Kathon Induces Fibrotic Inflammation in Lungs: The First Animal Study Revealing a Causal Relationship between Humidifier Disinfectant Exposure and Eosinophil and Th2-Mediated Fibrosis Induction


Abstract
1. Introduction
2. Results
2.1. Changes in Organ Weight
2.2. Histological Changes in Lung Samples
2.3. Cellular Changes and Cytokine Levels in BALF
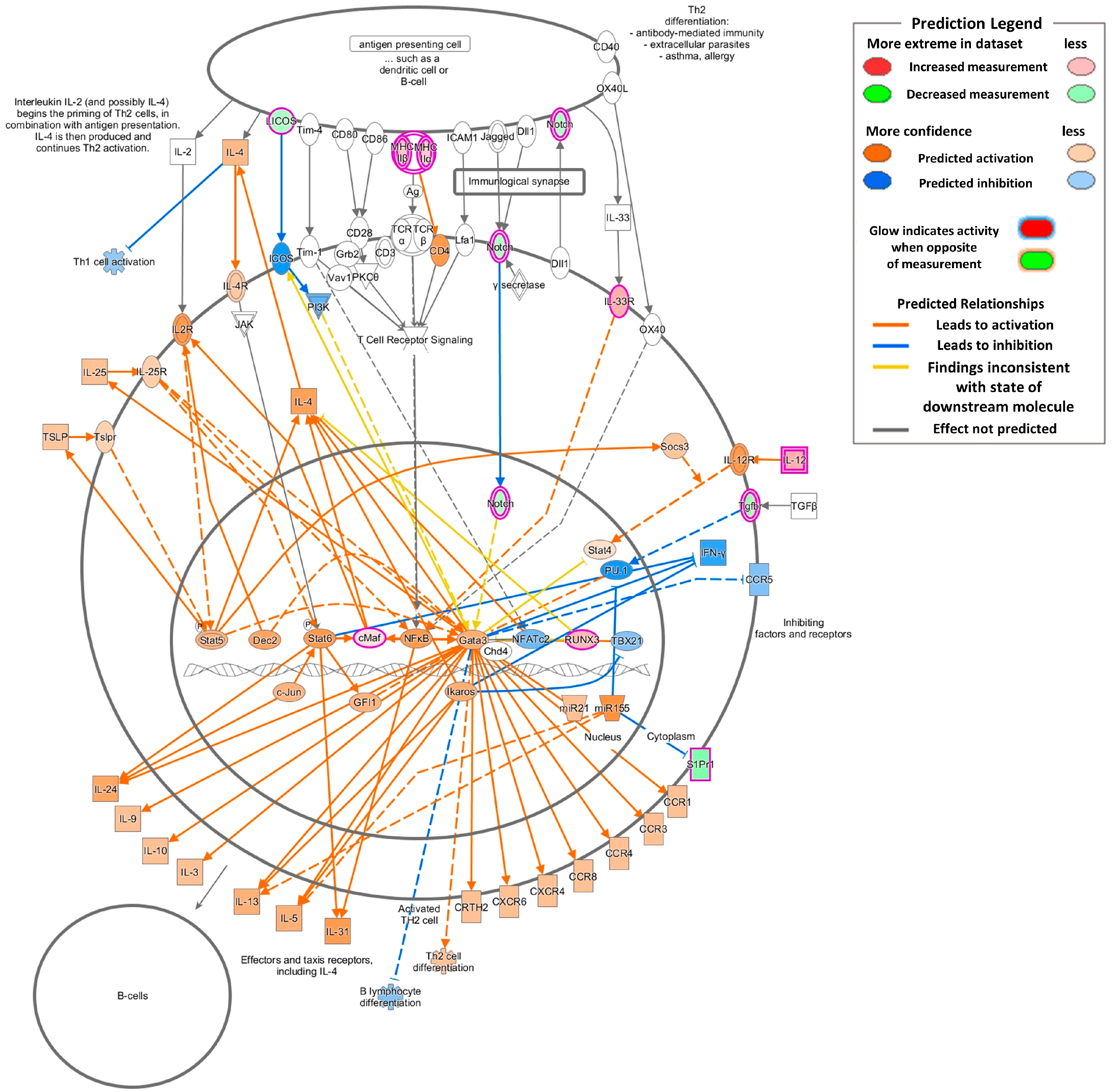
2.4. Gene Expression Changes Associated with Inflammation and Asthmatic Responses
3. Discussion
4. Materials and Methods
4.1. Animals and Exposure Protocol
4.2. BALF Preparation
4.3. Measurement of Cytokine Levels
4.4. Histological Analysis
4.5. RNA Isolation, Library Preparation, and Sequencing
4.6. Data Analysis
4.7. GO Category and Pathway Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Korea Centers for Disease Control and Prevention (KCDC). Interim report of epidemiologic investigation of lung injury with unknown causes in Korea. Public Health Wkly. Rep. 2011, 4, 817–818. [Google Scholar]
- Jeon, B.-H.; Park, Y.J. Frequency of Humidifier and Humidifier Disinfectant Usage in Gyeonggi Provine. Environ. Health Toxicol. 2012, 27, 2012002. [Google Scholar] [CrossRef] [PubMed]
- Bourke, S.J.; Convery, R.P.; Stenton, S.C.; Malcolm, R.M.; Hendrick, D.J. Occupational asthma in an isothiazolinone manufacturing plant. Thorax 1997, 52, 746–748. [Google Scholar] [CrossRef] [PubMed]
- Nagorka, R.; Gleue, C.; Scheller, C.; Moriske, H.-J.; Straff, W. Isothiazolone emissions from building products. Indoor Air 2014, 25, 68–78. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Kim, H.H.; Cho, K.-H. Acute Cardiovascular Toxicity of Sterilizers, PHMG, and PGH: Severe Inflammation in Human Cells and Heart Failure in Zebrafish. Cardiovasc. Toxicol. 2012, 13, 148–160. [Google Scholar] [CrossRef]
- Park, S.; Lee, K.; Lee, E.J.; Lee, S.Y.; In, K.H.; Kim, H.K.; Kang, M.-S. Humidifier Disinfectant–associated Interstitial Lung Disease in an Animal Model Induced by Polyhexamethylene Guanidine Aerosol. Am. J. Respir. Crit. Care Med. 2014, 190, 706–708. [Google Scholar] [CrossRef]
- Kim, H.-R.; Hwang, G.-W.; Naganuma, A.; Chung, K.-H. Adverse health effects of humidifier disinfectants in Korea: Lung toxicity of polyhexamethylene guanidine phosphate. J. Toxicol. Sci. 2016, 41, 711–717. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Park, D.; Do, K.-H.; Jang, S.J.; Hong, S.-J. The Pathological Findings of Chloromethylisothiazolinone and Methylisothiazolinone-associated Lung Injury. J. Korean Med Sci. 2019, 34, e102. [Google Scholar] [CrossRef]
- Lee, E.; Son, S.K.; Yoon, J.; Cho, H.-J.; Yang, S.-I.; Jung, S.; Do, K.-H.; Cho, Y.A.; Lee, S.-Y.; Park, D.; et al. Two Cases of Chloromethylisothiazolinone and Methylisothiazolinone-associated Toxic Lung Injury. J. Korean Med. Sci. 2018, 33, 119. [Google Scholar] [CrossRef]
- Park, E.-J.; Han, J.-S.; Seong, E.; Lee, B.-S.; Lee, S.J.; Lee, K.; Park, E.-J. Inhaled Kathon may induce eosinophilia-mediated disease in the lung. Environ. Toxicol. 2019, 35, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Go, H.N.; Lee, S.H.; Cho, H.J.; Ahn, J.R.; Kang, M.J.; Lee, S.Y.; Hong, S.J. Effects of chloromethylisothiazolinone/methylisothiazolinone (CMIT/MIT) on Th2/Th17-related immune modulation in an atopic dermatitis mouse model. Sci. Rep. 2020, 10, 4099. [Google Scholar] [CrossRef] [PubMed]
