targeting Antibody–Drug Conjugate SYD985. Mol. Pharm. 2015, 12, 1813–1835. [Google Scholar] [CrossRef] [PubMed]
- Banerji, U.; van Herpen, C.M.L.; Saura, C.; Thistlethwaite, F.; Lord, S.; Moreno, V.; Macpherson, I.R.; Boni, V.; Rolfo, C.; de Vries, E.G.E.; et al. Trastuzumab duocarmazine in locally advanced and metastatic solid tumours and HER2-expressing breast cancer: A phase 1 dose-escalation and dose-expansion study. Lancet Oncol. 2019, 20, 1124–1135. [Google Scholar] [CrossRef]
- Mantaj, J.; Jackson, P.J.M.; Karu, K.; Rahman, K.M.; Thurston, D.E. Covalent Bonding of Pyrrolobenzodiazepines (PBDs) to Terminal Guanine Residues within Duplex and Hairpin DNA Fragments. PLoS ONE 2016, 11, e0152303. [Google Scholar] [CrossRef] [PubMed]
- Gerratana, B. Biosynthesis, synthesis, and biological activities of pyrrolobenzodiazepines. Med. Res. Rev. 2012, 32, 254–293. [Google Scholar] [CrossRef]
- Kahl, B.S.; Hamadani, M.; Radford, J.; Carlo-Stella, C.; Caimi, P.; Reid, E.; Feingold, J.M.; Ardeshna, K.M.; Solh, M.; Heffner, L.T.; et al. A phase I study of ADCT-402 (loncastuximab tesirine), a novel pyrrolobenzodiazepine-based antibody–drug conjugate, in relapsed/refractory B-cell non-Hodgkin lymphoma. Clin. Cancer Res. 2019, 25, 6986–6994. [Google Scholar] [CrossRef]
- Starodub, A.N.; Ocean, A.J.; Shah, M.A.; Guarino, M.J.; Picozzi, V.J., Jr.; Vahdat, L.T.; Thomas, S.S.; Govindan, S.V.; Maliakal, P.P.; Wegener, W.A.; et al. First-in-Human Trial of a Novel Anti-Trop-2 Antibody-SN-38 Conjugate, Sacituzumab Govitecan, for the Treatment of Diverse Metastatic Solid Tumors. Clin. Cancer Res. 2015, 21, 3870–3878. [Google Scholar] [CrossRef]
- Ogitani, Y.; Aida, T.; Hagihara, K.; Yamaguchi, J.; Ishii, C.; Harada, N.; Soma, M.; Okamoto, H.; Oitate, M.; Arakawa, S.; et al. DS-8201a, A Novel HER2-Targeting ADC with a Novel DNA Topoisomerase I Inhibitor, Demonstrates a Promising Antitumor Efficacy with Differentiation from T-DM1. Clin. Cancer Res. 2016, 22, 5097–5108. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef]
- Trendowski, M. Recent Advances in the Development of Antineoplastic Agents Derived from Natural Products. Drugs 2015, 75, 1993–2016. [Google Scholar] [CrossRef]
- Chen, H.; Lin, Z.; Arnst, K.E.; Miller, D.D.; Li, W. Tubulin Inhibitor-Based Antibody-Drug Conjugates for Cancer Therapy. Molecules 2017, 22, 1281. [Google Scholar] [CrossRef]
- Sehn, L.H.; Herrera, A.F.; Flowers, C.R.; Kamdar, M.K.; McMillan, A.; Hertzberg, M.; Assouline, S.; Kim, T.M.; Kim, W.S.; Ozcan, M.; et al. Polatuzumab Vedotin in Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2020, 38, 155–165. [Google Scholar] [CrossRef]
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab Vedotin Antibody–Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models. Cancer Res. 2016, 76, 3003–3013. [Google Scholar] [CrossRef]
- AbbVie News Center. AbbVie Provides Update on Depatuxizumab Mafodotin Depatux-M, An Investigational Medicine for Newly Diagnosed Glioblastoma, an Aggressive Form of Brain Cancer; AbbVie News Center: Chicago, IL, USA, 2019; Volume 3. [Google Scholar]
- Kupchan, S.M.; Komoda, Y.; Court, W.A.; Thomas, G.J.; Smith, R.M.; Karim, A.; Gilmore, C.J.; Haltiwanger, R.C.; Bryan, R.F. Maytansine, a novel antileukemic ansa macrolide from Maytenus ovatus. J. Am. Chem. Soc. 1972, 94, 1354–1356. [Google Scholar] [CrossRef]
- Lopus, M.; Oroudjev, E.; Wilson, L.; Wilhelm, S.; Widdison, W.; Chari, R.; Jordan, M.A. Maytansine and cellular metabolites of antibody-maytansinoid conjugates strongly suppress microtubule dynamics by binding to microtubules. Mol. Cancer Ther. 2010, 9, 2689–2699. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hamid, O.; Pavlick, A.C.; Kluger, H.; Kim, K.B.; Boasberg, P.D.; Simantov, R.; Crowley, E.; Green, J.A.; Hawthorne, T.; et al. Phase I/II Study of the Antibody-Drug Conjugate Glembatumumab Vedotin in Patients with Advanced Melanoma. J. Clin. Oncol. 2014, 32, 3659–3666. [Google Scholar] [CrossRef]
- Amiri-Kordestani, L.; Blumenthal, G.M.; Xu, Q.C.; Zhang, L.; Tang, S.W.; Ha, L.; Weinberg, W.C.; Chi, B.; Candau-Chacon, R.; Hughes, P.; et al. FDA approval: Ado-trastuzumab emtansine for the treatment of patients with HER2-positive metastatic breast cancer. Clin. Cancer Res. 2014, 20, 4436–4441. [Google Scholar] [CrossRef] [PubMed]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Dowell, J.A.; Korth-Bradley, J.; Liu, H.; King, S.P.; Berger, M.S. Pharmacokinetics of gemtuzumab ozogamicin, an antibody-targeted chemotherapy agent for the treatment of patients with acute myeloid leukemia in first relapse. J. Clin. Pharm. 2001, 41, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Hamann, P.R.; Hinman, L.M.; Hollander, I.; Beyer, C.F.; Lindh, D.; Holcomb, R.; Hallett, W.; Tsou, H.R.; Upeslacis, J.; Shochat, D.; et al. Gemtuzumab ozogamicin, a potent and selective anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Bioconjug. Chem. 2002, 13, 47–58. [Google Scholar] [CrossRef]
- Wynne, J.; Wright, D.; Stock, W. Inotuzumab: From preclinical development to success in B-cell acute lymphoblastic leukemia. Blood Adv. 2019, 3, 96. [Google Scholar] [CrossRef] [PubMed]
- Gymnopoulos, M.; Betancourt, O.; Blot, V.; Fujita, R.; Galvan, D.; Lieuw, V.; Nguyen, S.; Snedden, J.; Stewart, C.; Villicana, J.; et al. TR1801-ADC: A highly potent cMet antibody-drug conjugate with high activity in patient-derived xenograft models of solid tumors. Mol. Oncol. 2020, 14, 54–68. [Google Scholar] [CrossRef]
- Balendiran, G.K.; Dabur, R.; Fraser, D. The role of glutathione in cancer. Cell Biochem. Funct. 2004, 22, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.N.; Martin, L.P.; O’Malley, D.M.; Matulonis, U.A.; Konner, J.A.; Vergote, I.; Ponte, J.F.; Birrer, M.J. A review of mirvetuximab soravtansine in the treatment of platinum-resistant ovarian cancer. Future Oncol. 2018, 14, 123–136. [Google Scholar] [CrossRef]
