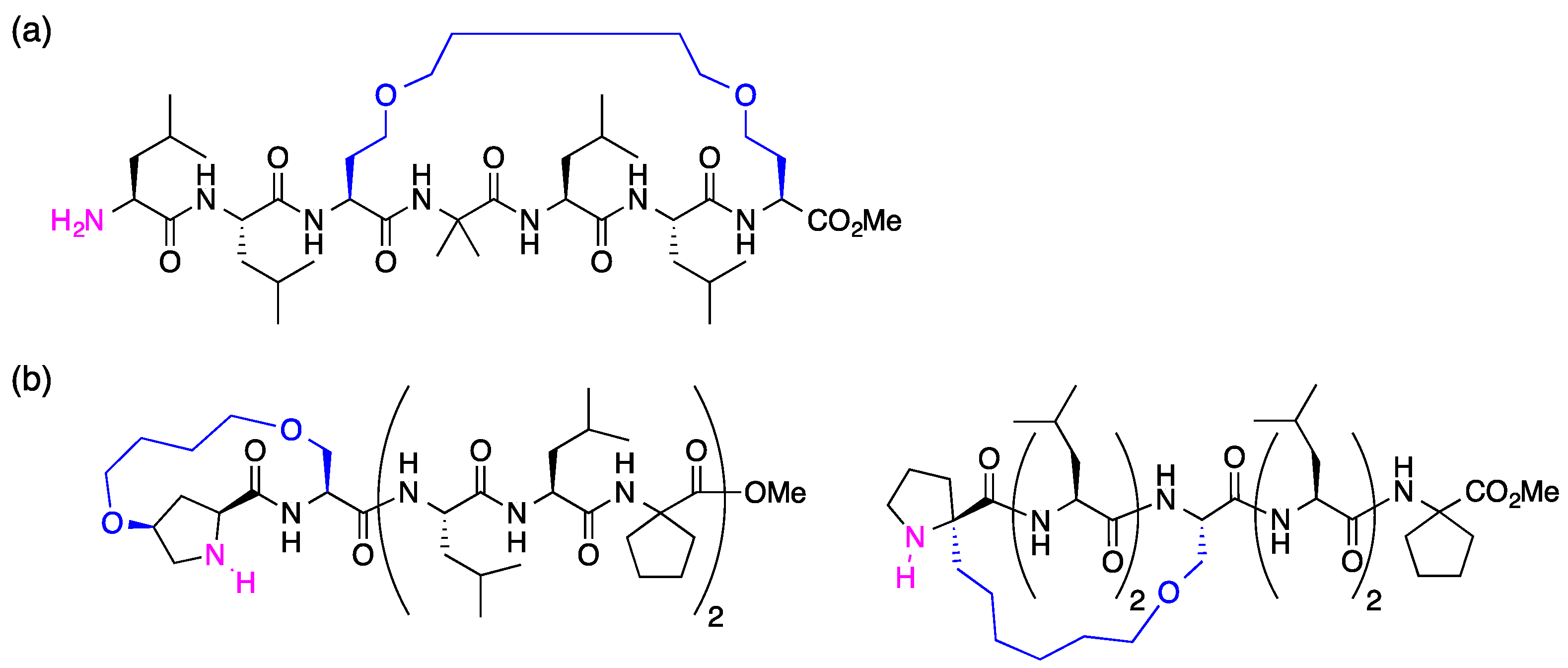
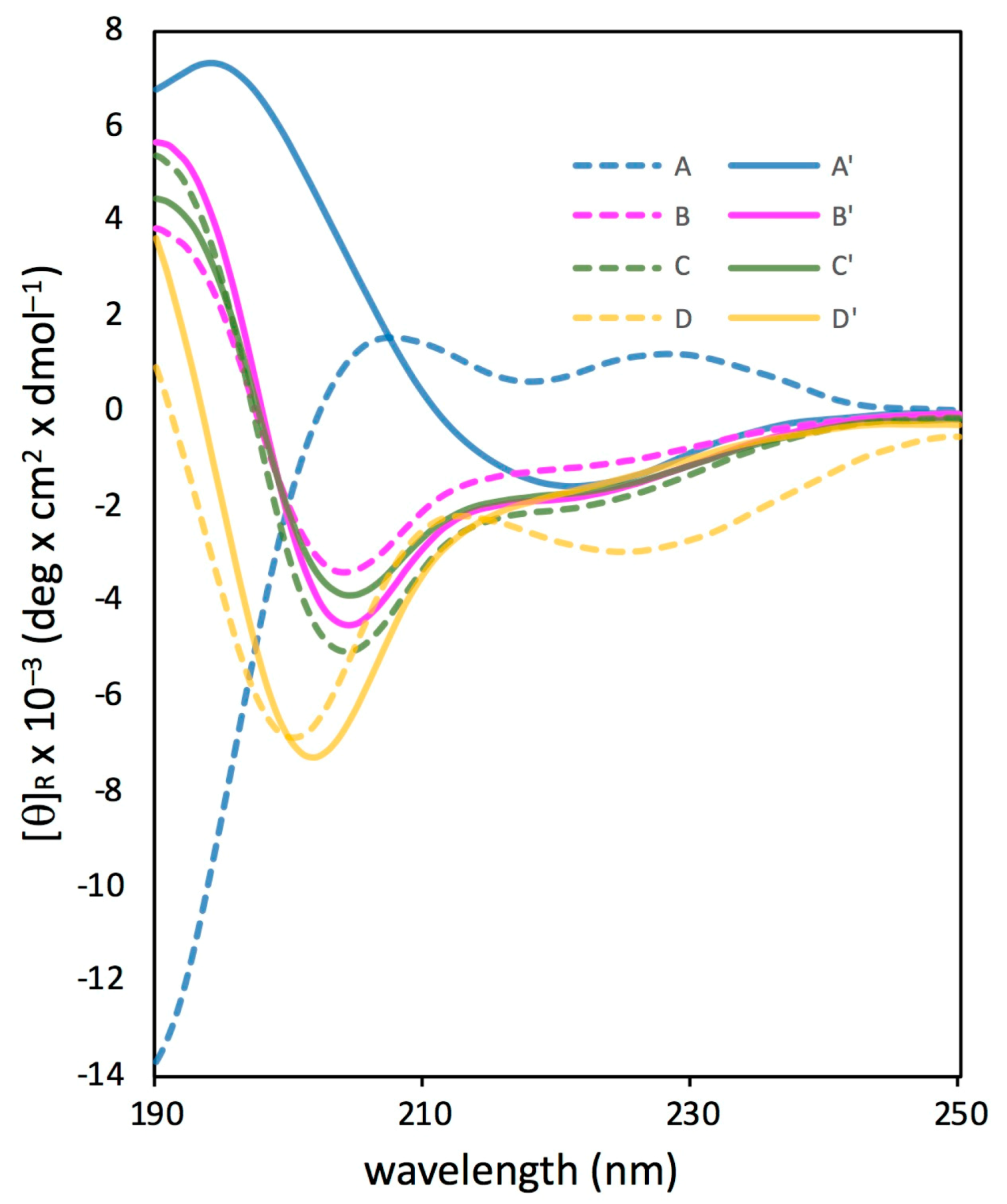
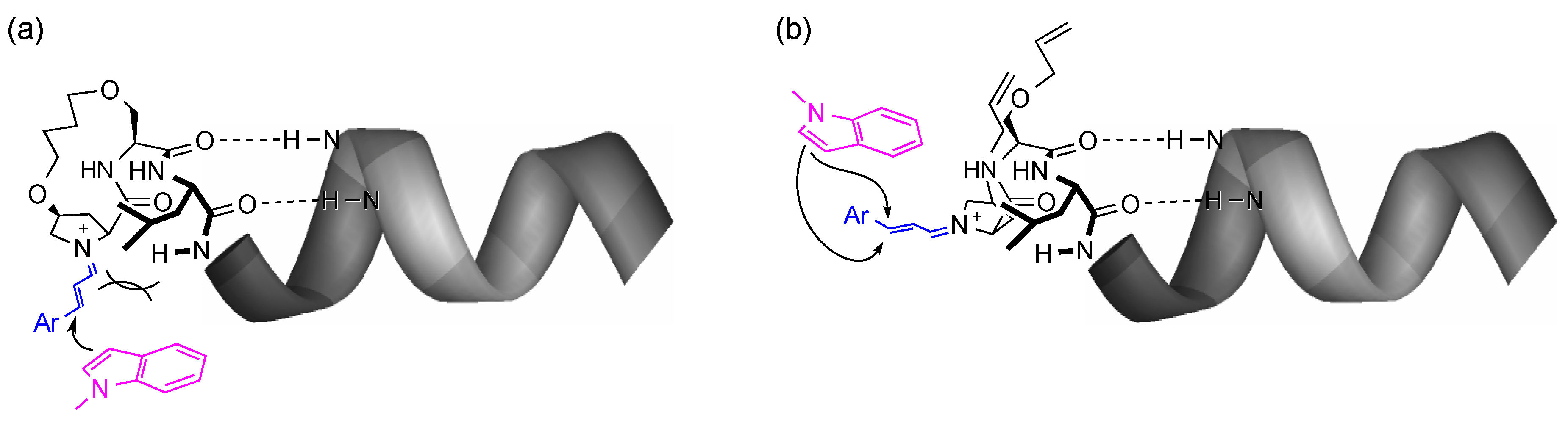
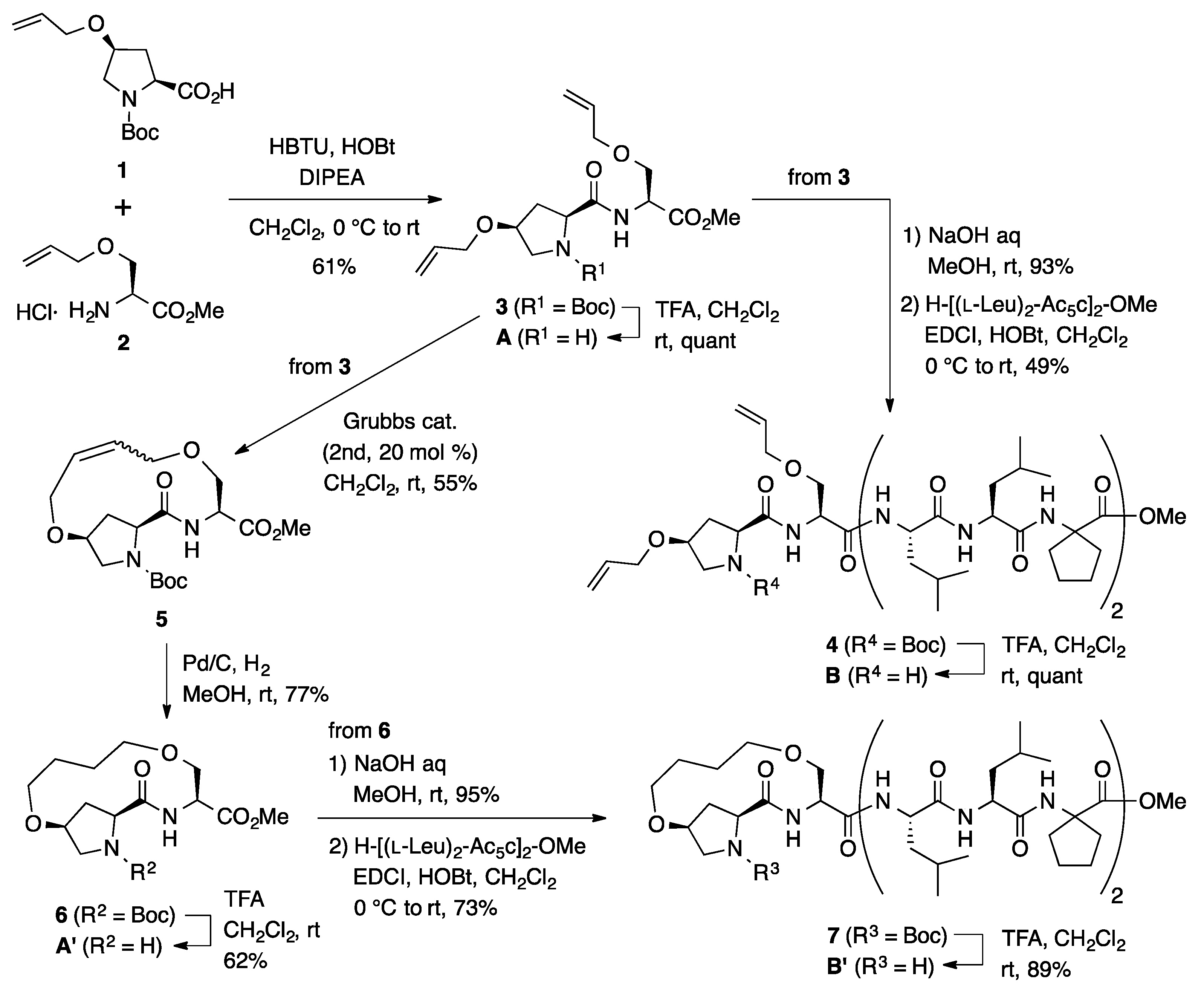
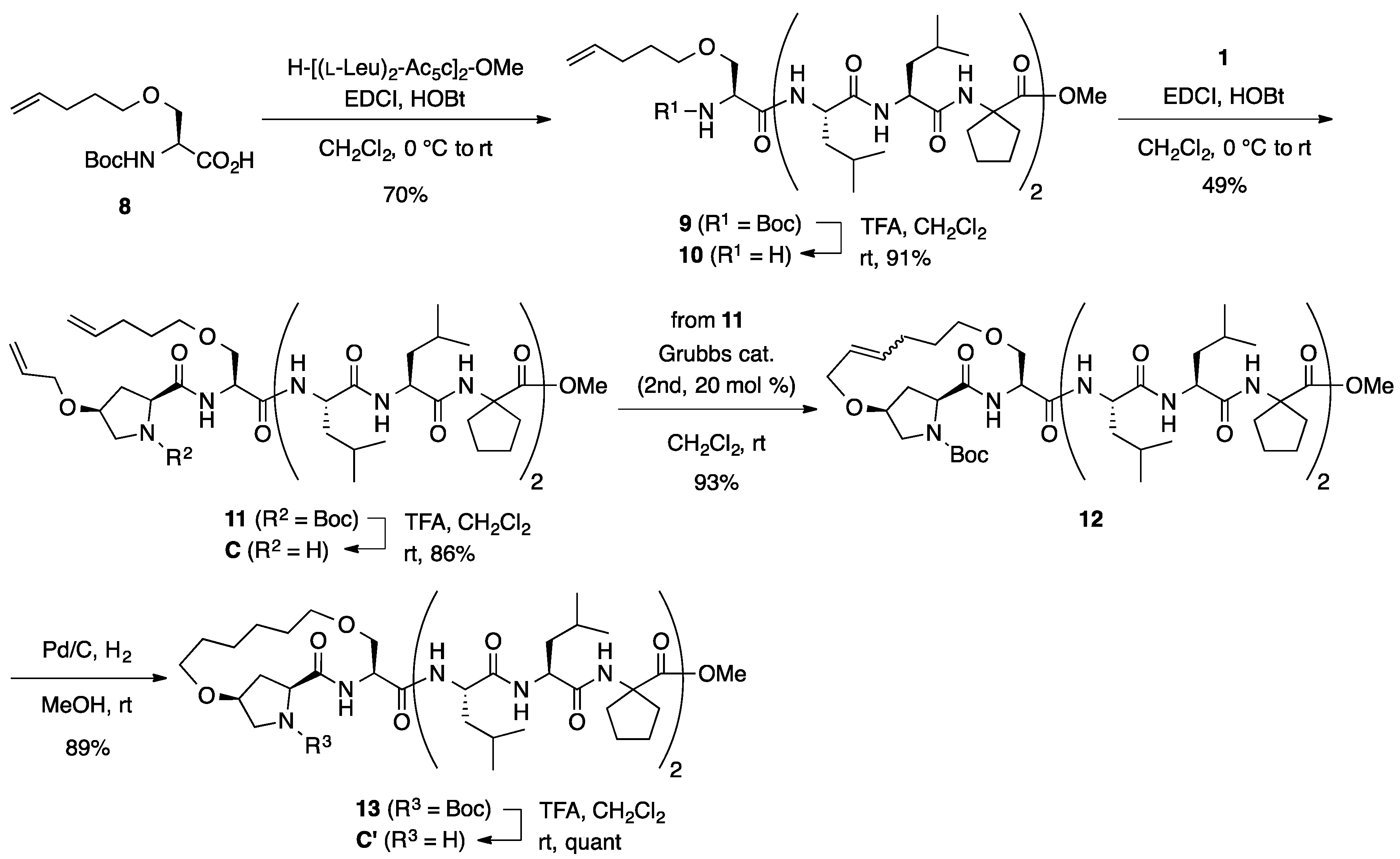
3.2. Synthesis of Unstapled Peptides A and B and Stapled Peptides A’ and B’
Boc-l-HypOAll-l-SerOAll-OMe (
3): To a solution of Boc-
l-Hyp
OAll-OH (
1 [
49,
50]; 4.22 g, 15.5 mmol) in CH
2Cl
2 (52 mL) were added 3-[bis(dimethylamino)methyliumyl]-3
H-benzotriazol-1-oxide hexafluorophosphate (HBTU; 6.48 g, 17.1 mmol) and 1-hydroxybenzotriazole hydrate (HOBt·H
2O; 2.62 g, 17.1 mmol) at 0 °C, and the solution was stirred for 30 min. Then, a solution of HCl·H-
l-Ser
OAll-OMe (
2 [
24,
25].; 2.47 g, 15.5 mmol) in CH
2Cl
2 (52 mL) and
N,
N-diisopropylethylamine (DIPEA; 5.41 mL, 31.1 mmol) was added to the reaction mixture at the same temperature, and the resultant mixture was gradually warmed to room temperature. After stirring overnight, the CH
2Cl
2 was removed and the residue was diluted with EtOAc. The solution was washed successively with 1 M of HCl, water, sat. aq NaHCO
3, and brine. The organic layer was dried over anhydrous MgSO
4 and concentrated in vacuo to give a crude product, which was purified by flash column chromatography on silica gel (40% EtOAc in
n-hexane) to give
3 (3.89 g, 61%) as a yellow oil.