- Song, J.A.; Park, H.-J.; Yang, M.-J.; Jung, K.J.; Yang, H.-S.; Song, C.W.; Lee, K. Polyhexamethyleneguanidine phosphate induces severe lung inflammation, fibrosis, and thymic atrophy. Food Chem. Toxicol. 2014, 69, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-Y.; Kim, M.-S.; Kim, S.-H.; Joen, D.; Lee, K. Protective Effects of Nintedanib against Polyhexamethylene Guanidine Phosphate-Induced Lung Fibrosis in Mice. Molecules 2018, 23, 1974. [Google Scholar] [CrossRef] [PubMed]
- Gross, T.J.; Hunninghake, G.W. Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2001, 345, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Crystal, R.G.; Bitterman, P.B.; Mossman, B.; Schwarz, M.I.; Sheppard, D.; Almasy, L.; Chapman, H.A.; Friedman, S.L.; King, T.E.; Leinwand, L.A.; et al. Future Research Directions in Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2002, 166, 236–246. [Google Scholar] [CrossRef]
- Sharma, S.K.; A MacLean, J.; Pinto, C.; Kradin, R.L. The effect of an anti-CD3 monoclonal antibody on bleomycin-induced lymphokine production and lung injury. Am. J. Respir. Crit. Care Med. 1996, 154, 193–200. [Google Scholar] [CrossRef]
- Helene, M.; Lake-Bullock, V.; Zhu, J.; Hao, H.; Cohen, D.A.; Kaplan, A.M. T cell independence of bleomycin-induced pulmonary fibrosis. J. Leukoc. Biol. 1999, 65, 187–195. [Google Scholar] [CrossRef]
- Gharaee-Kermani, M.; McGarry, B.; Lukacs, N.; Huffnagle, G.; Egan, R.W.; Phan, S.H. The role of IL-5 in bleomycin-induced pulmonary fibrosis. J. Leukoc. Biol. 1998, 64, 657–666. [Google Scholar] [CrossRef]
- Hao, H.; A Cohen, D.; Jennings, C.D.; Bryson, J.S.; Kaplan, A.M. Bleomycin-induced pulmonary fibrosis is independent of eosinophils. J. Leukoc. Biol. 2000, 68, 515. [Google Scholar]
- Zhang, K.; Gharaee-Kermani, M.; McGarry, B.; Remick, D.; Phan, S.H. TNF-alpha-mediated lung cytokine networking and eosinophil recruitment in pulmonary fibrosis. J. Immunol. 1997, 158, 954–959. [Google Scholar]
- Gharaee-Kermani, M.; Phan, S.H. The role of eosinophils in pulmonary fibrosis (Review). Int. J. Mol. Med. 1998, 1, 43–53. [Google Scholar] [CrossRef]
- Dunnill, M.S. Pulmonary fibrosis. Histopathology 1990, 16, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Thrall, R.S.; Barton, R.W.; A D’Amato, D.; Sulavik, S.B. Differential cellular analysis of bronchoalveolar lavage fluid obtained at various stages during the development of bleomycin-induced pulmonary fibrosis in the rat. Am. Rev. Respir. Dis. 1982, 126, 488–492. [Google Scholar] [PubMed]
- Zhang, K.; Gharaee-Kermani, M.; Jones, M.L.; Warren, J.S.; Phan, S.H. Lung monocyte chemoattractant protein-1 gene expression in bleomycin-induced pulmonary fibrosis. J. Immunol. 1994, 153, 4733–4741. [Google Scholar] [PubMed]
- Wynn, T.A. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat. Rev. Immunol. 2004, 4, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Lukacs, N.W.; Hogaboam, C.; Chensue, S.W.; Blease, K.; Kunkel, S.L. Type 1/type 2 cytokine paradigm and the progression of pulmonary fibrosis. Chest 2001, 120, 5S–8S. [Google Scholar] [CrossRef] [PubMed]
- Salmon-Ehr, V.; Serpier, H.; Nawrocki, B.; Gillery, P.; Clavel, C.; Kalis, B.; Birembaut, P.; Maquart, F.-X. Expression of Interleukin-4 in Scleroderma Skin Specimens and Scleroderma Fibroblast Cultures. Arch. Derm. 1996, 132, 802–806. [Google Scholar] [CrossRef] [PubMed]