- Erickson, H.K.; Park, P.U.; Widdison, W.C.; Kovtun, Y.V.; Garrett, L.M.; Hoffman, K.; Lutz, R.J.; Goldmacher, V.S.; Blättler, W.A. Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res. 2006, 66, 4426–4433. [Google Scholar] [CrossRef]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab emtansine: Mechanisms of action and drug resistance. Breast Cancer Res. 2014, 16, 209. [Google Scholar] [CrossRef]
- Lee, L.; Bounds, D.; Paterson, J.; Herledan, G.; Sully, K.; Seestaller-Wehr, L.M.; Fieles, W.E.; Tunstead, J.; McCahon, L.; Germaschewski, F.M.; et al. Evaluation of B cell maturation antigen as a target for antibody drug conjugate mediated cytotoxicity in multiple myeloma. Br. J. Haematol. 2016, 174, 911–922. [Google Scholar] [CrossRef]
- Shefet-Carasso, L.; Benhar, I. Antibody-targeted drugs and drug resistance—Challenges and solutions. Drug Resist. Updates 2015, 18, 36–46. [Google Scholar] [CrossRef]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Mark, S.D.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925. [Google Scholar] [CrossRef] [PubMed]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.C.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 7063. [Google Scholar] [CrossRef] [PubMed]
- Hallam, T.J.; Wold, E.; Wahl, A.; Smider, V.V. Antibody Conjugates with Unnatural Amino Acids. Mol. Pharm. 2015, 12, 1848–1862. [Google Scholar] [CrossRef] [PubMed]
- Junutula, J.R.; Bhakta, S.; Raab, H.; Ervin, K.E.; Eigenbrot, C.; Vandlen, R.; Scheller, R.H.; Lowman, H.B. Rapid identification of reactive cysteine residues for site-specific labeling of antibody-Fabs. J. Immunol. Methods 2008, 332, 41–52. [Google Scholar] [CrossRef]
- David, R.; Jason, S.R.; Gregory, W.D.; Peng, W.; Carolyn, R.B. Site-specific chemical protein conjugation using genetically encoded aldehyde tags. Nat. Protoc. 2012, 7, 1052. [Google Scholar] [CrossRef]
- Sunbul, M.; Yin, J. Site specific protein labeling by enzymatic posttranslational modification. Org. Biomol. Chem. 2009, 7, 3361–3371. [Google Scholar] [CrossRef] [PubMed]
- Gauzy-Lazo, L.; Sassoon, I.; Brun, M.-P. Advances in Antibody–Drug Conjugate Design: Current Clinical Landscape and Future Innovations. SLAS Discov. Adv. Sci. Drug Discov. 2020, 25, 843–868. [Google Scholar] [CrossRef]
- Sean, D.C.; Sandra, L.S. Regulated portals of entry into the cell. Nature 2003, 422, 37. [Google Scholar] [CrossRef]
- Kalim, M.; Chen, J.; Wang, S.; Lin, C.; Ullah, S.; Liang, K.; Ding, Q.; Chen, S.; Zhan, J. Intracellular trafficking of new anticancer therapeutics: Antibody-drug conjugates. Drug Des. Dev. Ther. 2017, 11, 2265–2276. [Google Scholar] [CrossRef]
- Chalouni, C.; Doll, S. Fate of Antibody-Drug Conjugates in Cancer Cells. J. Exp. Clin. Cancer Res. 2018, 37, 20. [Google Scholar] [CrossRef]
- Alexander, H.S.; Michael, P.B. Antibody drug conjugates and bystander killing: Is antigen-dependent internalisation required? Br. J. Cancer 2017, 117, 1736. [Google Scholar] [CrossRef]
- Khera, E.; Thurber, G. Pharmacokinetic and Immunological Considerations for Expanding the Therapeutic Window of Next-Generation Antibody–Drug Conjugates. BioDrugs 2018, 32, 465–480. [Google Scholar] [CrossRef]
- Fuh, F.K.; Looney, C.; Li, D.; Poon, K.A.; Dere, R.C.; Danilenko, D.M.; McBride, J.; Reed, C.; Chung, S.; Zheng, B.; et al. Anti-CD22 and anti-CD79b antibody-drug conjugates preferentially target proliferating B cells. Br. J. Pharm. 2017, 174, 628–640. [Google Scholar] [CrossRef] [PubMed]
- Deckert, J.; Park, P.U.; Chicklas, S.; Yi, Y.; Li, M.; Lai, K.C.; Mayo, M.F.; Carrigan, C.N.; Erickson, H.K.; Pinkas, J.; et al. A novel anti-CD37 antibody-drug conjugate with multiple anti-tumor mechanisms for the treatment of B-cell malignancies. Blood 2013, 122, 3500–3510. [Google Scholar] [CrossRef]
- Pro, B.; Advani, R.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.; Connors, J.M.; et al. Brentuximab Vedotin (SGN-35) in Patients With Relapsed or Refractory Systemic Anaplastic Large-Cell Lymphoma: Results of a Phase II Study. J. Clin. Oncol. 2012, 30, 2190–2196. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wang, F.; Zhang, H.; Chen, B. Brentuximab vedotin for treatment of relapsed or refractory malignant lymphoma: Results of a systematic review and meta-analysis of prospective studies. Drug Des. Devel. Ther. 2015, 9, 2277–2283. [Google Scholar] [CrossRef]
- LaCasce, A.S.; Bociek, R.G.; Sawas, A.; Caimi, P.; Agura, E.; Matous, J.; Ansell, S.M.; Crosswell, H.E.; Islas-Ohlmayer, M.; Behler, C.; et al. Brentuximab vedotin plus bendamustine: A highly active first salvage regimen for relapsed or refractory Hodgkin lymphoma. Blood 2018, 132, 40–48. [Google Scholar] [CrossRef]
- Horwitz, S.M.; Advani, R.H.; Bartlett, N.L.; Jacobsen, E.D.; Sharman, J.P.; O’Connor, O.A.; Siddiqi, T.; Kennedy, D.A.; Oki, Y. Objective responses in relapsed T-cell lymphomas with single-agent brentuximab vedotin. Blood 2014, 123, 3095–3100. [Google Scholar] [CrossRef]
- Prince, H.M.; Kim, Y.H.; Horwitz, S.M.; Dummer, R.; Scarisbrick, J.; Quaglino, P.; Zinzani, P.L.; Wolter, P.; Sanches, J.A.; Ortiz-Romero, P.L.; et al. Brentuximab vedotin or physician’s choice in CD30-positive cutaneous T-cell lymphoma (ALCANZA): An international, open-label, randomised, phase 3, multicentre trial. Lancet 2017, 390, 555–566. [Google Scholar] [CrossRef]
- Connors, J.M.; Jurczak, W.; Straus, D.J.; Ansell, S.M.; Kim, W.S.; Gallamini, A.; Younes, A.; Alekseev, S.; Illés, Á.; Picardi, M.; et al. Brentuximab Vedotin with Chemotherapy for Stage III or IV Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 378, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, S.; O’Connor, O.A.; Pro, B.; Illidge, T.; Fanale, M.; Advani, R.; Bartlett, N.L.; Christensen, J.H.; Morschhauser, F.; Domingo-Domenech, E.; et al. Brentuximab vedotin with chemotherapy for CD30-positive peripheral T-cell lymphoma (ECHELON-2): A global, double-blind, randomised, phase 3 trial. Lancet 2019, 393, 229–240. [Google Scholar] [CrossRef]