Rf = 0.69 (EtOAc).
−3.2 (
c 1.02, CHCl
3).
1H NMR (500 MHz, CDCl
3, VT = 50 °C) δ: 5.98–5.77 (m, 2H), 5.28–5.11 (m, 4H), 4.77–4.66 (m, 1H), 4.33 (d,
J = 8.8 Hz, 1H), 4.07–4.04 (m, 1H), 4.00–3.86 (m, 4H), 3.82 (dd,
J = 9.7, 3.5, Hz, 1H), 3.78–3.70 (m, 1H), 3.74 (s, 3H), 3.61–3.49 (m, 3H), 2.56–2.44 (m, 1H), 2.23–2.09 (m, 1H), 1.48 (s, 9H).
13C NMR (125 MHz, CDCl
3, VT = 50 °C) δ: 172.0, 170.5, 156.0, 134.6, 134.3, 117.0, 116.9, 81.0, 76.4, 72.2, 69.9, 69.6, 58.6, 53.0, 52.8, 52.2, 37.0, 28.3 (3C). IR (film): 3304, 2978, 2933, 1751, 1701 cm
−1. HRMS (DART)
m/
z: [M + H]
+ calcd for C
20H
33N
2O
7, 413.2288; found, 413.2282.
H-l-HypOAll-l-SerOAll-OMe (A): To a solution of Boc-protected dipeptide 3 (100 mg, 0.242 mmol) in CH2Cl2 (2.4 mL) was added trifluoroacetic acid (0.24 mL) dropwise at room temperature, and the reaction mixture was stirred for 2 days at the same temperature. The reaction mixture was neutralized by adding sat. aq NaHCO3 and the aqueous phase was extracted with CHCl3 three times. The combined organic extracts were dried over anhydrous MgSO4 and concentrated under a vacuum to give amine A (75.2 mg, quant) as an amorphous solid. Rf = 0.29 (EtOAc). Mp 81–83 °C. −39.4 (c 1.00, CHCl3). 1H NMR (400 MHz, CDCl3) δ: 8.27 (d, J = 8.6 Hz, 1H), 5.93–5.78 (m, 2H), 5.29–5.21 (m, 2H), 5.21–5.11 (m, 2H), 4.72 (dt, J = 8.3, 3.5 Hz, 1H), 4.08–4.00 (m, 1H), 4.00–3.96 (m, 2H), 3.94 (dt, J = 5.4, 1.5 Hz, 1H), 3.92–3.85 (m, 2H), 3.82 (dd, J = 8.3, 4.9 Hz, 1H), 3.76 (s, 3H), 3.58 (dd, J = 9.5, 3.7 Hz, 1H), 3.17 (dd, J = 11.2, 5.4 Hz, 1H), 3.05 (dd, J = 11.2, 2.9 Hz, 1H), 2.28–2.16 (m, 2H). 13C NMR (125 MHz, CDCl3) δ: 168.4, 163.9, 134.1, 133.6, 118.0, 117.4, 74.6, 72.3, 70.1, 68.5, 57.2, 54.1, 52.5, 50.7, 33.6. IR (KBr): 3215, 2874, 1692, 1450 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C15H24N2O5Na, 335.1583; found, 335.1573.
Boc-l-HypOAll-l-SerOAll-[(l-Leu)2-Ac5c]2-OMe (4): To a solution of dipeptide 3 (50.0 mg, 0.121 mmol) in MeOH (1.2 mL) was added 1 M of aqueous NaOH (0.121 mL, 0.121 mmol) at room temperature, and the mixture was stirred overnight at the same temperature. The solution was acidified with 1 M of aqueous HCl and the MeOH was removed in vacuo. The resulting aqueous solution was extracted with EtOAc three times. The combined organic extracts were washed with brine, dried over Na2SO4, and concentrated in vacuo to give a carboxylic acid (44.7 mg, 93%). To a solution of the acid (206 mg, 0.500 mmol) in CH2Cl2 (2.5 mL) was added EDCI·HCl (96.0 mg, 0.500 mmol) and HOBt·H2O (92.0 mg, 0.600 mmol) at 0 °C, and the mixture was stirred at the same temperature for 30 min. Then, a solution of H-[(l-Leu)2-Ac5c]2-OMe (354 mg, 0.500 mmol) in CH2Cl2 (2.5 mL) was added dropwise to the reaction mixture at 0 °C. The reaction mixture was gradually warmed to room temperature and stirred overnight. After the removal of CH2Cl2, the residue was diluted with EtOAc. The solution was washed successively with 1 M of HCl, water, sat. aq NaHCO3, and brine. The organic layer was dried over anhydrous MgSO4 and concentrated in vacuo to give crude product, which was purified by flash column chromatography on silica gel (70% EtOAc in n-hexane) to give 4 (269 mg, 49%) as a white solid. Rf = 0.20 (60% EtOAc in n-hexane). Mp 76–79 °C. +2.2 (c 1.02, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.49 (d, J = 5.6 Hz, 1H), 7.45 (d, J = 7.8 Hz, 1H), 7.39 (d, J = 4.9 Hz, 1H), 7.28–7.23 (m, 4H), 5.96–5.74 (m, 2H), 5.37–5.16 (m, 4H), 4.34 (t, J = 8.6 Hz, 1H), 4.25–4.12 (m, 5H), 4.05–3.90 (m, 5H), 3.84–3.73 (m, 2H), 3.72–3.65 (m, 1H), 3.67 (s, 3H), 3.48 (dd, J = 12.0, 3.4 Hz, 1H), 2.70–2.60 (m, 1H), 2.39–2.22 (m, 3H), 2.22–2.11 (m, 3H), 2.11–2.02 (m, 1H), 1.96–1.57 (m, 22H), 1.50 (s, 9H), 1.02–0.83 (m, 24H). 13C NMR (125 MHz, CDCl3) δ: 175.6, 175.2, 174.6, 174.2, 174.1, 173.2, 173.1, 171.8, 155.7, 133.9, 133.5, 117.8, 117.5, 81.7, 72.2, 69.6, 67.6, 66.7, 65.7, 60.1, 56.2, 54.8, 54.1, 54.0, 53.3, 52.23, 52.22, 52.1, 39.6, 39.4, 39.1, 38.3, 37.3, 36.74, 36.70, 35.5, 34.4, 28.32, 28.26 (3C), 25.2, 25.0, 24.73, 24.65, 24.54, 24.53, 24.46, 23.5, 23.4, 22.99, 22.97, 21.1, 21.0, 20.90, 20.87. IR (CDCl3): 3325, 2961, 1732, 1661, 1530 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C56H94N8O13Na, 1109.6838; found, 1109.6808.
H-l-HypOAll-l-SerOAll-[(l-Leu)2-Ac5c]2-OMe (B): To a solution of Boc-protected peptide 4 (135 mg, 0.124 mmol) in CH2Cl2 (1 mL) was added trifluoroacetic acid (0.12 mL) dropwise at room temperature, and the reaction mixture was stirred overnight at the same temperature. The reaction mixture was neutralized by adding sat. aq NaHCO3 and the aqueous phase was extracted with CHCl3 four times. The combined organic extracts were dried over anhydrous MgSO4 and concentrated under vacuum to give amine product B (124 mg, quant). Rf = 0.10 (80% EtOAc in n-hexane). Mp 107–108 °C. −1.9 (c 0.99, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 9.04 (br s, 1H), 8.06 (br s, 1H), 7.64 (d, J = 6.8 Hz, 1H), 7.57 (br s, 1H), 7.52–7.42 (m, 2H), 7.35 (d, J = 4.9 Hz, 1H), 5.92–5.75 (m, 2H), 5.32–5.14 (m, 4H), 4.60 (br s, 1H), 4.35 (d, J = 4.2 Hz, 1H), 4.29 (br s, 1H), 4.21–4.04 (m, 3H), 4.04–3.95 (m, 4H), 3.95–3.88 (m, 1H), 3.86–3.74 (m, 2H), 3.68 (s, 3H), 3.60 (d, J = 11.7 Hz, 1H), 3.47 (d, J = 8.8 Hz, 1H), 2.65–2.44 (m, 3H), 2.29–2.16 (m, 2H), 2.12 (br s, 2H), 2.03 (dd, J = 11.7, 6.1 Hz, 1H), 1.92–1.53 (m, 22H), 1.03–0.78 (m, 24H). 13C NMR (125 MHz, CDCl3) δ: 175.7, 175.5, 175.4, 174.7, 174.4, 174.3, 173.9, 171.8, 133.8, 133.5, 118.0, 117.9, 76.1, 72.3, 70.0, 68.1, 66.7, 66.0, 59.3, 56.4, 55.0, 54.9, 54.4, 53.3, 52.4, 51.3, 40.1, 39.7, 39.4, 39.2, 37.9, 36.89, 36.85, 35.2, 35.0, 29.7, 25.0, 24.78, 24.76, 24.58, 24.56, 24.4, 24.3, 24.23, 24.17, 23.2, 23.1, 22.5, 21.8, 21.6, 21.2. IR (KBr): 3329, 2959, 1736, 1655, 1535 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C51H86N8O11Na, 1009.6314; found, 1009.6288.