- Doucet, C.; Brouty-Boyé, D.; Pottin-Clémenceau, C.; Canonica, G.W.; Jasmin, C.; Azzarone, B. Interleukin (IL) 4 and IL-13 act on human lung fibroblasts. Implication in asthma. J. Clin. Investig. 1998, 101, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Gon, Y.; Takeshita, I.; Maruoka, S.; Horie, T. IL-4 and IL-13 induce myofibroblastic phenotype of human lung fibroblasts through c-Jun NH2-terminal kinase–dependent pathway. J. Allergy Clin. Immunol. 2001, 107, 1001–1008. [Google Scholar] [CrossRef]
- Rankin, J.A.; Picarella, D.E.; Geba, G.P.; Temann, U.A.; Prasad, B.; DiCosmo, B.; Tarallo, A.; Stripp, B.; Whitsett, J.; Flavell, R.A. Phenotypic and physiologic characterization of transgenic mice expressing interleukin 4 in the lung: Lymphocytic and eosinophilic inflammation without airway hyperreactivity. Proc. Natl. Acad. Sci. USA 1996, 93, 7821–7825. [Google Scholar] [CrossRef]
- Zhu, Z.; Homer, R.J.; Wang, Z.; Chen, Q.; Geba, G.P.; Wang, J.; Zhang, Y.; Elias, J.A. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J. Clin. Investig. 1999, 103, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Huaux, F.; Liu, T.; McGarry, B.; Ullenbruch, M.; Xing, Z.; Phan, S.H. Eosinophils and T Lymphocytes Possess Distinct Roles in Bleomycin-Induced Lung Injury and Fibrosis. J. Immunol. 2003, 171, 5470–5481. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.Y.; Miller, M.; Baek, K.J.; Han, J.W.; Nayar, J.; Lee, S.Y.; McElwain, K.; McElwain, S.; Friedman, S.; Broide, D.H. Inhibition of airway remodeling in IL-5–deficient mice. J. Clin. Investig. 2004, 113, 551–560. [Google Scholar] [CrossRef] [PubMed]
- Fulkerson, P.C.; Fischetti, C.A.; Rothenberg, M.E. Eosinophils and CCR3 Regulate Interleukin-13 Transgene-Induced Pulmonary Remodeling. Am. J. Pathol. 2006, 169, 2117–2126. [Google Scholar] [CrossRef]
- Reiman, R.M.; Thompson, R.W.; Feng, C.G.; Hari, D.; Knight, R.; Cheever, A.W.; Rosenberg, H.F.; Wynn, T.A. Interleukin-5 (IL-5) Augments the Progression of Liver Fibrosis by Regulating IL-13 Activity. Infect. Immun. 2006, 74, 1471–1479. [Google Scholar] [CrossRef]
- Gharaee-Kermani, M.; Phan, S.H. Lung interleukin-5 expression in murine bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 1997, 16, 438–447. [Google Scholar] [CrossRef]
- Hauber, H.-P.; Manoukian, J.J.; Nguyen, L.H.P.; Sobol, S.E.; Levitt, R.C.; Holroyd, K.J.; McElvaney, N.G.; Griffin, S.; Hamid, Q. Increased Expression of Interleukin-9, Interleukin-9 Receptor, and the Calcium-Activated Chloride Channel hCLCA1 in the Upper Airways of Patients With Cystic Fibrosis. Laryngoscope 2003, 113, 1037–1042. [Google Scholar] [CrossRef]
- Hauber, H.; Tsicopoulos, A.; Wallaert, B.; Griffin, S.; McElvaney, N.; Daigneault, P.; Mueller, Z.; Olivenstein, R.; Holroyd, K.; Levitt, R.; et al. Expression of HCLCA1 in cystic fibrosis lungs is associated with mucus overproduction. Eur. Respir. J. 2004, 23, 846–850. [Google Scholar] [CrossRef]
- Nakanishi, A.; Morita, S.; Iwashita, H.; Sagiya, Y.; Ashida, Y.; Shirafuji, H.; Fujisawa, Y.; Nishimura, O.; Fujino, M. Role of gob-5 in mucus overproduction and airway hyperresponsiveness in asthma. Proc. Natl. Acad. Sci. USA 2001, 98, 5175–5180. [Google Scholar] [CrossRef]
- Toda, M.; Tulić, M.K.; Levitt, R.C.; Hamid, Q. A calcium-activated chloride channel (HCLCA1) is strongly related to IL-9 expression and mucus production in bronchial epithelium of patients with asthma. J. Allergy Clin. Immunol. 2002, 109, 246–250. [Google Scholar] [CrossRef]
- Hauber, H.-P.; Lavigne, F.; Hung, H.-L.; Levitt, R.C.; Hamid, Q. Effect of Th2 type cytokines on hCLCA1 and mucus expression in cystic fibrosis airways. J. Cyst. Fibros. 2010, 9, 277–279. [Google Scholar] [CrossRef]