- Wagner-Johnston, N.D.; Bartlett, N.L.; Cashen, A.; Berger, J.R. Progressive multifocal leukoencephalopathy in a patient with Hodgkin lymphoma treated with brentuximab vedotin. Leuk. Lymphoma 2012, 53, 2283–2286. [Google Scholar] [CrossRef]
- Urru, S.A.; Mariotti, E.; Carta, P.; Massidda, S.; Marcias, M.; Murru, R.; Sanna, P.; Angelucci, E. Acute pancreatitis following brentuximab vedotin therapy for refractory Hodgkin lymphoma: A case report. Drugs R&D 2014, 14, 9–11. [Google Scholar] [CrossRef][Green Version]
- Krop, I.E.; Beeram, M.; Modi, S.; Jones, S.F.; Holden, S.N.; Yu, W.; Girish, S.; Tibbitts, J.; Yi, J.H.; Sliwkowski, M.X.; et al. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J. Clin. Oncol. 2010, 28, 2698–2704. [Google Scholar] [CrossRef]
- Krop, I.E.; Kim, S.-B.; Martin, A.G.; Lorusso, P.M.; Ferrero, J.-M.; Badovinac-Crnjevic, T.; Hoersch, S.; Smitt, M.; Wildiers, H. Trastuzumab emtansine versus treatment of physician’s choice in patients with previously treated HER2-positive metastatic breast cancer (TH3RESA): Final overall survival results from a randomised open-label phase 3 trial. Lancet Oncol. 2017, 18, 743–754. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DeAngelo, D.J.; Advani, A.S.; Stelljes, M.; Kebriaei, P.; Cassaday, R.D.; Merchant, A.A.; Fujishima, N.; Uchida, T.; Calbacho, M.; et al. Hepatic adverse event profile of inotuzumab ozogamicin in adult patients with relapsed or refractory acute lymphoblastic leukaemia: Results from the open-label, randomised, phase 3 INO-VATE study. Lancet Haematol. 2017, 4, e387–e398. [Google Scholar] [CrossRef]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2001, 7, 1490. [Google Scholar]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef]
- Castaigne, S.; Pautas, C.; Terré, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.-N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet 2012, 379, 1508–1516. [Google Scholar] [CrossRef]
- Amadori, S.; Suciu, S.; Selleslag, D.; Aversa, F.; Gaidano, G.; Musso, M.; Annino, L.; Venditti, A.; Voso, M.T.; Mazzone, C.; et al. Gemtuzumab Ozogamicin Versus Best Supportive Care in Older Patients with Newly Diagnosed Acute Myeloid Leukemia Unsuitable for Intensive Chemotherapy: Results of the Randomized Phase III EORTC-GIMEMA AML-19 Trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 972. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; O’Donnell, P.H.; Balar, A.V.; McGregor, B.A.; Heath, E.I.; Yu, E.Y.; Galsky, M.D.; Hahn, N.M.; Gartner, E.M.; Pinelli, J.M.; et al. Pivotal Trial of Enfortumab Vedotin in Urothelial Carcinoma After Platinum and Anti-Programmed Death 1/Programmed Death Ligand 1 Therapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 2592. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Mayer, I.A.; Vahdat, L.T.; Tolaney, S.M.; Isakoff, S.J.; Diamond, J.R.; O’Shaughnessy, J.; Moroose, R.L.; Santin, A.D.; Abramson, V.G.; et al. Sacituzumab Govitecan-hziy in Refractory Metastatic Triple-Negative Breast Cancer. N. Engl. J. Med. 2019, 380, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Trudel, S.; Lendvai, N.; Popat, R.; Voorhees, P.M.; Reeves, B.; Libby, E.N.; Richardson, P.G.; Hoos, A.; Gupta, I.; Bragulat, V.; et al. Antibody–drug conjugate, GSK2857916, in relapsed/refractory multiple myeloma: An update on safety and efficacy from dose expansion phase I study. Blood Cancer J. 2019, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.; Li, M.; Kitto, A.; Li, J.; Wang, C.S.; Kirk, D.T.; Yellin, O.; Nichols, C.M.; Dreyer, M.P.; Ahles, C.P.; et al. Serum B-cell maturation antigen is elevated in multiple myeloma and correlates with disease status and survival. Br. J. Haematol. 2012, 158, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.-O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): A two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef]
- Snyder, J.T.; Malinao, M.-C.; Dugal-Tessier, J.; Atkinson, J.E.; Anand, B.S.; Okada, A.; Mendelsohn, B.A. Metabolism of an Oxime-Linked Antibody Drug Conjugate, AGS62P1, and Characterization of its Identified Metabolite. Mol. Pharm. 2018, 15, 2384–2390. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Calvo, E.; Moreno, V.; Chung, H.C.; Park, Y.H.; Bang, Y.-J.; Rosen, L.S.; Mita, M.M.; Garrido-Laguna, I.; Leung, A.C.F.; et al. A phase I dose escalation study evaluating the safety and tolerability of a novel anti-HER2 antibody-drug conjugate (PF-06804103) in patients with HER2-positive solid tumors. J. Clin. Oncol. 2020, 38, 1039. [Google Scholar] [CrossRef]
- Sachdev, J.C.; Maitland, M.L.; Sharma, M.; Moreno, V.; Boni, V.; Kummar, S.; Stringer-Reasor, E.M.; Forero-Torres, A.; Lakhani, N.J.; Gibson, B.; et al. PF-06647020 (PF-7020), an antibody-drug conjugate (ADC) targeting protein tyrosine kinase 7 (PTK7), in patients (pts) with advanced solid tumors: Results of a phase I dose escalation and expansion study. J. Clin. Oncol. 2018, 36, 5565. [Google Scholar] [CrossRef]
- Akla, B.; Broussas, M.; Loukili, N.; Robert, A.; Beau-Larvor, C.; Malissard, M.; Boute, N.; Champion, T.; Haeuw, J.-F.; Beck, A.; et al. Efficacy of the Antibody-Drug Conjugate W0101 in Preclinical Models of IGF-1 Receptor Overexpressing Solid Tumors. Mol. Cancer Ther. 2020, 19, 168–177. [Google Scholar] [CrossRef]
- Hamblett, K.; Barnscher, S.D.; Davies, R.H.; Hammond, P.; Hernandez, A.; Wickman, G.; Fung, V.; Ding, T.; Garnett, G.; Galey, A.; et al. Abstract P6-17-13: ZW49, a HER2 targeted biparatopic antibody drug conjugate for the treatment of HER2 expressing cancers. Cancer Res. 2019, 79 (Suppl. 4), P6-7-13–P16-17-13. [Google Scholar] [CrossRef]
- Hamilton, E.B.M.; Tolcher, A.; Buscema, J.; Papadopoulos, K.P.; Zarwan, C.; Anderson, C.; Doroshow, D. Mersana Therapeutics Reports Updated Interim Data from the Ovarian Cancer Cohort of the XMT-1536 Phase 1 Expansion Study; News Bites Pty Ltd.: Melbourne, Australia, 2020. [Google Scholar]
- Gourd, K. San Antonio Breast Cancer Symposium 2019. Lancet Oncol. 2020, 21, 28. [Google Scholar] [CrossRef]