Stapled Boc-l-Hyp-l-Ser-OMe (6): Under an argon atmosphere, to a solution of 3 (90.0 mg, 0.218 mmol) in CH2Cl2 (11 mL) was added second-generation Grubbs catalyst (37.0 mg, 0.0436 mmol) at room temperature, and the mixture was stirred for 2 h at the same temperature. The reaction mixture was filtered through a short pad of silica gel (60% EtOAc in n-hexane) and concentrated. The crude material was purified by flash chromatography on silica gel (60% EtOAc in n-hexane) to provide a stapled peptide 5 (46.2 mg, 55%) as a mixture of E- and Z-isomers (E/Z = 1.0:5.6). Rf = 0.30 (EtOAc). Next, to a solution of stapled peptides 5 (46.2 mg, 0.120 mmol) in MeOH (12 mL) was added 10% Pd-C (23 mg, 50 wt %) under a nitrogen atmosphere. After being vigorously stirred under a hydrogen atmosphere for 19 h at room temperature, the reaction mixture was passed through a short plug of Celite. The filtrate was concentrated under vacuum to give a crude product, which was purified by flash column chromatography on silica gel (70% EtOAc in n-hexane) to give 6 (35.5 mg, 77%) as an amber oil. Rf = 0.29 (EtOAc). −3.5 (c 1.07, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.27 (br s, 1H), 4.76–4.56 (m, 1H), 4.39–4.21 (m, 1H), 3.97 (br s, 2H), 3.87–3.78 (m, 1H), 3.76 (s, 3H), 3.59 (br s, 3H), 3.54–3.39 (m, 2H), 3.38–3.29 (m, 1H), 2.40–2.17 (m, 2H), 1.92–1.77 (m, 2H), 1.77–1.54 (m, 2H), 1.54–1.38 (m, 9H). 13C NMR (125 MHz, CDCl3) δ: 172.5, 170.3, 154.9, 80.8, 70.8, 69.7, 69.5, 69.1, 60.6, 53.0, 52.4, 52.0, 37.2, 28.1 (3C), 26.9, 25.5. IR (film): 3422, 2934, 1751, 1697 cm−1. HRMS (DART) m/z: [M + H]+ calcd for C18H31N2O7, 387.2131; found, 387.2130.
Stapled H-l-Hyp-l-Ser-OMe (A’): To a solution of Boc-protected dipeptide 6 (45.0 mg, 0.116 mmol) in CH2Cl2 (1 mL) was added trifluoroacetic acid (0.2 mL) dropwise at room temperature, and the reaction mixture was stirred for 24 h at the same temperature. The reaction mixture was neutralized by adding sat. aq NaHCO3, and the aqueous phase was extracted with CHCl3 three times. The combined organic extracts were dried over anhydrous MgSO4 and concentrated under vacuum to give crude product A’ (20.5 mg, 62%) as an amber oil, which was used for the next step without further purification. Rf = 0.30 (EtOAc). +11.6 (c 1.00, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 8.52 (d, J = 7.6 Hz, 1H), 4.69–4.63 (m, 1H), 3.95 (t, J = 3.8 Hz, 1H), 3.91–3.82 (m, 2H), 3.78 (s, 3H), 3.79–3.74 (m, 1H), 3.61 (dt, J = 9.8, 5.9 Hz, 1H), 3.53–3.43 (m, 2H), 3.43–3.31 (m, 2H), 2.99 (dd, J = 10.5, 3.2 Hz, 1H), 2.27 (d, J = 14.2 Hz, 1H), 2.15 (ddd, J = 14.2, 11.0, 4.2 Hz, 1H), 1.78–1.46 (m, 4H). 13C NMR (125 MHz, CDCl3) δ: 175.1, 171.0, 78.6, 71.1, 69.0, 68.3, 58.9, 53.4, 52.5, 51.3, 37.0, 26.5, 26.1. IR (KBr): 3345, 2920, 2868, 1748, 1658, 1526, 1441 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C13H22N2O5Na, 309.1426; found, 309.1428.
Stapled Boc-l-Hyp-l-Ser-[(l-Leu)2-Ac5c]2-OMe (
7): To a solution of stapled dipeptide
6 (104 mg, 0.269 mmol) in MeOH (3 mL) was added 1 M of aqueous NaOH (0.270 mL, 0.270 mmol) at room temperature, and the mixture was stirred overnight at the same temperature. The solution was acidified with 1 M of aqueous HCl and the MeOH was removed in vacuo. The resulting aqueous solution was extracted with EtOAc three times. The combined organic extracts were washed with brine, dried over Na
2SO
4, and concentrated in vacuo to give a crude product (95.0 mg, 95%), which was used for the next step without further purification.
Rf = 0.27 (EtOAc). To a solution of the crude acid (85.6 mg, 0.230 mmol) in CH
2Cl
2 (2.3 mL) was added
N-(3-dimethylaminopropyl)-
N’-ethylcarbodiimide hydrochloride (EDCI·HCl; 44.0 mg, 0.230 mmol) and HOBt·H
2O (42.0 mg, 0.276 mmol) at 0 °C, and the mixture was stirred at the same temperature for 30 min. Then, a solution of H-[(
l-Leu)
2-Ac
5c]
2-OMe [
44] (163 mg, 0.230 mmol) in CH
2Cl
2 (1 mL) was added dropwise to the reaction mixture at 0 °C. The reaction mixture was gradually warmed to room temperature and stirred for 2 days. After the removal of CH
2Cl
2, the residue was diluted with EtOAc. The solution was washed successively with 1 M of HCl, water, sat. aq NaHCO
3, and brine. The organic layer was dried over anhydrous MgSO
4 and concentrated in vacuo to give a crude product, which was purified by flash column chromatography on silica gel (80% EtOAc in
n-hexane) to give
7 (178 mg, 73%) as a yellow oil.
Rf = 0.46 (EtOAc).
−4.1 (
c 1.07, CHCl
3).
1H NMR (500 MHz, CDCl
3) δ: 7.75 (br s, 1H), 7.54–7.41 (m, 2H), 7.31 (s, 1H), 7.28–7.25 (m, 2H), 7.22 (d,
J = 6.1 Hz, 1H), 4.37–4.30 (m, 1H), 4.28 (dd,
J = 10.5, 4.2 Hz, 1H), 4.24–4.16 (m, 2H), 4.14 (d,
J = 10.3 Hz, 1H), 4.08–4.03 (m, 1H), 3.98 (dd,
J = 11.0, 4.9 Hz, 1H), 3.96–3.90 (m, 1H), 3.84 (d,
J = 11.7 Hz, 1H), 3.75–3.69 (m, 1H), 3.67 (s, 3H), 3.65–3.50 (m, 3H), 3.46–3.36 (m, 2H), 2.70–2.60 (m, 1H), 2.39 (ddd,
J = 14.6, 10.9, 4.0 Hz, 1H), 2.31–2.03 (m, 8H), 1.94–1.59 (m, 24H), 1.52 (s, 9H), 1.01–0.84 (m, 24H).
13C NMR (125 MHz, CDCl
3) δ: 175.4, 175.2 (2C), 174.4, 174.2, 173.2, 173.1, 171.0, 155.4, 81.4, 78.9, 70.8, 68.5, 68.4, 66.7, 65.7, 60.4, 54.9, 54.7, 54.0, 53.9, 53.4, 52.2, 52.0, 40.0, 39.6, 39.4, 39.2, 38.2, 37.2, 36.7, 35.4, 35.3, 28.3 (3C), 27.4, 26.3, 25.0, 24.82, 24.81, 24.6, 24.50 (2C), 24.48, 24.4, 23.5, 23.4, 22.9, 22.6, 21.5, 21.2, 21.0, 20.9. IR (KBr): 3329, 2957, 1736, 1647, 1522 cm
−1. HRMS (ESI)
m/
z: [M + Na]
+ calcd for C
54H
92N
8O
13Na, 1083.6682; found, 1083.6685.