- Lee, C.G.; Hartl, D.; Lee, G.R.; Koller, B.; Matsuura, H.; Da Silva, C.A.; Sohn, M.H.; Cohn, L.; Homer, R.J.; Kozhich, A.A.; et al. Role of breast regression protein 39 (BRP-39)/chitinase 3-like-1 in Th2 and IL-13-induced tissue responses and apoptosis. J. Exp. Med. 2009, 206, 1149–1166. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.; Lee, C.G.; A Da Silva, C.; Chupp, G.L.; A Elias, J. Novel biomarkers in asthma: Chemokines and chitinase-like proteins. Curr. Opin. Allergy Clin. Immunol. 2009, 9, 60–66. [Google Scholar] [CrossRef]
- Chupp, G.; Lee, C.G.; Jarjour, N.; Shim, Y.M.; Holm, C.T.; He, S.; Dziura, J.D.; Reed, J.; Coyle, A.J.; Kiener, P.; et al. A Chitinase-like Protein in the Lung and Circulation of Patients with Severe Asthma. N. Engl. J. Med. 2007, 357, 2016–2027. [Google Scholar] [CrossRef] [PubMed]
- Furuhashi, K.; Suda, T.; Nakamura, Y.; Inui, N.; Hashimoto, D.; Miwa, S.; Hayakawa, H.; Kusagaya, H.; Nakano, Y.; Nakamura, H.; et al. Increased expression of YKL-40, a chitinase-like protein, in serum and lung of patients with idiopathic pulmonary fibrosis. Respir. Med. 2010, 104, 1204–1210. [Google Scholar] [CrossRef] [PubMed]
- Sokai, A.; Handa, T.; Tanizawa, K.; Oga, T.; Uno, K.; Tsuruyama, T.; Kubo, T.; Ikezoe, K.; Nakatsuka, Y.; Tanimura, K.; et al. Matrix metalloproteinase-10: A novel biomarker for idiopathic pulmonary fibrosis. Respir. Res. 2015, 16, 120. [Google Scholar] [CrossRef]
- Allen, J.T.; A Spiteri, M. Growth factors in idiopathic pulmonary fibrosis: Relative roles. Respir. Res. 2001, 3, 13. [Google Scholar] [CrossRef] [PubMed]
- Bartram, U.; Speer, C.P. The Role of Transforming Growth Factor β in Lung Development and Disease. Chest 2004, 125, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Sime, P.J.; Marr, R.A.; Gauldie, D.; Xing, Z.; Hewlett, B.R.; Graham, F.L.; Gauldie, J. Transfer of Tumor Necrosis Factor-α to Rat Lung Induces Severe Pulmonary Inflammation and Patchy Interstitial Fibrogenesis with Induction of Transforming Growth Factor-β1 and Myofibroblasts. Am. J. Pathol. 1998, 153, 825–832. [Google Scholar] [CrossRef]
- Piguet, P.F.; A Collart, M.; E Grau, G.; Kapanci, Y.; Vassalli, P. Tumor necrosis factor/cachectin plays a key role in bleomycin-induced pneumopathy and fibrosis. J. Exp. Med. 1989, 170, 655–663. [Google Scholar] [CrossRef]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef]
- Hayashida, T.; Decaestecker, M.; Schnaper, H.W. Cross-talk between ERK MAP kinase and Smad signaling pathways enhances TGF-b– dependent responses in human mesangial cells. FASEB J. 2003, 17, 1576–1578. [Google Scholar] [CrossRef]
- Asano, Y.; Ihn, H.; Yamane, K.; Jinnin, M.; Mimura, Y.; Tamaki, K. Phosphatidylinositol 3-Kinase Is Involved in α2(I) Collagen Gene Expression in Normal and Scleroderma Fibroblasts. J. Immunol. 2004, 172, 7123–7135. [Google Scholar] [CrossRef] [PubMed]