- Tang, W.; Huang, X.; Ou, Z.; Yan, H.; Gan, J.; Dong, Q.; Tan, B.; Yang, Y.; Guo, Y.; Li, S.; et al. Abstract P6-20-16: BAT8003, a potent anti-Trop-2 antibody-drug conjugate, for the treatment of triple negative breast cancer. Cancer Res. 2019, 79 (Suppl. 4), P6-20-16–P26-20-16. [Google Scholar] [CrossRef]
- Ko, A.H.; Coveler, A.L.; Schlechter, B.L.; Bekaii-Saab, T.S.; Wolpin, B.M.; Clark, J.W.; Cheng, Y.-L.; Cheng, T.-Y.; Langecker, P.J.; Lin, S.-Y. Phase Ia, first-in-human study of AbGn-107, a novel antibody-drug conjugate (ADC), in patients with gastric, colorectal, pancreatic, or biliary cancers. J. Clin. Oncol. 2020, 38, 713. [Google Scholar] [CrossRef]
- Shen, Y.; Yang, T.; Cao, X.; Zhang, Y.; Zhao, L.; Li, H.; Zhao, T.; Xu, J.; Zhang, H.; Guo, Q.; et al. Conjugation of DM1 to anti-CD30 antibody has potential antitumor activity in CD30-positive hematological malignancies with lower systemic toxicity. mAbs 2019, 11, 1149–1161. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Flinn, I.W.; Madan, S.; Maddocks, K.; Freedman, A.; Weitman, S.; Zucca, E.; Munteanu, M.C.; Palomba, M.L. Safety, tolerability, and preliminary activity of IMGN529, a CD37-targeted antibody-drug conjugate, in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: A dose-escalation, phase I study. Investig. New Drugs 2018, 36, 869–876. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Ailawadhi, S.; Weitman, S.D.; Zildjian, S.; O’Leary, J.J.; O’Keeffe, J.; Guild, R.; Whiteman, K.; Chanan-Khan, A.A.A. Phase I study of lorvotuzumab mertansine (LM, IMGN901) in combination with lenalidomide (Len) and dexamethasone (Dex) in patients with CD56-positive relapsed or relapsed/refractory multiple myeloma (MM). J. Clin. Oncol. 2011, 29, 8013. [Google Scholar] [CrossRef]
- Socinski, M.A.; Kaye, F.J.; Spigel, D.R.; Kudrik, F.J.; Ponce, S.; Ellis, P.M.; Majem, M.; Lorigan, P.; Gandhi, L.; Gutierrez, M.E.; et al. Phase 1/2 Study of the CD56-Targeting Antibody-Drug Conjugate Lorvotuzumab Mertansine (IMGN901) in Combination With Carboplatin/Etoposide in Small-Cell Lung Cancer Patients With Extensive-Stage Disease. Clin. Lung. Cancer 2017, 18, 68–76.e62. [Google Scholar] [CrossRef]
- Whiteman, K.R.; Johnson, H.A.; Mayo, M.F.; Audette, C.A.; Carrigan, C.N.; LaBelle, A.; Zukerberg, L.; Lambert, J.M.; Lutz, R.J. Lorvotuzumab mertansine, a CD56-targeting antibody-drug conjugate with potent antitumor activity against small cell lung cancer in human xenograft models. mAbs 2014, 6, 556–566. [Google Scholar] [CrossRef]
- Tolcher, A.; Papadopoulos, K.; Cole, Y.; Rivas, K.; Chandana, S.; Sinclair, S.; Wood, D.; Nadler, P.I.; Lakhani, N. A Phase 1a/2a trial of AVID100, an anti-EGFR antibody-drug conjugate. Ann. Oncol. 2018, 29, iii8. [Google Scholar] [CrossRef]
- Merlino, G.; Fiascarelli, A.; Bigioni, M.; Bressan, A.; Carrisi, C.; Bellarosa, D.; Salerno, M.; Bugianesi, R.; Manno, R.; Bernadó Morales, C.; et al. MEN1309/OBT076, a First-In-Class Antibody–Drug Conjugate Targeting CD205 in Solid Tumors. Mol. Cancer Ther. 2019, 18, 1533–1543. [Google Scholar] [CrossRef]
- Jagannath, S.; Heffner, L.T., Jr.; Ailawadhi, S.; Munshi, N.C.; Zimmerman, T.M.; Rosenblatt, J.; Lonial, S.; Chanan-Khan, A.; Ruehle, M.; Rharbaoui, F.; et al. Indatuximab Ravtansine (BT062) Monotherapy in Patients with Relapsed and/or Refractory Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2019, 19, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Hideshima, T.; Fulciniti, M.; Lutz, R.J.; Yasui, H.; Okawa, Y.; Kiziltepe, T.; Vallet, S.; Pozzi, S.; Santo, L.; et al. The Monoclonal Antibody nBT062 Conjugated to Cytotoxic Maytansinoids Has Selective Cytotoxicity Against CD138-Positive Multiple Myeloma Cells In vitro and In vivo. Clin. Cancer Res. 2009, 15, 4028–4037. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Corbacho, J.; Spira, A.; Boni, V.; Feliu, J.; Middleton, M.; Burris, H.; Yang Weaver, A.; Will, M.; Harding, J.; Meric-Bernstam, F.; et al. PROCLAIM-CX-2009: A first-in-human trial to evaluate CX-2009 in adults with metastatic or locally advanced unresectable solid tumors. Ann. Oncol. 2017, 28, v140. [Google Scholar] [CrossRef]
- Gazzah, A.; Cousin, S.; Boni, V.; Ricordel, C.; Kim, T.M.; Kim, J.-S.; Helissey, C.; Gardeazabal, I.; Chadjaa, M.; Allard, A.; et al. First-in-human phase 1 study of the antibody-drug conjugate (ADC) SAR408701 in advanced solid tumors: Dose-expansion cohort of patients (pts) with non-squamous non-small cell lung cancer (NSQ NSCLC). J. Clin. Oncol. 2019, 37, 9072. [Google Scholar] [CrossRef]
- Moore, K.N.; Matulonis, U.A.; O’Malley, D.M.; Konner, J.A.; Martin, L.P.; Perez, R.P.; Bauer, T.M.; Gilbert, L.; Seward, S.M.; Oza, A.M.; et al. Mirvetuximab soravtansine (IMGN853), a folate receptor alpha (FRα)-targeting antibody-drug conjugate (ADC), in platinum-resistant epithelial ovarian cancer (EOC) patients (pts): Activity and safety analyses in phase I pooled expansion cohorts. J. Clin. Oncol. 2017, 35, 5547. [Google Scholar] [CrossRef]
- Bendell, J.; Blumenschein, G.; Zinner, R.; Hong, D.; Jones, S.; Infante, J.; Burris, H.; Rajagopalan, P.; Kornacker, M.; Henderson, D.; et al. Abstract LB-291: First-in-human phase I dose escalation study of a novel anti-mesothelin antibody drug conjugate (ADC), BAY 94-9343, in patients with advanced solid tumors. Cancer Res. 2013, 73 (Suppl. 8), LB-291. [Google Scholar] [CrossRef]
- Hassan, R.; Jr, G.R.B.; Moore, K.N.; Santin, A.D.; Kindler, H.L.; Nemunaitis, J.J.; Seward, S.M.; Thomas, A.; Kim, S.K.; Rajagopalan, P.; et al. First-in-Human, Multicenter, Phase I Dose-Escalation and Expansion Study of Anti-Mesothelin Antibody–Drug Conjugate Anetumab Ravtansine in Advanced or Metastatic Solid Tumors. J. Clin. Oncol. 2020, 38, 1824–1835. [Google Scholar] [CrossRef]
- Scribner, J.A.; Brown, J.G.; Sharma, S.; Li, H.; Chiechi, M.; Li, P.; Son, T.; Costa, A.D.; Chen, Y.; Chen, F.; et al. Abstract 820: Preclinical development of MGC018, a duocarmycin-based antibody-drug conjugate targeting B7-H3 for solid cancer. Cancer Res. 2018, 78, 820. [Google Scholar] [CrossRef]