Stapled H-l-Hyp-l-Ser-[(l-Leu)2-Ac5c]2-OMe (B’): To a solution of Boc-protected peptide 7 (120 mg, 0.113 mmol) in CH2Cl2 (2 mL) was added trifluoroacetic acid (0.113 mL) dropwise at room temperature, and the reaction mixture was stirred overnight at the same temperature. The reaction mixture was neutralized by adding sat. aq NaHCO3, and the aqueous phase was extracted with CHCl3 three times. The combined organic extracts were dried over anhydrous MgSO4 and concentrated under a vacuum to give crude product B’ (96.9 mg, 89%), which was used for the next step without further purification. Rf = 0.25 (EtOAc). Mp 117–118 °C. −5.2 (c 0.95, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 8.83 (d, J = 5.4 Hz, 1H), 7.58 (d, J = 4.6 Hz, 1H), 7.45 (d, J = 7.8 Hz, 1H), 7.34 (s, 1H), 7.32–7.24 (m, 3H), 4.37–4.27 (m, 2H), 4.23–4.16 (m, 1H), 4.11 (br s, 2H), 4.02 (dd, J = 10.9, 5.0 Hz, 1H), 3.94 (dt, J = 9.7, 4.8 Hz, 1H), 3.89 (dd, J = 8.8, 3.4 Hz, 1H), 3.78 (dd, J = 10.9, 3.1 Hz, 1H), 3.67 (s, 3H), 3.66–3.61 (m, 1H), 3.60–3.50 (m, 3H), 3.34 (d, J = 10.5 Hz, 1H), 3.06 (dd, J = 10.5, 2.4 Hz, 1H), 2.69–2.60 (m, 1H), 2.36 (br s, 3H), 2.30–2.21 (m, 4H), 2.20–2.03 (m, 3H), 1.97–1.55 (m, 24H), 1.03–0.80 (m, 24H). 13C NMR (125 MHz, CDCl3) δ: 178.1, 175.6, 175.1, 174.2, 173.9, 173.2, 173.1, 172.4, 80.0, 70.3, 69.8, 68.7, 66.8, 65.7, 59.8, 55.2, 54.8, 54.1, 54.0, 52.3, 52.1, 51.5, 39.6, 39.3, 38.2, 37.2, 36.7, 36.3, 35.4, 29.6, 28.4, 27.2, 26.5, 25.2, 25.12, 25.07, 24.7, 24.50, 24.49, 24.4 (2C), 23.5, 23.4, 23.1, 22.8, 21.3, 21.1, 21.0, 20.8. IR (CDCl3): 3325, 2958, 1655, 1526 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C49H84N8O11Na, 983.6157; found, 983.6142.
3.3. Synthesis of Unstapled Peptide C and Stapled Peptide C’
Boc-l-SerOPte-[(l-Leu)2-Ac5c]2-OMe (
9): To a solution of Boc-
l-Ser
OPte-OH
8 [
51] (193 mg, 0.707 mmol) in CH
2Cl
2 (2.5 mL) were added EDCI·HCl (136 mg, 0.707 mmol) and HOBt·H
2O (130 mg, 0.848 mmol) at 0 °C, and the solution was stirred for 30 min. Then, a solution of H-[(
l-Leu)
2-Ac
5c]
2-OMe [
44] (500 mg, 0.707 mmol) in CH
2Cl
2 (2.5 mL) was added to the reaction mixture at the same temperature, and the resultant mixture was gradually warmed to room temperature. After stirring overnight, CH
2Cl
2 was removed, and the residue was diluted with EtOAc. The solution was washed successively with 1 M of HCl, water, sat. aq NaHCO
3, and brine. The organic layer was dried over anhydrous MgSO
4 and concentrated in vacuo to give a crude product, which was purified by flash column chromatography on silica gel (60% EtOAc in
n-hexane) to give
9 (478 mg, 70%) as a white solid.
Rf = 0.66 (EtOAc). Mp 109–115 °C.
−4.3 (
c 1.00, CHCl
3).
1H NMR (500 MHz, CDCl
3) δ: 7.42 (d,
J = 8.1 Hz, 1H), 7.33 (s, 1H), 7.29 (d,
J = 4.4 Hz, 1H), 7.25–7.19 (m, 2H), 6.60 (d,
J = 3.7 Hz, 1H), 5.80 (ddt,
J = 17.0, 10.3, 6.6 Hz, 1H), 5.50 (d,
J = 2.4 Hz, 1H), 5.08–4.96 (m, 2H), 4.39–4.32 (m, 1H), 4.20 (dd,
J = 11.2, 6.1 Hz, 1H), 4.14–4.07 (m, 1H), 4.04 (q,
J = 3.7 Hz, 1H), 3.98 (dt,
J = 9.7, 5.0, Hz, 1H), 3.77–3.70 (m, 2H), 3.67 (s, 3H), 3.55–3.44 (m, 2H), 2.70–2.60 (m, 1H), 2.32–2.23 (m, 1H), 2.23–2.02 (m, 6H), 1.96–1.54 (m, 24H), 1.50 (s, 9H), 1.05–0.82 (m, 24H).
13C NMR (125 MHz, CDCl
3) δ: 175.5, 175.1, 173.6, 173.4, 173.0, 172.9, 172.0, 156.9, 137.7, 115.1, 81.7, 71.0, 68.8, 66.8, 65.7, 56.9, 54.7, 54.1, 54.0, 52.2, 52.1, 40.1, 39.8, 39.6, 39.4, 38.1, 37.3, 36.7, 35.5, 30.2, 28.5, 28.1 (3C), 25.3, 25.1, 24.8, 24.7, 24.53, 24.51, 24.43, 24.40, 23.5, 23.4, 23.0, 22.9, 21.4, 21.3, 21.1, 20.9. IR (KBr): 3329, 2957, 1701, 1632, 1524 cm
−1. HRMS (ESI)
m/
z: [M + Na]
+ calcd for C
50H
87N
7O
11Na, 984.6361; found, 984.6386.
Boc-l-HypOAll-l-SerOPte-[(l-Leu)2-Ac5c]2-OMe (11): To a solution of Boc-protected peptide 9 (480 mg, 0.499 mmol) in CH2Cl2 (5 mL) was added trifluoroacetic acid (0.749 mL) dropwise at room temperature, and the reaction mixture was stirred overnight at the same temperature. The reaction mixture was neutralized by adding sat. aq NaHCO3, and the aqueous phase was extracted with CHCl3 three times. The combined organic extracts were dried over anhydrous MgSO4 and concentrated under a vacuum to give crude product 10 (391 mg, 91%), which was used for the next step without further purification. Rf = 0.20 (60% EtOAc in n-hexane). To a solution of Boc-l-HypOAll-OH (123 mg, 0.454 mmol) in CH2Cl2 (2.5 mL) were added EDCI·HCl (87.0 mg, 0.454 mmol) and HOBt·H2O (84.0 mg, 0.545 mmol) at 0 °C, and the solution was stirred for 30 min. Then, a solution of amine 10 (391 mg, 0.454 mmol) in CH2Cl2 (2.5 mL) was added to the reaction mixture at the same temperature, and the resultant mixture was gradually warmed to room temperature. After stirring overnight, the CH2Cl2 was removed and the residue was diluted with EtOAc. The solution was washed successively with 1 M of HCl, water, sat. aq NaHCO3, and brine. The organic layer was dried over anhydrous MgSO4 and concentrated in vacuo to give crude product, which was purified by flash column chromatography on silica gel (70% EtOAc in n-hexane) to give 11 (248 mg, 49%) as a white solid. Rf = 0.56 (EtOAc). Mp 75–85 °C. −3.2 (c 1.03, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.49–7.41 (m, 2H), 7.37 (d, J = 4.9 Hz, 1H), 7.26–7.20 (m, 4H), 5.93–5.72 (m, 2H), 5.33–5.19 (m, 2H), 5.06–4.95 (m, 2H), 4.39–4.30 (m, 1H), 4.25–4.09 (m, 5H), 4.05–3.90 (m, 3H), 3.83–3.72 (m, 2H), 3.67 (s, 3H), 3.67–3.63 (m, 1H), 3.52–3.38 (m, 3H), 2.70–2.61 (m, 1H), 2.36–2.32 (m, 1H), 2.31–2.11 (m, 4H), 2.10–2.02 (m, 3H), 1.96–1.66 (m, 21 H), 1.66–1.56 (m, 4H), 1.51 (s, 9H), 1.03–0.80 (m, 24H). 13C NMR (125 MHz, CDCl3) δ: 175.6, 175.2, 174.6, 174.2, 174.1, 173.1, 173.0, 171.9, 155.7, 137.5, 133.9, 117.5, 115.2, 81.6, 77.2, 70.8, 69.6, 68.3, 66.8, 65.7, 60.2, 56.2, 54.8, 54.1, 54.0, 53.4, 52.2, 52.1, 39.7, 39.6, 39.4, 39.2, 38.3, 37.3, 36.7, 35.5, 34.3, 30.0, 28.4, 28.3 (3C), 25.2, 25.1, 24.8, 24.7, 24.57, 24.55 (2C), 24.47, 23.5, 23.4, 23.00, 22.97, 21.12, 21.06, 21.0, 20.9. IR (CDCl3): 3325, 2959, 2872, 1732, 1661, 1530 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C58H98N8O13Na, 1137.7151; found, 1137.7201.