- Manickam, N.; Patel, M.; Griendling, K.K.; Gorin, Y.; Barnes, J.L. RhoA/Rho kinase mediates TGF-b1–induced kidney myofibroblast activation through Poldip2/Nox4–derived reactive oxygen species. Am. J. Physiol. Ren. Physiol. 2014, 307, F159–F171. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Molina-Molina, M.; Abdul-Hafez, A.; Uhal, V.; Xaubet, A.; Uhal, B.D. Angiotensin converting enzyme-2 is protective but downregulated in human and experimental lung fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2008, 295, L178–L185. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.-R.; Guo, Y.; Wan, H.; Fan, L.; Feng, Y.; Ni, L.; Xiang, Y.; Li, Q.-Y. Angiotensin-converting enzyme 2 attenuates the metastasis of non-small cell lung cancer through inhibition of epithelial-mesenchymal transition. Oncol. Rep. 2013, 29, 2408–2414. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Eickelberg, O. The Impact of TGF-β on Lung Fibrosis. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef]
- Bowler, R.P.; Nicks, M.; Warnick, K.; Crapo, J.D. Role of extracellular superoxide dismutase in bleomycin-induced pulmonary fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2002, 282, L719–L726. [Google Scholar] [CrossRef]
- Fattman, C.L.; Chang, L.-Y.; A Termin, T.; Petersen, L.; Enghild, J.J.; Oury, T.D. Enhanced bleomycin-induced pulmonary damage in mice lacking extracellular superoxide dismutase. Free. Radic. Biol. Med. 2003, 35, 763–771. [Google Scholar] [CrossRef]
- Rabbani, Z.N.; Anscher, M.S.; Folz, R.J.; Archer, E.; Huang, H.; Chen, L.; Golson, M.L.; Samulski, T.S.; Dewhirst, M.W.; Vujaskovic, Z. Overexpression of extracellular superoxide dismutase red uces acute radiation induced lung toxicity. BMC Cancer 2005, 5, 59. [Google Scholar] [CrossRef]
- Shackelford, C.; Long, G.; Wolf, J.; Okerberg, C.; Herbert, R. Qualitative and Quantitative Analysis of Nonneoplastic Lesions in Toxicology Studies. Toxicol. Pathol. 2002, 30, 93–96. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples used and/or analyzed during the current study are available from the corresponding author on reasonable request. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | Annotated Genes Quantity | p Value | Annotated Genes (p Value) |
|---|---|---|---|
| Biological Processes | |||
| Inflammatory response | 36 | 0.000 | GM21541(0.01), RARRES2(0.02), CYSLTR1(0.01), PF4(0.03), NLRP1A(0.02), CX3CL1(0.01), FPR2(0.03), IL15(0.03), TLR6(0.02), TLR7(0.01), CCRL2(0.00), CHIL4(0.00), HRH1(0.01), REL(0.02), ITGB6(0.01), REG3G(0.03), NOS2(0.03), FAS(0.01), HYAL1(0.03), HC(0.03), CCL21C(0.03), EPHX2(0.02), CCL21A(0.01), AXL(0.03), CCL21B(0.02), CHST4(0.01), GM13304(0.01), GAL(0.01), CHST1(0.00), TNFRSF10B(0.02), RELT(0.03), GM10591(0.01), PTGDR(0.01), GM1987(0.01), CAMK1D(0.01), BMP6(0.00) |
| Immune response | 28 | 0.001 | GM21541(0.01), PF4(0.03), CX3CL1(0.01), OAS2(0.01), IL15(0.03), TLR6(0.02), TLR7(0.01), CD74(0.02), LIF(0.04), FAS(0.01), SMAD6(0.01), CCL21C(0.03), CTLA4(0.01), CCL21B(0.02), GM13304(0.01), TNFSF8(0.01), TNFRSF10B(0.02), CD36(0.02), CXCL14(0.01), H2-EB2(0.02), RELT(0.03), PPBP(0.01), GM10591(0.01), H2-EB1(0.01), TGFBR3(0.04), H2-AA(0.02), EDA(0.00), BMP6(0.00) |
| Cellular response to interleukin-1 | 17 | 0.000 | GM21541(0.01), HYAL1(0.03), HYAL2(0.03), CCL21C(0.03), MYLK3(0.00), CCL21A(0.01), CCL21B(0.02), CX3CL1(0.01), GM13304(0.01), SOX9(0.05), PCK1(0.00), PTGIS(0.02), GM10591(0.01), ADAMTS12(0.01), GM1987(0.01), MYC(0.05), FN1(0.04) |
| Positive regulation of extracellular signal regulated kinase (ERK)1 and ERK2 cascade | 17 | 0.043 | GM21541(0.01), BMP4(0.01), FGFR3(0.04), CCL21C(0.03), CCL21A(0.01), CCL21B(0.02), FGF10(0.03), CX3CL1(0.01), GM13304(0.01), CD74(0.02), KDR(0.02), CD36(0.02), GM10591(0.01), VEGFA(0.03), TEK(0.01), ADRA1A(0.03), GM1987(0.01) |