- Masuda, N.; Yonemori, K.; Takahashi, S.; Kogawa, T.; Nakayama, T.; Iwase, H.; Takahashi, M.; Toyama, T.; Saeki, T.; Saji, S.; et al. Abstract PD1-03: Single agent activity of U3-1402, a HER3-targeting antibody-drug conjugate, in HER3-overexpressing metastatic breast cancer: Updated results of a phase 1/2 trial. Cancer Res. 2019, 79 (Suppl. 4), PD1-03. [Google Scholar] [CrossRef]
- Iida, K.; Abdelhamid, A.H.; Nagatsuma, A.K.; Shibutani, T.; Yasuda, S.; Kitamura, M.; Hattori, C.; Abe, M.; Hasegawa, J.; Iguchi, T.; et al. Abstract 5181: Therapeutic targeting of GPR20, selectively expressed in gastrointestinal stromal tumor (GIST), with DS-6157a, an antibody-drug conjugate (ADC). Cancer Res. 2020, 80, 5181. [Google Scholar] [CrossRef]
- Sands, J.M.; Shimizu, T.; Garon, E.B.; Greenberg, J.; Guevara, F.M.; Heist, R.S.; Kobayashi, F.; Noguchi, Y.; Okajima, D.; Tajima, N.; et al. First-in-human phase 1 study of DS-1062a in patients with advanced solid tumors. J. Clin. Oncol. 2019, 37, 9051. [Google Scholar] [CrossRef]
- Cheng, X.; Li, J.; Tanaka, K.; Majumder, U.; Milinichik, A.Z.; Verdi, A.C.; Maddage, C.J.; Rybinski, K.A.; Fernando, S.; Fernando, D.; et al. MORAb-202, an Antibody-Drug Conjugate Utilizing Humanized Anti-human FRα Farletuzumab and the Microtubule-targeting Agent Eribulin, has Potent Antitumor Activity. Mol. Cancer Ther. 2018, 17, 2665–2675. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.G.; Erba, H.P.; Papadantonakis, N.; DeAngelo, D.J.; Wang, E.S.; Konopleva, M.Y.; Sloss, C.M.; Culm-Merdek, K.; Zweidler-McKay, P.A.; Kantarjian, H.M. A Phase I, First-in-Human Study Evaluating the Safety and Preliminary Antileukemia Activity of IMGN632, a Novel CD123-Targeting Antibody-Drug Conjugate, in Patients with Relapsed/Refractory Acute Myeloid Leukemia and Other CD123-Positive Hematologic Malignancies. Blood 2018, 132, 27. [Google Scholar] [CrossRef]
- Koopman, L.A.; Janmaat, M.L.; Jacobsen, K.; Terp, M.G.; Heuvel, E.G.-V.D.; Forssman, U.; Lingnau, A.; Parren, P.W.; Ditzel, H.; Breij, E.C. Abstract 832: An AXL-specific antibody-drug conjugate shows preclinical anti-tumor activity in non-small cell lung cancer, including EGFR-inhibitor resistant NSCLC. Cancer Res. 2018, 78, 832. [Google Scholar] [CrossRef]
- Ahnert, J.R.; Taylor, M.H.; O’Reilly, E.M.; Zhang, J.; Doebele, R.C.; Ben, Y.; Sharp, L.L.; Boyle, W.J.; Chang, C.; Frey, G.; et al. A phase 1/2 dose-escalation and expansion study of a conditionally active anti-AXL humanized monoclonal antibody (BA3011) in patients with advanced solid tumors. J. Clin. Oncol. 2018, 36, TPS12126. [Google Scholar] [CrossRef]
- Johnson, M.L.; El-Khoueiry, A.B.; Hafez, N.; Lakhani, N.J.; Mamdani, H.; Ahnert, J.R.; Sanborn, R.E.; Ho, T.; Li, R.; Waldes, J.; et al. CX-2029, a PROBODY drug conjugate targeting CD71 (transferrin receptor): Results from a first-in-human study (PROCLAIM-CX-2029) in patients (Pts) with advanced cancer. J. Clin. Oncol. 2020, 38, 3502. [Google Scholar] [CrossRef]
- Patnaik, A.; Meric-Bernstam, F.; Lima, C.M.S.P.R.; Robert, F.; Dowlati, A.; Kindler, H.L.; Davar, D.; Powell, S.F.; Garfin, P.M.; Balmanoukian, A.S. SGN228-001: A phase I open-label dose-escalation, and expansion study of SGN-CD228A in select advanced solid tumors. J. Clin. Oncol. 2020, 38, TPS3652. [Google Scholar] [CrossRef]
- Goldman, J.; Angevin, E.; Strickler, J.; Camidge, D.; Heist, R.; Morgensztern, D.; Barve, M.; Yue, H.; Beaulieu, J.; Motwani, M.; et al. Phase I Study of ABBV-399 (Telisotuzumab Vedotin) as Monotherapy and in Combination with Erlotinib in NSCLC. J. Thorac. Oncol. 2017, 12, S1805–S1806. [Google Scholar] [CrossRef]
- Xu, B.; Wang, J.; Fang, J.; Chen, X.; Han, Y.; Li, Q.; Zhang, P.; Yuan, P.; Ma, F.; Luo, Y.; et al. Abstract PD4-06: Early clinical development of RC48-ADC in patients with HER2 positive metastatic breast cancer. Cancer Res. 2020, 80, PD4-06. [Google Scholar] [CrossRef]
- Jiang, J.; Li, S.; Shan, X.; Wang, L.; Ma, J.; Huang, M.; Dong, L.; Chen, F. Preclinical safety profile of disitamab vedotin: A novel anti-HER2 antibody conjugated with MMAE. Toxicol. Lett. 2020, 324, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Alteogen Secures. FDA Approval for Orphan Drug Designation with An Antibody-Drug Conjugate for Gastric Cancer. In Health & Beauty Close-Up; Alteogen Inc.: Daejeon, Korea, 2018. [Google Scholar]
- Li, J.; Guo, Y.; Xue, J.; Peng, W.; Ge, X.; Zhao, W.; Dai, C.; Xue, L.; Tang, W.; Hu, C. First-in-human phase I study of anti-HER2 ADC MRG002 in patients with relapsed/refractory solid tumors. J. Clin. Oncol. 2020, 38, TPS1101. [Google Scholar] [CrossRef]
- OBI Pharma, Inc. Press Release: OBI Pharma Granted FDA Orphan Drug Designation for OBI-888 for the Treatment of Pancreatic Cancer; OBI Pharma, Inc.: New York, NY, USA, 2018. [Google Scholar]
- Vaisitti, T.; Jessen, K.; Vo, T.-T.; Ko, M.; Arruga, F.; Vitale, N.; Braggio, E.; Di Napoli, A.; Chadburn, A.; Allan, J.N.; et al. Vls-101 is a Novel Therapeutic Antibody-Drug Conjugate (ADC) Targeting Receptor Tyrosine Kinase-like Orphan Receptor 1 (ROR1) in Richter’s Syndrome (RS). Blood 2019, 134, 2856. [Google Scholar] [CrossRef]
- Han, H.; Diab, S.; Alemany, C.; Basho, R.; Brown-Glaberman, U.; Meisel, J.; Pluard, T.; Cortes, J.; Dillon, P.; Ettl, J.; et al. Abstract PD1-06: Open label phase 1b/2 study of ladiratuzumab vedotin in combination with pembrolizumab for first-line treatment of patients with unresectable locally-advanced or metastatic triple-negative breast cancer. Cancer Res. 2020, 80, PD1-06-PD01-06. [Google Scholar] [CrossRef]
- De Bono, J.S.; Concin, N.; Hong, D.S.; Thistlethwaite, F.C.; Machiels, J.-P.; Arkenau, H.-T.; Plummer, R.; Jones, R.H.; Nielsen, D.; Windfeld, K.; et al. Tisotumab vedotin in patients with advanced or metastatic solid tumours (InnovaTV 201): A first-in-human, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 383–393. [Google Scholar] [CrossRef]