H-l-HypOAll-l-SerOPte-[(l-Leu)2-Ac5c]2-OMe (C): To a solution of Boc-protected peptide 11 (30.0 mg, 0.0269 mmol) in CH2Cl2 (1 mL) was added trifluoroacetic acid (0.03 mL) dropwise at room temperature, and the reaction mixture was stirred overnight at the same temperature. The reaction mixture was neutralized by adding sat. aq NaHCO3, and the aqueous phase was extracted with CHCl3 three times. The combined organic extracts were dried over anhydrous MgSO4 and concentrated under a vacuum to give crude product C (23.5 mg, 86%), which was used for the next step without further purification. Rf = 0.31 (EtOAc). Mp 79–81 °C. −7.8 (c 1.00, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 8.45 (br s, 1H), 7.65 (d, J = 5.1 Hz, 1H), 7.46 (d, J = 7.8 Hz, 1H), 7.32 (s, 1H), 7.26–7.21 (m, 2H), 6.59 (br s, 1H), 5.90–5.74 (m, 2H), 5.27–5.12 (m, 2H), 5.08–4.95 (m, 2H), 4.37–4.26 (m, 1H), 4.25–4.15 (m, 1H), 4.15–4.08 (m, 2H), 4.04 (br s, 1H), 3.98–3.83 (m, 4H), 3.76 (dd, J = 10.0, 4.0 Hz, 1H), 3.70 (dd, J = 10.0, 3.9 Hz, 1H), 3.67 (s, 3H), 3.55–3.43 (m, 2H), 3.20 (dd, J = 11.3, 4.2 Hz, 1H), 3.13 (d, J = 11.3 Hz, 1H), 2.64 (dt, J = 13.6, 8.4 Hz, 1H), 2.37 (d, J = 13.7 Hz, 1H), 2.30–2.02 (m, 8H), 1.98–1.50 (m, 25H), 1.03–0.81 (m, 24H). 13C NMR (125 MHz, CDCl3) δ: 178.3, 175.6, 175.2, 174.2, 174.1, 173.3, 173.2, 172.1, 137.7, 134.6, 116.8, 115.1, 78.7, 70.7, 69.6, 68.8, 66.8, 65.7, 59.2, 56.5, 54.8, 54.1 (2C), 52.4, 52.3, 52.1, 39.64, 39.62, 39.4, 39.1, 38.2, 37.2, 36.7, 35.7, 35.3, 30.1, 28.6, 25.2, 25.0 (2C), 24.7, 24.52, 24.49, 24.4, 24.3, 23.45, 23.36, 22.99, 22.96, 21.13, 21.08 (2C), 20.9. IR (KBr): 3343, 2957, 1639, 1547 cm−1. HRMS (ESI) m/z: [M + H]+ calcd for C53H91N8O11, 1015.6807; found, 1015.6843.
Stapled Boc-l-Hyp-l-Ser-[(l-Leu)2-Ac5c]2-OMe (13): Under an argon atmosphere, to a solution of 11 (70.0 mg, 0.0628 mmol) in CH2Cl2 (3 mL) was added second-generation Grubbs catalyst (10.7 mg, 0.0126 mmol) at room temperature, and the mixture was stirred for 2 h at the same temperature. The reaction mixture was filtered through short pad of silica gel (EtOAc) and concentrated. The crude material was purified by flash chromatography on silica gel (70% EtOAc in n-hexane) to provide a stapled peptide 12 (63.2 mg, 93%) as a mixture of E- and Z-isomers (E/Z = 5.5:1). Rf = 0.43 (EtOAc). Next, to a solution of stapled peptides 12 (52.9 mg, 0.0486 mmol) in MeOH (4 mL) was added 10% Pd-C (26 mg, 50 wt %) under a nitrogen atmosphere. After being vigorously stirred under a hydrogen atmosphere for 23 h at room temperature, the reaction mixture was passed through a short plug of Celite. The filtrate was concentrated under vacuum to give a crude product, which was purified by flash column chromatography on silica gel (4% MeOH in CHCl3) to give 13 (46.9 mg, 89%) as a colorless oil. Rf = 0.13 (3% MeOH in CHCl3). −7.8 (c 1.00, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.58 (d, J = 5.9 Hz, 1H), 7.50 (d, J = 4.9 Hz, 1H), 7.45 (d, J = 7.8 Hz, 1H), 7.31 (s, 1H), 7.27–7.22 (m, 2H), 7.17 (d, J = 2.0 Hz, 1H), 4.37–4.30 (m, 2H), 4.23–4.13 (m, 3H), 4.02 (t, J = 3.4 Hz, 1H), 3.97–3.91 (m, 1H), 3.83–3.76 (m, 2H), 3.67 (s, 3H), 3.63–3.51 (m, 4H), 3.50–3.44 (m, 1H), 3.33 (dd, J = 12.0, 2.9 Hz, 1H), 2.65 (dt, J = 13.6, 8.2 Hz, 1H), 2.46–2.37 (m, 1H), 2.31–2.03 (m, 6H), 2.00 (br s, 1H), 1.95–1.54 (m, 26H), 1.52 (s, 9H), 1.51–1.42 (m, 3H), 1.01–0.82 (m, 24H). 13C NMR (125 MHz, CDCl3) δ: 175.6, 175.2, 174.5, 174.4, 174.1, 173.1, 173.0, 171.1, 155.8, 81.6, 78.0, 71.8, 69.7, 69.4, 66.7, 65.7, 60.5, 56.2, 54.8, 54.2, 54.0, 52.7, 52.2, 52.0, 39.7, 39.6, 39.4, 39.1, 38.3, 37.3, 36.7, 35.6, 35.4, 29.1, 28.2 (3C), 27.0, 26.9, 25.4, 25.1, 25.0, 24.8, 24.7, 24.54, 24.52 (2C), 24.4, 23.5, 23.4, 23.0, 22.8, 21.14, 21.05, 21.0, 20.9. IR (CDCl3): 3321, 2959, 1732, 1661, 1530 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C56H96N8O13Na, 1111.6995; found, 1111.7016.
Stapled H-l-Hyp-l-Ser-[(l-Leu)2-Ac5c]2-OMe (C’): To a solution of Boc-protected peptide 13 (11.5 mg, 0.0110 mmol) in CH2Cl2 (1 mL) was added trifluoroacetic acid (0.0110 mL) dropwise at room temperature, and the reaction mixture was stirred for 2 days at the same temperature. The reaction mixture was neutralized by adding sat. aq NaHCO3, and the aqueous phase was extracted with CHCl3 three times. The combined organic extracts were dried over anhydrous MgSO4 and concentrated under a vacuum to give crude product C’ (11.6 mg, quant), which was used for the next step without further purification. Rf = 0.20 (EtOAc). Mp 105–107 °C. −8.7 (c 1.00, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 8.27 (d, J = 5.1 Hz, 1H), 7.44 (d, J = 7.8 Hz, 1H), 7.33 (d, J = 4.6 Hz, 1H), 7.26–7.21 (m, 3H), 7.00 (d, J = 4.2 Hz, 1H), 4.37–4.27 (m, 2H), 4.19 (dd, J = 10.8, 5.9 Hz, 1H), 4.13–4.04 (m, 2H), 3.97–3.86 (m, 2H), 3.84 (dd, J = 10.5, 5.9 Hz, 1H), 3.70 (dd, J = 10.6, 2.8 Hz, 1H), 3.67 (s, 3H), 3.64–3.59 (m, 1H), 3.59–3.53 (m, 1H), 3.48 (t, J = 8.9 Hz, 1H), 3.43–3.37 (m, 1H), 3.35 (d, J = 10.5 Hz, 1H), 3.00 (dd, J = 10.5, 2.9 Hz, 1H), 2.69–2.60 (m, 1H), 2.37–2.03 (m, 7H), 1.97–1.49 (m, 30H), 1.03–0.78 (m, 24H). 13C NMR (125 MHz, CDCl3) δ: 175.7, 175.3, 174.2, 174.0, 173.23, 173.20 (2C), 172.0, 79.5, 71.5, 69.3, 69.0, 66.8, 65.7, 59.6, 55.9, 54.8, 54.1 (2C), 52.4, 52.2, 51.0, 39.6, 39.5, 39.4, 39.2, 38.3, 37.2, 36.7, 36.0, 35.4, 28.9, 28.3, 27.5, 26.4, 25.23, 25.20, 25.1, 24.8, 24.6, 24.52, 24.49, 24.40, 23.5, 23.4, 23.1, 22.9, 21.3, 21.1, 21.0, 20.9. IR (KBr): 3337, 2957, 1736, 1655, 1535 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C51H88N8O11Na, 1011.6470; found, 1011.6467.