| Positive regulation of mitogen-activated protein kinase (MAPK) cascade | 15 | 0.001 | CAV2(0.02), FGFR3(0.04), FGF10(0.03), IGF2(0.04), PRKCE(0.01), KDR(0.02), LIF(0.04), ADRB3(0.03), DUSP19(0.01), ADRB2(0.00), CD36(0.02), RELT(0.03), BNIP2(0.01), ADRA1A(0.03), FAS(0.01) |
| Cellular response to tumor necrosis factor | 15 | 0.002 | GM21541(0.01), HYAL1(0.03), HYAL2(0.03), CCL21C(0.03), CCL21A(0.01), CCL21B(0.02), CX3CL1(0.01), GM13304(0.01), DCSTAMP(0.02), PCK1(0.00), GM10591(0.01), SLC2A4(0.00), COL1A1(0.05), ADAMTS12(0.01), GM1987(0.01) |
| Notch signaling pathway | 15 | 0.007 | HP(0.03), KCNA5(0.03), SOX9(0.05), RCAN2(0.05), HES1(0.00), ASCL1(0.04), PTP4A3(0.00), HEY1(0.00), DLL4(0.00), MIB2(0.04), PLN(0.02), NOTCH4(0.05), FOXC1(0.00), TMEM100(0.01), CFD(0.01) |
| Extracellular matrix organization | 15 | 0.003 | B4GALT1(0.05), COL18A1(0.01), RECK(0.04), COL4A1(0.04), EGFL6(0.01), ELN(0.05), SOX9(0.05), NDNF(0.03), LAMB3(0.03), HPSE2(0.05), CRISPLD2(0.04), FOXF1(0.01), COL6A4(0.05), LAMC2(0.05), FN1(0.04) |
| Chemotaxis | 14 | 0.010 | RARRES2(0.02), CYSLTR1(0.01), FGF10(0.03), PF4(0.03), FPR2(0.03), ACKR4(0.04), CX3CL1(0.01), LSP1(0.00), CCRL2(0.00), CXCL17(0.01), S1PR1(0.01), ECSCR(0.02), CX3CR1(0.01), XCR1(0.02) |
| Neutrophil chemotaxis | 10 | 0.011 | GM21541(0.01), ITGA9(0.01), PPBP(0.01), GM10591(0.01), CCL21C(0.03), CCL21A(0.01), CCL21B(0.02), GM13304(0.01), CX3CL1(0.01), GM1987(0.01) |
| Cellular response to interferon-gamma | 14 | 0.000 | WNT5A(0.02), GM21541(0.01), CCL21C(0.03), CCL21A(0.01), CCL21B(0.02), GM13304(0.01), CX3CL1(0.01), DAPK3(0.03), H2-Q7(0.01), GM10591(0.01), NOS2(0.03), IL12B(0.02), GM1987(0.01), MYC(0.05) |
| Chemokine-mediated signaling pathway | 11 | 0.001 | GM21541(0.01), CXCL17(0.01), PPBP(0.01), GM10591(0.01), CCL21C(0.03), CCL21A(0.01), CCL21B(0.02), PF4(0.03), GM13304(0.01), CX3CL1(0.01), GM1987(0.01) |
| Positive regulation of canonical Wnt signaling pathway | 11 | 0.003 | WNT2(0.03), CAV1(0.01), FGFR3(0.04), SULF2(0.05), RSPO1(0.02), FGF10(0.03), SOX4(0.02), COL1A1(0.05), EDA(0.00), DAPK3(0.03), WNT2B(0.03) |
| Transforming growth factor beta receptor signaling pathway | 11 | 0.006 | ACVRL1(0.01), SMAD9(0.03), TGFBR1(0.03), SMAD6(0.01), COL3A1(0.03), CLDN5(0.02), COL1A2(0.04), TGFBR3(0.04), ENG(0.01), HPGD(0.00), CDH5(0.03) |
| Collagen fibril organization | 10 | 0.000 | FMOD(0.03), P3H1(0.02), TGFBR1(0.03), COL3A1(0.03), COL1A2(0.04), FOXC1(0.00), COL1A1(0.05), LOXL2(0.03), ADAMTS2(0.03), COL5A1(0.04) |
| Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathways | |||
| PI3K-Akt signaling pathway | 35 | 0.000 | FGFR3(0.04), TNC(0.03), COL3A1(0.03), FGF10(0.03), GNG11(0.05), BCL2L1(0.03), LAMB3(0.03), COL6A4(0.05), TEK(0.01), ITGB6(0.01), COL6A3(0.03), COL6A2(0.02), TNN(0.00), CREB3L3(0.01), PRKAA2(0.02), PPP2R2B(0.05), MYC(0.05), PPP2R2C(0.00), AKT3(0.01), FN1(0.04), GHR(0.02), COL4A1(0.04), KITL(0.02), COL5A1(0.04), PCK1(0.00), KDR(0.02), VWF(0.03), ITGA9(0.01), CD19(0.04), ITGA5(0.00), VEGFA(0.03), COL1A2(0.04), LAMC2(0.05), RELN(0.04), COL1A1(0.05) |
| Cytokine-cytokine receptor interaction | 26 | 0.001 | GM21541(0.01), TNFRSF12A(0.05), BMPR2(0.04), PF4(0.03), IL15(0.03), CX3CL1(0.01), LIF(0.04), FAS(0.01), XCR1(0.02), GHR(0.02), TGFBR1(0.03), CCL21C(0.03), CCL21A(0.01), EDA2R(0.02), CCL21B(0.02), GM13304(0.01), TNFSF8(0.01), TNFRSF10B(0.02), CXCL14(0.01), GM10591(0.01), RELT(0.03), PPBP(0.01), CX3CR1(0.01), IL12B(0.02), EDA(0.00), GM1987(0.01) |