- Skidmore, L.; Sakamuri, S.; Knudsen, N.A.; Hewet, A.G.; Milutinovic, S.; Barkho, W.; Biroc, S.L.; Kirtley, J.; Marsden, R.; Storey, K.; et al. ARX788, a Site-specific Anti-HER2 Antibody-Drug Conjugate, Demonstrates Potent and Selective Activity in HER2-low and T-DM1-resistant Breast and Gastric Cancers. Mol. Cancer Ther. 2020, 19, 1833–1843. [Google Scholar] [CrossRef]
- Lopez, D.M.; Barve, M.; Wang, J.; Bullock, A.J.; Pectasides, E.; Vaishampayan, U.; Spira, A.I.; Ulahannan, S.; Patnaik, A.; Sanborn, R.E.; et al. Abstract B005: A phase I study of A166, a novel anti-HER2 antibody-drug conjugate (ADC), in patients with locally advanced/metastatic solid tumors. Mol. Cancer Ther. 2019, 18, B005. [Google Scholar] [CrossRef]
- Maclaren, A.P.; Levin, N.; Lowman, H. Abstract 835: TRPH-222, a novel anti-CD22 antibody drug conjugate (ADC), has significant anti-tumor activity in NHL xenografts and reduces B cells in monkeys. Cancer Res. 2018, 78, 835. [Google Scholar] [CrossRef]
- Radford, J.; Kahl, B.S.; Hamadani, M.; Carlo-Stella, C.; Caimi, P.; Ardeshna, K.M.; Feingold, J.; He, S.; Reid, E.; Solh, M.; et al. Interim Results from the First-in-Human Clinical Trial of Adct-402 (Loncastuximab Tesirine), a Novel Pyrrolobenzodiazepine-Based Antibody Drug Conjugate, in Relapsed/Refractory Diffuse Large B-Cell Lymphoma. Blood 2018, 132, 398. [Google Scholar] [CrossRef]
- Hamadani, M.; Collins, G.P.; Samaniego, F.; Spira, A.I.; Davies, A.; Radford, J.; Caimi, P.; Menne, T.; Boni, J.; Cruz, H.; et al. Phase 1 Study of Adct-301 (Camidanlumab Tesirine), a Novel Pyrrolobenzodiazepine-Based Antibody Drug Conjugate, in Relapsed/Refractory Classical Hodgkin Lymphoma. Blood 2018, 132, 928. [Google Scholar] [CrossRef]
- Anderson, M.G.; Falls, H.D.; Mitten, M.J.; Oleksijew, A.; Vaidya, K.S.; Boghaert, E.R.; Gao, W.; Palma, J.P.; Cao, D.; Chia, P.-L.; et al. Targeting Multiple EGFR Expressing Tumors with a Highly Potent Tumor-Selective Antibody Drug Conjugate. Mol. Cancer Ther. 2020. [Google Scholar] [CrossRef]
- Abu-Yousif, A.O.; Bannerman, B.M.; Cvet, D.; Gallery, M.; Ganno, M.L.; Smith, M.D.; Lai, K.C.; Keating, T.A.; Bolleddula, J.; Stringer, B.; et al. Abstract B120: TAK-164, a GCC-targeted antibody-drug conjugate (ADC) for the treatment of colorectal cancers and other GI malignancies. Mol. Cancer Ther. 2018, 17, B120. [Google Scholar] [CrossRef]
- Shah, N.N.; Krishnan, A.Y.; Shah, N.D.; Burke, J.M.; Melear, J.M.; Spira, A.I.; Popplewell, L.L.; Andreadis, C.B.; Chhabra, S.; Sharman, J.P.; et al. Preliminary Results of a Phase 1 Dose Escalation Study of the First-in-Class Anti-CD74 Antibody Drug Conjugate (ADC), STRO-001, in Patients with Advanced B-Cell Malignancies. Blood 2019, 134, 5329. [Google Scholar] [CrossRef]
- Li, X.; Abrahams, C.; Zhou, S.; Krimm, S.; Henningsen, R.; Stephenson, H.; Hanson, J.; Masikat, M.R.; Bajjuri, K.; Heibeck, T.; et al. Abstract 1782: Discovery and activity of STRO-002, a novel ADC targeting folate receptor alpha for ovarian and endometrial cancer. Cancer Res. 2018, 78, 1782. [Google Scholar] [CrossRef]
- Yang, C.-Y.; Wang, L.; Sun, X.; Tang, M.; Quan, H.-T.; Zhang, L.-S.; Lou, L.-G.; Gou, S.-H. SHR-A1403, a novel c-Met antibody-drug conjugate, exerts encouraging anti-tumor activity in c-Met-overexpressing models. Acta Pharmacol. Sin. 2019, 40, 971–979. [Google Scholar] [CrossRef]
- García-Alonso, S.; Ocaña, A.; Pandiella, A. Resistance to Antibody-Drug Conjugates. Cancer Res. 2018, 78, 2159. [Google Scholar] [CrossRef] [PubMed]
- Loganzo, F.; Tan, X.; Sung, M.; Jin, G.; Myers, J.S.; Melamud, E.; Wang, F.; Diesl, V.; Follettie, M.T.; Musto, S.; et al. Tumor Cells Chronically Treated with a Trastuzumab–Maytansinoid Antibody–Drug Conjugate Develop Varied Resistance Mechanisms but Respond to Alternate Treatments. Mol. Cancer Ther. 2015, 14, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Al-Rohil, R.N.; Torres-Cabala, C.A.; Patel, A.; Tetzlaff, M.T.; Ivan, D.; Nagarajan, P.; Curry, J.L.; Miranda, R.N.; Duvic, M.; Prieto, V.G.; et al. Loss of CD30 expression after treatment with brentuximab vedotin in a patient with anaplastic large cell lymphoma: A novel finding. J. Cutan. Pathol. 2016, 43, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- Phillips, G.D.L.; Fields, C.T.; Li, G.; Dowbenko, D.; Schaefer, G.; Miller, K.; Andre, F.; Burris, H.A.; Albain, K.S.; Harbeck, N.; et al. Dual targeting of HER2-positive cancer with trastuzumab emtansine and pertuzumab: Critical role for neuregulin blockade in antitumor response to combination therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 456. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.; Golas, J.; Wang, F.; King, L.; Myers, J.; Rosfjord, E.; Lucas, J. Caveolae-Mediated Endocytosis as a Novel Mechanism of Resistance to Trastuzumab Emtansine (T-DM1). Mol. Cancer Ther. 2018, 17, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Ríos-Luci, C.; García-Alonso, S.; Díaz-Rodríguez, E.; Nadal-Serrano, M.; Arribas, J.; Ocaña, A.; Pandiella, A. Resistance to the Antibody-Drug Conjugate T-DM1 Is Based in a Reduction in Lysosomal Proteolytic Activity. Cancer Res. 2017, 77, 4639. [Google Scholar] [CrossRef] [PubMed]
- Kinneer, K.; Meekin, J.; Tiberghien, A.C.; Tai, Y.-T.; Phipps, S.; Kiefer, C.M.; Rebelatto, M.C.; Dimasi, N.; Moriarty, A.; Papadopoulos, K.P.; et al. SLC46A3 as a Potential Predictive Biomarker for Antibody-Drug Conjugates Bearing Noncleavable Linked Maytansinoid and Pyrrolobenzodiazepine Warheads. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 6570. [Google Scholar] [CrossRef] [PubMed]
- Hunter, F.W.; Barker, H.R.; Lipert, B.; Rothé, F.; Gebhart, G.; Piccart-Gebhart, M.J.; Sotiriou, C.; Jamieson, S.M.F. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br. J. Cancer 2020, 122, 603–612. [Google Scholar] [CrossRef]
- Collins, D.M.; Bossenmaier, B.; Kollmorgen, G.; Niederfellner, G. Acquired Resistance to Antibody-Drug Conjugates. Cancers 2019, 11, 394. [Google Scholar] [CrossRef] [PubMed]