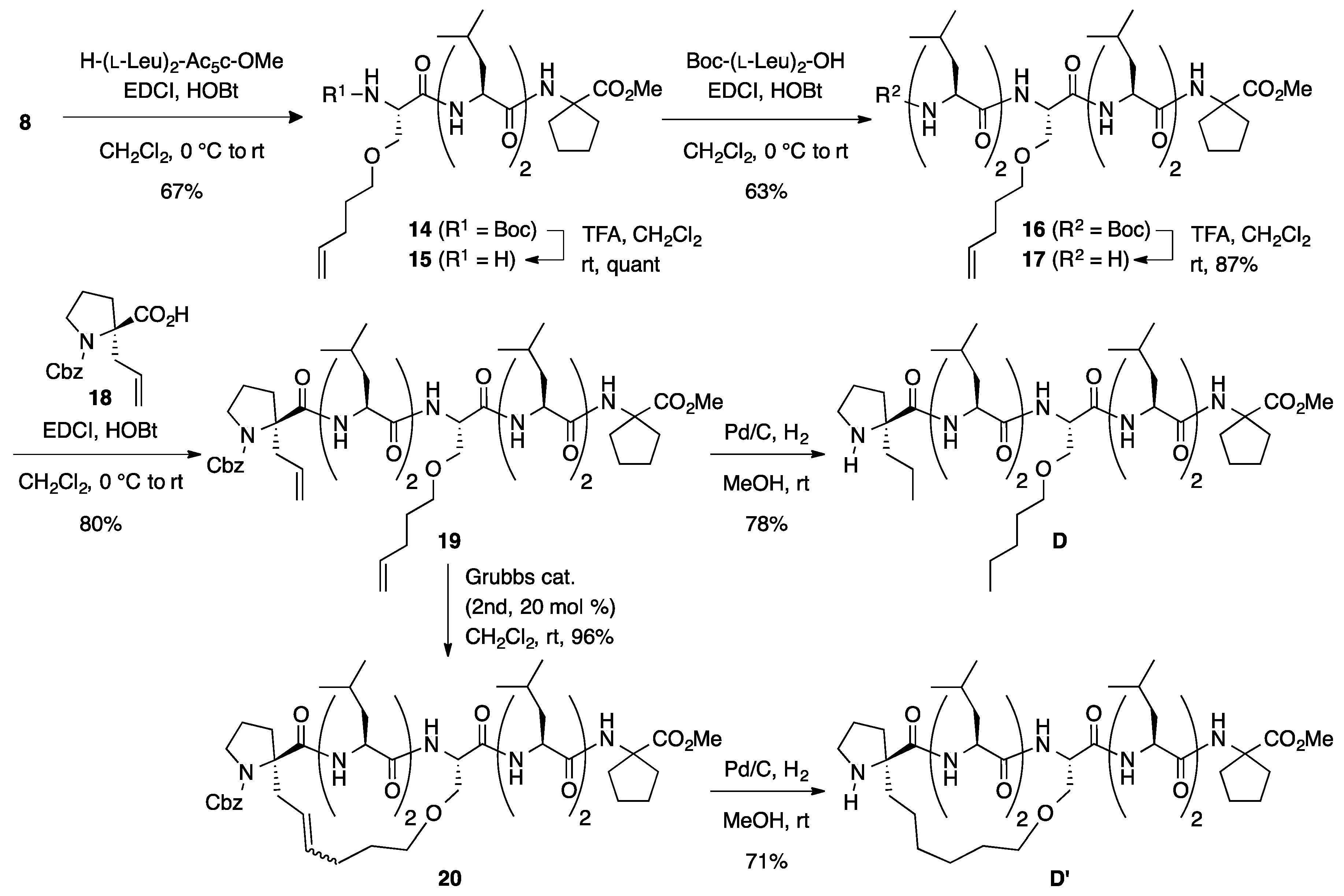
3.4. Synthesis of Unstapled Peptide D and Stapled Peptide D’
Boc-l-SerOPte-(l-Leu)2-Ac5c-OMe (
14): To a solution of Boc-
l-Ser
OPte-OH
8 [
51] (200 mg, 0.732 mmol) in CH
2Cl
2 (2.5 mL) were added EDCI·HCl (140 mg, 0.732 mmol) and HOBt·H
2O (135 g, 0.878 mmol) at 0 °C, and the solution was stirred for 30 min. Then, a solution of H-(
l-Leu)
2-Ac
5c-OMe [
44] (270 mg, 0.732 mmol) in CH
2Cl
2 (2.5 mL) was added to the reaction mixture at the same temperature, and the resultant mixture was gradually warmed to room temperature. After stirring overnight, the CH
2Cl
2 was removed and the residue was diluted with EtOAc. The solution was washed successively with 1 M of HCl, water, sat. aq NaHCO
3, and brine. The organic layer was dried over anhydrous MgSO
4 and concentrated in vacuo to give a crude product, which was purified by flash column chromatography on silica gel (50% EtOAc in
n-hexane) to give
14 (307 mg, 67%) as a white solid.
Rf = 0.52 (60% EtOAc in
n-hexane). Mp 109–115 °C.
−40.4 (
c 0.995, CHCl
3).
1H NMR (500 MHz, CDCl
3) δ: 6.84 (br s, 2H), 6.57 (d,
J = 4.9 Hz, 1H), 5.79 (ddt,
J = 17.0, 10.3, 6.6 Hz, 1H), 5.38 (br s, 1H), 5.06–4.95 (m, 2H), 4.44 (td,
J = 9.2, 4.9 Hz, 1H), 4.37–4.29 (m, 1H), 4.12 (q,
J = 4.9 Hz, 1H), 3.76 (dd,
J = 9.5, 4.6 Hz, 1H), 3.68 (s, 3H), 3.62 (dd,
J = 9.2, 5.3 Hz, 1H), 3.53–3.44 (m, 2H), 2.25 (dt,
J = 13.2, 7.7 Hz, 1H), 2.17 (dt,
J = 13.1, 7.8 Hz, 1H), 2.10 (q,
J = 7.3 Hz, 2H), 2.07–1.95 (m, 2H), 1.87–1.63 (m, 9H), 1.63–1.49 (m, 3H), 1.47 (s, 9H), 1.00–0.85 (m, 12H).
13C NMR (125 MHz, CDCl
3) δ: 174.6, 171.6, 171.5, 171.1, 156.3, 137.8, 115.1, 81.1, 71.0, 69.4, 65.9, 55.6, 53.0, 52.3, 51.5, 40.4, 39.9, 37.2, 36.9, 30.2, 28.6, 28.2 (3C), 25.1, 24.8, 24.5, 24.4, 23.1, 23.0, 21.6, 21.4. IR (KBr): 3277, 2957, 1719, 1670, 1560 cm
−1. HRMS (ESI)
m/
z: [M + Na]
+ calcd for C
32H
56N
4O
8Na, 647.3996; found, 647.3991.
Boc-(l-Leu)2-l-SerOPte-(l-Leu)2-Ac5c-OMe (16): To a solution of Boc-protected peptide 14 (307 mg, 0.491 mmol) in CH2Cl2 (5 mL) was added trifluoroacetic acid (0.982 mL) dropwise at room temperature, and the reaction mixture was stirred overnight at the same temperature. The reaction mixture was neutralized by adding sat. aq NaHCO3, and the aqueous phase was extracted with CHCl3 four times. The combined organic extracts were dried over anhydrous MgSO4 and concentrated under a vacuum to give crude product 15 (295 mg, quant), which was used for the next step without further purification. Rf = 0.20 (60% EtOAc in n-hexane). To a solution of Boc-(l-Leu)2-OH (194 mg, 0.562 mmol) in CH2Cl2 (2 mL) were added EDCI·HCl (108 mg, 0.562 mmol) and HOBt·H2O (103 mg, 0.674 mmol) at 0 °C, and the solution was stirred for 30 min. Then, a solution of amine 15 (295 mg, 0.562 mmol) in CH2Cl2 (2 mL) was added to the reaction mixture at the same temperature, and the resultant mixture was gradually warmed to room temperature. After stirring for 5 days, the CH2Cl2 was removed and the residue was diluted with EtOAc. The solution was washed successively with 1 M of HCl, water, sat. aq NaHCO3, and brine. The organic layer was dried over anhydrous MgSO4 and concentrated in vacuo to give a crude product, which was purified by flash column chromatography on silica gel (60% EtOAc in n-hexane) to give 16 (300 mg, 63%) as a white solid. Rf = 0.30 (60% EtOAc in n-hexane). Mp 243–246 °C. −43.2 (c 1.02, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 7.41 (d, J = 4.4 Hz, 1H), 7.30 (d, J = 6.6 Hz, 1H), 7.13 (d, J = 8.1 Hz, 1H), 7.02 (s, 1H), 6.84 (d, J = 3.7 Hz, 1H), 5.76 (ddt, J = 17.1, 10.3, 6.6 Hz, 1H), 5.05–4.92 (m, 2H), 5.00 (s, 1H), 4.42–4.34 (m, 1H), 4.34–4.27 (m, 1H), 4.25–4.20 (m, 1H), 4.06–3.96 (m, 2H), 3.86 (dd, J = 10.0, 5.4 Hz, 1H), 3.67 (s, 3H), 3.67–3.64 (m, 1H), 3.50–3.38 (m, 2H), 2.27–2.00 (m, 6H), 1.87–1.51 (m, 18H), 1.48 (s, 9H), 1.02–0.83 (m, 24H). 13C NMR (125 MHz, CDCl3) δ: 174.9, 174.3, 173.4, 172.47, 172.45, 170.9, 156.6, 137.8, 114.9, 81.5, 70.5, 68.7, 65.7, 56.3, 54.3, 54.0, 53.4, 52.2, 52.0, 40.2, 39.62 (2C), 39.59, 39.4, 37.2, 36.8, 30.1, 28.7, 28.22 (3C), 28.17 (2C), 25.0, 24.9, 24.8, 24.7, 24.4, 24.3, 23.4, 22.9, 21.5, 21.2, 20.7. IR (KBr): 3277, 2957, 1719, 1630, 1560 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C44H78N6O10Na, 873.5677; found, 873.5658.