| Extracellular matrix (ECM)-receptor interaction | 20 | 0.043 | COL4A1(0.04), TNC(0.03), COL3A1(0.03), COL5A1(0.04), GP9(0.00), VWF(0.03), ITGA9(0.01), LAMB3(0.03), CD36(0.02), ITGA5(0.00), COL6A4(0.05), ITGB6(0.01), COL6A3(0.03), COL6A2(0.02), COL1A2(0.04), RELN(0.04), TNN(0.00), LAMC2(0.05), COL1A1(0.05), FN1(0.04) |
| Calcium signaling pathway | 17 | 0.001 | CCKAR(0.04), ADCY4(0.05), ADORA2B(0.01), CYSLTR1(0.01), MYLK3(0.00), MYLK4(0.00), EDNRB(0.01), ADRB3(0.03), ATP2B2(0.03), HRH1(0.01), ADRB2(0.00), PDE1B(0.03), PLN(0.02), PLCG2(0.03), AVPR1A(0.03), ADRA1A(0.03), NOS2(0.03) |
| Metabolism of xenobiotics by cytochrome P450 | 13 | 0.000 | GSTA3(0.00), CYP2F2(0.02), ALDH3B2(0.01), EPHX1(0.05), DHDH(0.00), MGST3(0.01), GSTM1(0.00), GSTM2(0.01), GSTM3(0.03), GSTK1(0.02), HSD11B1(0.02), GSTP2(0.01), GSTP1(0.00) |
| Peroxisome proliferator-activated receptors (PPAR) signaling pathway | 10 | 0.002 | 1700061G19RIK(0.04), ACSL1(0.03), CD36(0.02), SORBS1(0.02), FABP3(0.01), FABP1(0.00), AQP7(0.05), FABP7(0.01), ADIPOQ(0.01), PCK1(0.01) |
| Disease Name | Annotated Genes Quantity | p Value | Annotated Genes(p Value) |
|---|---|---|---|
| Respiratory tract diseases | 107 | 2.36 × 10−32 | ACE2(0.01), ACTA2(0.04), ACVRL1(0.01), ADAMTS2(0.03), ADCYAP1R1(0.01), ADGRE5(0.01), ADIPOQ(0.01), ADRB2(0.00), ALDH2(0.01), AREG(0.04), ASCL1(0.04), AXL(0.03), BCHE(0.00), BCL2L1(0.03), BMPR2(0.04), BTK(0.04), CAT(0.01), CAV1(0.01), CCDC40(0.03), CD74(0.02), CDH13(0.01), CDH23(0.00), CEACAM1(0.02), CFD(0.01), CLDN5(0.02), COL3A1(0.03), CPE(0.00), CRISPLD2(0.04), CTLA4(0.01), CX3CL1(0.01), CXCL14(0.01), CYB5A(0.00), CYSLTR1(0.01), DNAAF3(0.05), DNAAF5(0.02), DNAH5(0.05), DNMT3A(0.02), EDNRB(0.01), EFEMP1(0.03), EFNB2(0.03), ELN(0.05), ENG(0.01), EPHX1(0.05), FAS(0.01), FGD6(0.01), FN1(0.04), FOXF1(0.01), GJA1(0.02), GPX3(0.02), GSTM1(0.00), GSTM2(0.01), GSTP1(0.00), HES1(0.00), HEY1(0.00), HILPDA(0.00), HRH1(0.01), ICAM2(0.01), IER2(0.03), IGFBP6(0.00), IL12B(0.02), IL15(0.03), IL1RL1(0.02), ITGB6(0.01), KCNA5(0.03), LAMC2(0.05), LYST(0.01), MAPT(0.01), MERTK(0.05), MMP10(0.04), MSLN(0.01), MYC(0.05), MYCL(0.01), NEK2(0.01), NNAT(0.00), NOS2(0.03), NQO1(0.01), OAS2(0.01), PF4(0.03), PLLP(0.01), PON1(0.01), PPBP(0.01), PRDX6(0.01), PTGDR(0.01), PTGIS(0.02), RAMP2(0.02), RECK(0.04), RIMS2(0.01), RUNX3(0.05), SCGB1A1(0.03), SCNN1B(0.01), SELENBP1(0.00), SLC34A2(0.02), SLC6A4(0.01), SMAD9(0.03), SMARCAL1(0.01), SOD1(0.01), SOX9(0.05), TFF1(0.05), THBD(0.05), TMPRSS4(0.05), TNC(0.03), TNFSF8(0.01), TNNT2(0.03), UBE2L6(0.03), UCHL1(0.05), VEGFA(0.03), WNT5A(0.02) |
| Lung diseases, obstructive | 25 | 1.52 × 10−12 | ADCYAP1R1(0.01), ADRB2(0.00), ALDH2(0.01), AREG(0.04), CAT(0.01), CTLA4(0.01), CXCL14(0.01), DNAH5(0.05), ELN(0.05), EPHX1(0.05), FOXF1(0.01), GSTM1(0.00), GSTP1(0.00), IL1RL1(0.02), ITGB6(0.01), MMP10(0.04), NOS2(0.03), NQO1(0.01), PTGDR(0.01), SCNN1B(0.01), SOD1(0.01), TNC(0.03), TNFSF8(0.01), TNNT2(0.03), VEGFA(0.03) |
| Respiratory hypersensitivity | 22 | 7.48 × 10−10 | ADCYAP1R1(0.01), ADRB2(0.00), ALDH2(0.01), AREG(0.04), CAT(0.01), CLDN5(0.02), CTLA4(0.01), CXCL14(0.01), CYSLTR1(0.01), DNAH5(0.05), GSTM1(0.00), GSTP1(0.00), HRH1(0.01), IL15(0.03), IL1RL1(0.02), MMP10(0.04), NOS2(0.03), NQO1(0.01), PTGDR(0.01), SOD1(0.01), TNC(0.03), VEGFA(0.03) |
| Carcinoma, bronchogenic | 20 | 2.03 × 10−5 | AREG(0.04), ASCL1(0.04), AXL(0.03), CAT(0.01), CD74(0.02), CDH13(0.01), GSTM2(0.01), GSTP1(0.00), HES1(0.00), LAMC2(0.05), MYC(0.01), MYCL(0.04), NEK2, NNAT(0.00), NQO1(0.01), RECK(0.04), RIMS2(0.01), SLC34A2(0.02), UCHL1(0.05), VEGFA(0.03) |