- Haag, P.; Viktorsson, K.; Lindberg, M.L.; Kanter, L.; Lewensohn, R.; Stenke, L. Deficient activation of Bak and Bax confers resistance to gemtuzumab ozogamicin-induced apoptotic cell death in AML. Exp. Hematol. 2009, 37, 755–766. [Google Scholar] [CrossRef]
- Walter, R.B.; Raden, B.W.; Cronk, M.R.; Bernstein, I.D.; Appelbaum, F.R.; Banker, D.E. The peripheral benzodiazepine receptor ligand PK11195 overcomes different resistance mechanisms to sensitize AML cells to gemtuzumab ozogamicin. Blood 2004, 103, 4276–4284. [Google Scholar] [CrossRef] [PubMed]
- Loganzo, F.; Sung, M.; Gerber, H.-P. Mechanisms of Resistance to Antibody–Drug Conjugates. Mol. Cancer Ther. 2016, 15, 2825–2834. [Google Scholar] [CrossRef] [PubMed]
- Kovtun, Y.V.; Audette, C.A.; Mayo, M.F.; Jones, G.E.; Doherty, H.; Maloney, E.K.; Erickson, H.K.; Sun, X.; Wilhelm, S.; Ab, O.; et al. Antibody-Maytansinoid Conjugates Designed to Bypass Multidrug Resistance. Cancer Res. 2010, 70, 2528–2537. [Google Scholar] [CrossRef] [PubMed]
- Kaitin, K.I.; Dimasi, J.A. Pharmaceutical Innovation in the 21st Century: New Drug Approvals in the First Decade, 2000–2009. Clin. Pharmacol. Ther. 2010, 89, 183. [Google Scholar] [CrossRef]
- DiMasi, J.A.; Grabowski, H.G. The cost of biopharmaceutical R&D: Is biotech different? Manag. Decis. Econ. 2007, 28, 469–479. [Google Scholar] [CrossRef]
- Dammes, N.; Peer, D. Monoclonal antibody-based molecular imaging strategies and theranostic opportunities. Theranostics 2020, 10, 938–955. [Google Scholar] [CrossRef]
- Carmon, K.S.; Azhdarinia, A. Application of Immuno-PET in Antibody–Drug Conjugate Development. Mol. Imaging 2018, 17, 1536012118801223. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, G.A.M.S.; Visser, G.W.M.; Lub-De Hooge, M.N.; De Vries, E.G.; Perk, L.R. Immuno-PET: A Navigator in Monoclonal Antibody Development and Applications. Oncologist 2007, 12, 1379–1389. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Rosenkrans, Z.T.; Liu, J.; Huang, G.; Luo, Q.-Y.; Cai, W. ImmunoPET: Concept, Design, and Applications. Chem. Rev. 2020, 120, 3787–3851. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Jiang, D.; Ehlerding, E.B.; Barnhart, T.E.; Ni, D.; Engle, J.W.; Wang, R.; Huang, P.; Xu, X.; Cai, W. Noninvasive Trafficking of Brentuximab Vedotin and PET Imaging of CD30 in Lung Cancer Murine Models. Mol. Pharm. 2018, 15, 1627–1634. [Google Scholar] [CrossRef]
- Wang, R.; Li, L.; Zhang, S.; Li, Y.; Wang, X.; Miao, Q.; Zhen, Y. A novel enediyne-integrated antibody–drug conjugate shows promising antitumor efficacy against CD30+ lymphomas. Mol. Oncol. 2018, 12, 339–355. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, L.; Duan, A.; Li, Y.; Liu, X.; Miao, Q.; Gong, J.; Zhen, Y. Crizotinib enhances anti-CD30-LDM induced antitumor efficacy in NPM-ALK positive anaplastic large cell lymphoma. Cancer Lett. 2019, 448, 84–93. [Google Scholar] [CrossRef]
- Gong, J.; Guo, F.; Cheng, W.; Fan, H.; Miao, Q.; Yang, J. Preliminary biological evaluation of 123I-labelled anti-CD30-LDM in CD30-positive lymphomas murine models. Artif. Cells Nanomed. Biotechnol. 2020, 48, 408–414. [Google Scholar] [CrossRef]
- Glynne-Jones, E.; Harper, M.E.; Seery, L.T.; James, R.; Anglin, I.; Morgan, H.E.; Taylor, K.M.; Gee, J.M.; Nicholson, R.I. TENB2, a proteoglycan identified in prostate cancer that is associated with disease progression and androgen independence. Int. J. Cancer 2001, 94, 178–184. [Google Scholar] [CrossRef]
- Hubert, R.S.; Vivanco, I.; Chen, E.; Rastegar, S.; Leong, K.; Mitchell, S.C.; Madraswala, R.; Zhou, Y.; Kuo, J.; Raitano, A.B.; et al. STEAP: A prostate-specific cell-surface antigen highly expressed in human prostate tumors. Proc. Natl. Acad. Sci. USA 1999, 96, 14523. [Google Scholar] [CrossRef]
- Williams, S.; Ogasawara, A.; Tinianow, J.; Flores, J.; Kan, D.; Lau, J.; Go, M.; Vanderbilt, A.; Gill, H.; Miao, L.; et al. ImmunoPET helps predicting the efficacy of antibody-drug conjugates targeting TENB2 and STEAP1. Oncotarget 2016, 7, 25103–25112. [Google Scholar] [CrossRef] [PubMed]
- Servais, E.L.; Colovos, C.; Rodriguez, L.; Bograd, A.J.; Nitadori, J.-I.; Sima, C.; Rusch, V.W.; Sadelain, M.; Adusumilli, P.S. Mesothelin overexpression promotes mesothelioma cell invasion and MMP-9 secretion in an orthotopic mouse model and in epithelioid pleural mesothelioma patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 2478–2489. [Google Scholar] [CrossRef] [PubMed]
- Lamberts, L.E.; Menke-van Der Houven van Oordt, C.W.; Ter Weele, E.J.; Bensch, F.; Smeenk, M.M.; Voortman, J.; Hoekstra, O.S.; Williams, S.P.; Fine, B.M.; Maslyar, D.; et al. ImmunoPET with Anti-Mesothelin Antibody in Patients with Pancreatic and Ovarian Cancer before Anti-Mesothelin Antibody-Drug Conjugate Treatment. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 1642. [Google Scholar] [CrossRef]
- Azhdarinia, A.; Voss, J.; Ghosh, S.C.; Simien, J.A.; Hernandez Vargas, S.; Cui, J.; Yu, W.A.; Liu, Q.; Carmon, K.S. Evaluation of Anti-LGR5 Antibodies by ImmunoPET for Imaging Colorectal Tumors and Development of Antibody-Drug Conjugates. Mol. Pharm. 2018, 15, 2448–2454. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.G.; Mortensson, E.; Williams, A.C. Targeting LGR5 in Colorectal Cancer: Therapeutic gold or too plastic? Br. J. Cancer 2018, 118, 1410–1418. [Google Scholar] [CrossRef]
- Gong, X.; Azhdarinia, A.; Ghosh, S.C.; Xiong, W.; An, Z.; Liu, Q.; Carmon, K.S. LGR5-Targeted Antibody-Drug Conjugate Eradicates Gastrointestinal Tumors and Prevents Recurrence. Mol. Cancer Ther. 2016, 15, 1580–1590. [Google Scholar] [CrossRef]
- Scott, A.M.; Geleick, D.; Rubira, M.; Clarke, K.; Nice, E.C.; Smyth, F.E.; Stockert, E.; Richards, E.C.; Carr, F.J.; Harris, W.J.; et al. Construction, production, and characterization of humanized anti-Lewis Y monoclonal antibody 3S193 for targeted immunotherapy of solid tumors. Cancer Res. 2000, 60, 3254. [Google Scholar] [CrossRef]
- Boghaert, E.; Sridharan, L.; Armellino, D.C.; Khandke, K.; Dijoseph, J.; Kunz, A.; Dougher, M.M.; Jiang, F.; Kalyandrug, L.; Hamann, P.; et al. Antibody-targeted chemotherapy with the calicheamicin conjugate hu3S193-N-Acetyl gamma calicheamicin dimethyl hydrazide targets lewisy and eliminates Lewis(y)-positive human carcinoma cells and xenografts. Clin. Cancer Res. 2004, 10, 4538–4549. [Google Scholar] [CrossRef]
- Herbertson, R.A.; Tebbutt, N.C.; Lee, F.-T.; Macfarlane, D.J.; Chappell, B.; Micallef, N.; Lee, S.-T.; Saunder, T.; Hopkins, W.; Smyth, F.E.; et al. Phase I biodistribution and pharmacokinetic study of Lewis Y-targeting immunoconjugate CMD-193 in patients with advanced epithelial cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 6709. [Google Scholar] [CrossRef]
- Scott, A.M.; Tebbutt, N.; Lee, F.-T.; Cavicchiolo, T.; Liu, Z.; Gill, S.; Poon, A.M.T.; Hopkins, W.; Smyth, F.E.; Murone, C.; et al. A phase I biodistribution and pharmacokinetic trial of humanized monoclonal antibody Hu3s193 in patients with advanced epithelial cancers that express the Lewis-Y antigen. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 3286. [Google Scholar] [CrossRef]
- Gebhart, G.; Lamberts, L.E.; Wimana, Z.; Garcia, C.; Emonts, P.; Ameye, L.; Stroobants, S.; Huizing, M.; Aftimos, P.; Tol, J.; et al. Molecular imaging as a tool to investigate heterogeneity of advanced HER2-positive breast cancer and to predict patient outcome under trastuzumab emtansine (T-DM1): The ZEPHIR trial. Ann. Oncol. 2016, 27, 619–624. [Google Scholar] [CrossRef] [PubMed]




| Indication | Targets |
|---|---|
| Acute myeloid leukaemia | CD25, CD33, CD123 (IL-3Rα), FLT3 |
| Breast cancer | CD25, CD174, CD197 (CCR7), CD205 (Ly75), CD228 (P79, SEMF), c-MET, CRIPTO, ErbB2 (HER2), ErbB3 (HER3), FLOR1 (FRα), Globo H, GPNMB, IGF-1R, integrin β-6, PTK7 (CCK4), nectin-4 (PVRL4), ROR2, SLC39A6 (LIV1A ZIP6) |
| Bladder cancer | CD25, CD205(Ly75) |
| Colorectal cancer | CD74, CD174, CD166, CD227 (MUC-1), CD326 (Epcam), CEACAM5, CRIPTO, FAP, ED-B, ErbB3 (HER3) |
| Gastric cancer | CD25, CD197 (CCR7), CD228 (P79, SEMF), FLOR1(FRα), Globo H, GRP20, GCC, SLC39A6 (LIV1A ZIP6) |
| Gliomas GIII and GIV | CD25, EGFR |
| Head and neck cancer | CD71 (transferrin R), CD197 (CCR7), EGFR, SLC39A6 (LIV1A ZIP6) |
| Hodgkin’s lymphoma | CD25, CD30, CD197 (CCR7) |
| Lung cancer | Axl, alpha v beta6, CD25, CD56, CD71 (transferrin R), CD228 (P79, SEMF), CD326, CRIPTO, EGFR, ErbB3 (HER3), FAP, Globo H, GD2, IGF-1R, integrin β-6, mesothelin, PTK7 (CCK4), ROR2, SLC34A2 (NaPi2b), SLC39A6 (LIV1A ZIP6) |
| Liver cancer | CD276 (B7-H3), c-MET |
| Melanoma | CD276 (B7-H3), GD2, GPNMB, ED-B, PMEL 17, endothelin B receptor |
| Mesothelioma | Mesothelin, CD228 (P79, SEMF) |
| Multiple Myeloma | CD38, CD46 (MCP), CD56, CD74, CD138, CD269 (BCMA), endothelin B receptor |
| Non-Hodgkin Lymphoma | CD19, CD20, CD22, CD25, CD30, CD37, CD70, CD71 (transferrin R), CD72, CD79, CD180, CD205 (Ly75), ROR1 |
| Ovarian cancer | CA125(MUC16), CD142 (TF), CD205 (Ly75), FLOR1(FRα), Globo H, mesothelin, PTK7 (CCK4) |
| Pancreatic cancer | CD25, CD71 (transferrin R), CD74, CD227 (MUC1), CD228 (P79, SEMF), GRP20, GCC, IGF-1R, integrin β-6, nectin-4 (PVRL4), SLC34A2 (NaPi2b), SLC44A4, alpha v beta6, mesothelin |
| Prostate cancer | CD46 (MCP), PSMA, STEAP-1, SLC44A4, TENB2 |
| Renal cancer | AGS-16, EGFR, c-MET, CAIX, CD70, FLOR1 (FRα) |
| Payload | Target Antigen | Antibody–Drug Conjugates | Antibody | Drug to Antibody Ratio | Linker | Approved Indications | Year of FDA Approval | Year of EMA Approval |
|---|---|---|---|---|---|---|---|---|
| Calicheamicin derivative | CD22 | Inotuzumab ozogamicin (Besponsa) | Recombinant humanized IgG4 | 5–7 | Acid-labile hydrazone-based linker | B cell precursor ALL | 2017 | 2017 |
| CD33 | Gemtuzumab ozogamicin (Mylotarg) | Humanized IgG4 | 2–3 | Acid-labile hydrazone-based linker | CD33-positive AML | 2000 (withdrawn 2010); reapproved 2017 | 2018 | |
| DM1 | ErbB2 | Trastuzumab emtansine (T-DM1, Kadcyla), | Humanized IgG1 | 3.5 | Non-cleavable thioether linker | ErbB2-positive metastatic breast cancer | 2013 | 2013 |
| MMAE | CD30 | Brentuximab vedotin (SGN-35, Adcetris) | Chimeric IgG1 | 4 | Protease-cleavable linker | Hodgkin’s lymphoma, ALCL, PTCL, mycosis fungoides | 2011 | 2012 |
| CD79 | Polatuzumab vedotin (Polivy) | Humanized IgG1 | 4 | Protease-cleavable linker | DLBCL | 2019 | Not approved by the EMA | |
| Nectin-4 | Enfortumab vedotin (ASG-22ME, Padcev) | Human IgG1 | 4 | Cleavable valine-citrulline linker | Advanced urothelial cancer | 2019 | Not approved by the EMA | |
| MMAF | BCMA | Belantamab mafodotin (GSK2857916, Blenrep) | Humanized IgG1 | Unknown | Non-cleavable maleimidocaproyl (mc) linker | Relapsed/refractory multiple myeloma | 2020 | Orphan drug designation by the EMA, 2017 |
| DXd (DX-8951 derivative) | ErbB2 | Trastuzumab deruxtecan (DS-8201a, Enhertu) | Humanized IgG1 | 8 | Cleavable peptide linker | Metastatic ErbB2-positive breast cancer | 2019 | Not approved by the EMA |
| SN-38 | TROP2 | Sacituzumab govitecan (IMMU-132, Trodelvy) | Humanized IgG1 | 7.6 | Cleavable CL2A linker | Triple-negative breast cancer | 2020 | Not approved by the EMA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hafeez, U.; Parakh, S.; Gan, H.K.; Scott, A.M. Antibody–Drug Conjugates for Cancer Therapy. Molecules 2020, 25, 4764. https://doi.org/10.3390/molecules25204764
Hafeez U, Parakh S, Gan HK, Scott AM. Antibody–Drug Conjugates for Cancer Therapy. Molecules. 2020; 25(20):4764. https://doi.org/10.3390/molecules25204764
Chicago/Turabian StyleHafeez, Umbreen, Sagun Parakh, Hui K. Gan, and Andrew M. Scott. 2020. "Antibody–Drug Conjugates for Cancer Therapy" Molecules 25, no. 20: 4764. https://doi.org/10.3390/molecules25204764
APA StyleHafeez, U., Parakh, S., Gan, H. K., & Scott, A. M. (2020). Antibody–Drug Conjugates for Cancer Therapy. Molecules, 25(20), 4764. https://doi.org/10.3390/molecules25204764