| Asthma | 19 | 1.75 × 10−8 | ADCYAP1R1(0.01), ADRB2(0.00), ALDH2(0.01), AREG(0.04), CAT(0.01), CTLA4(0.01), CXCL14(0.01), CYSLTR1(0.01), DNAH5(0.05), GSTM1(0.00), GSTP1(0.00), IL1RL1(0.02), MMP10(0.04), NOS2(0.03), NQO1(0.01), PTGDR(0.01), SOD1(0.01), TNC(0.03), VEGFA(0.03) |
| Pulmonary fibrosis | 12 | 0.01161 | ACE2(0.01), ACTA2(0.04), ADIPOQ(0.01), AREG(0.04), CAT(0.01), CFD(0.01), COL3A1(0.03), ELN(0.05), FN1(0.04), IL12B(0.02), SOD1(0.01), WNT5A(0.02) |
| Pulmonary disease, chronic obstructive | 9 | 0.01999 | ELN(0.05), EPHX1(0.05), ITGB6(0.01), NOS2(0.03), SCNN1B(0.01), TNFSF8(0.01), TNNT2(0.03), VEGFA(0.03) |
| Th2 Pathway | |||
|---|---|---|---|
| Symbol | Entrez Gene Name | Fold Change | p Value |
| BMPR2 | bone morphogenetic protein receptor, type II (serine/threonine kinase) | 0.62 | 0.04 |
| H2-Eb2 | histocompatibility 2, class II antigen E beta2 | 2.95 | 0.02 |
| H2-Q7 | major histocompatibility complex, class I, A | 2.10 | 0.01 |
| H2-Aa | major histocompatibility complex, class II, DQ alpha 1 | 2.23 | 0.02 |
| H2-Eb1 | major histocompatibility complex, class II, DR beta 5 | 2.52 | 0.01 |
| Icosl | inducible T cell costimulator ligand | 0.62 | 0.04 |
| IL12B | interleukin 12b | 3.50 | 0.02 |
| IL1RL1 | interleukin 1 receptor-like 1 | 3.46 | 0.02 |
| MAF | avian musculoaponeurotic fibrosarcoma oncogene homolog | 1.71 | 0.05 |
| NOTCH4 | notch 4 | 0.62 | 0.05 |
| RUNX3 | runt related transcription factor 3 | 3.02 | 0.05 |
| S1PR1 | sphingosine-1-phosphate receptor 1 | 0.50 | 0.01 |
| TGFBR1 | transforming growth factor, beta receptor I | 2.09 | 0.03 |
| TGFBR3 | transforming growth factor, beta receptor III | 0.51 | 0.04 |
| IL-4 Signaling | |||
| Symbol | Entrez Gene Name | Fold Change | p Value |
| AKT3 | thymoma viral proto-oncogene 3 | 0.59 | 0.01 |
| H2-Eb2 | histocompatibility 2, class II antigen E beta2 | 2.95 | 0.02 |
| H2-Q7 | major histocompatibility complex, class I, A | 2.10 | 0.01 |
| H2-Aa | major histocompatibility complex, class II, DQ alpha 1 | 2.23 | 0.02 |
| H2-Eb1 | major histocompatibility complex, class II, DR beta 5 | 2.52 | 0.01 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, M.-K.; Kim, D.I.; Lee, K. Kathon Induces Fibrotic Inflammation in Lungs: The First Animal Study Revealing a Causal Relationship between Humidifier Disinfectant Exposure and Eosinophil and Th2-Mediated Fibrosis Induction. Molecules 2020, 25, 4684. https://doi.org/10.3390/molecules25204684
Song M-K, Kim DI, Lee K. Kathon Induces Fibrotic Inflammation in Lungs: The First Animal Study Revealing a Causal Relationship between Humidifier Disinfectant Exposure and Eosinophil and Th2-Mediated Fibrosis Induction. Molecules. 2020; 25(20):4684. https://doi.org/10.3390/molecules25204684
Chicago/Turabian StyleSong, Mi-Kyung, Dong Im Kim, and Kyuhong Lee. 2020. "Kathon Induces Fibrotic Inflammation in Lungs: The First Animal Study Revealing a Causal Relationship between Humidifier Disinfectant Exposure and Eosinophil and Th2-Mediated Fibrosis Induction" Molecules 25, no. 20: 4684. https://doi.org/10.3390/molecules25204684
APA StyleSong, M.-K., Kim, D. I., & Lee, K. (2020). Kathon Induces Fibrotic Inflammation in Lungs: The First Animal Study Revealing a Causal Relationship between Humidifier Disinfectant Exposure and Eosinophil and Th2-Mediated Fibrosis Induction. Molecules, 25(20), 4684. https://doi.org/10.3390/molecules25204684