Cbz-l-ProαAll-(l-Leu)2-l-SerOPte-(l-Leu)2-Ac5c-OMe (
19): To a solution of Boc-protected peptide
16 (112 mg, 0.132 mmol) in CH
2Cl
2 (1.3 mL) was added trifluoroacetic acid (0.264 mL) dropwise at room temperature, and the reaction mixture was stirred for 2 days at the same temperature. The reaction mixture was neutralized by adding sat. aq NaHCO
3, and the aqueous phase was extracted with CHCl
3 three times. The combined organic extracts were dried over anhydrous MgSO
4 and concentrated under a vacuum to give crude product
17 (85.8 mg, 87%), which was used for the next step without further purification.
Rf = 0.37 (EtOAc). To a solution of Cbz-
l-Pro
αAll-OH (
18 [
52,
53]; 27.2 mg, 0.0939 mmol) in CH
2Cl
2 (1 mL) were added EDCI·HCl (18.0 mg, 0.0939 mmol) and HOBt·H
2O (17.3 mg, 0.113 mmol) at 0 °C, and the solution was stirred for 30 min. Then, a solution of amine
17 (70.5 mg, 0.0939 mmol) in CH
2Cl
2 (1 mL) was added to the reaction mixture at the same temperature, and the resultant mixture was gradually warmed to room temperature. After stirring for 23 h, the CH
2Cl
2 was removed and the residue was diluted with EtOAc. The solution was washed successively with 1 M of HCl, water, sat. aq NaHCO
3, and brine. The organic layer was dried over anhydrous MgSO
4 and concentrated in vacuo to give a crude product, which was purified by flash column chromatography on silica gel (50% EtOAc in
n-hexane) to give
19 (77.0 mg, 80%) as a colorless oil.
Rf = 0.60 (60% EtOAc in
n-hexane).
−8.6 (
c 2.13, CHCl
3).
1H NMR (500 MHz, CDCl
3) δ: 7.65 (br s, 1H), 7.51 (br s, 1H), 7.42–7.35 (m, 3H), 7.35–7.31 (m, 2H), 7.29 (d,
J = 6.6 Hz, 1H), 7.19 (d,
J = 7.8 Hz, 1H), 7.08 (s, 1H), 6.33 (br s, 1H), 5.82–5.63 (m, 2H), 5.18 (s, 2H), 5.17–5.05 (m, 2H), 5.00–4.89 (m, 2H), 4.37 (q,
J = 7.5 Hz, 1H), 4.32–4.24 (m, 2H), 4.10–3.98 (m, 2H), 3.92–3.85 (m, 1H), 3.83–3.71 (m, 2H), 3.67 (s, 3H), 3.54–3.47 (m, 1H), 3.45 (t,
J = 6.6 Hz, 2H), 2.96 (dd,
J = 14.2, 7.3 Hz, 1H), 2.76 (dd,
J = 14.2, 7.6 Hz, 1H), 2.28–2.11 (m, 5H), 2.11–1.99 (m, 5H), 1.91–1.54 (m, 17H), 1.46–1.38 (m, 1H), 1.05–0.81 (m, 24H).
13C NMR (125 MHz, CDCl
3) δ: 175.3, 175.0, 174.6, 174.1, 172.62, 172.60, 171.0, 155.2, 138.1, 136.0, 132.0, 128.7 (2C), 128.5, 127.4 (2C), 120.5, 114.6, 70.2, 69.0, 68.9, 67.5, 65.7, 56.7, 54.6, 54.2, 53.5, 52.12, 52.11, 48.6, 39.7 (2C), 39.4, 39.3, 37.6, 37.1, 36.8, 35.7, 30.2, 28.7, 25.3, 25.0, 24.9, 24.8, 24.4 (2C), 24.3, 23.4 (2C), 23.3 (2C), 23.1, 22.9, 21.3, 20.9. IR (CDCl
3): 3323, 2959, 1663, 1531 cm
−1. HRMS (ESI)
m/
z: [M + Na]
+ calcd for C
55H
87N
7O
11Na, 1044.6361; found, 1044.6360.
H-l-ProαAll-(l-Leu)2-l-SerOPte-(l-Leu)2-Ac5c-OMe (D): To a solution of Cbz-protected peptide 19 (21.4 mg, 0.0209 mmol) in MeOH (2 mL) was added 10% Pd-C (10 mg, 50 wt %) under a nitrogen atmosphere. After being vigorously stirred under a hydrogen atmosphere for 2 days at room temperature, the reaction mixture was passed through a short plug of Celite. The filtrate was concentrated under a vacuum to give a crude product, which was purified by flash column chromatography on silica gel (70% EtOAc in n-hexane) to give D (14.5 mg, 78%) as a white solid. Rf = 0.38 (EtOAc). −54.3 (c 1.60, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 8.45 (d, J = 4.4 Hz, 1H), 7.32 (d, J = 5.1 Hz, 1H), 7.28 (d, J = 8.0 Hz, 1H), 7.19–7.10 (m, 2H), 6.99 (s, 1H), 4.43–4.29 (m, 2H), 4.22–4.14 (m, 2H), 4.00–3.92 (m, 1H), 3.85 (dd, J = 9.9, 4.8 Hz, 1H), 3.70 (dd, J = 9.8, 3.4 Hz, 1H), 3.67 (s, 3H), 3.48–3.37 (m, 2H), 3.16–3.07 (m, 1H), 2.84 (dt, J = 10.5, 5.4 Hz, 1H), 2.27–2.12 (m, 3H), 2.12–1.94 (m, 3H), 1.86 (d, J = 12.5 Hz, 2H), 1.83–1.61 (m, 18H), 1.59–1.46 (m, 4H), 1.41–1.22 (m, 4H), 1.04–0.81 (m, 30H). 13C NMR (125 MHz, CDCl3) δ: 179.4, 174.9, 173.9, 173.4, 172.5, 172.4, 171.0, 71.5, 70.0, 68.6, 65.7, 56.2, 54.5, 53.3, 52.3, 52.2, 52.0, 47.4, 41.3, 40.4, 39.6, 39.3, 38.5, 37.2, 37.0, 36.8, 29.3, 28.0, 26.3, 24.9, 24.8 (3C), 24.4, 24.3, 23.6, 23.4, 23.2, 22.9, 22.5, 21.3, 21.2, 21.1, 20.7, 18.6, 14.4, 14.0. IR (CDCl3): 3327, 2961, 1734, 1663, 1530 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C47H85N7O9Na, 914.6306; found, 914.6267.
Stapled H-l-Pro-(l-Leu)2-l-Ser-(l-Leu)2-Ac5c-OMe (D’): Under an argon atmosphere, to a solution of 19 (22.3 mg, 0.0218 mmol) in CH2Cl2 (1 mL) was added second-generation Grubbs catalyst (3.7 mg, 4.4 μmol) at room temperature, and the mixture was stirred for 2 h at the same temperature. The reaction mixture was filtered through a short pad of silica gel (EtOAc) and concentrated. The crude material was purified by flash chromatography on silica gel (80% EtOAc in n-hexane) to provide stapled peptide 20 (21.0 mg, 96%) as a mixture of E- and Z-isomers (E/Z = 1.1:1). Rf (E-form) = 0.35 (50% EtOAc in n-hexane), Rf (Z-form) = 0.26 (50% EtOAc in n-hexane). Next, to a solution of stapled peptides 20 (28.1 mg, 0.0283 mmol) in MeOH (1.5 mL) was added 10% Pd-C (15 mg, 50 wt %) under a nitrogen atmosphere. After being vigorously stirred under a hydrogen atmosphere for 21 h at room temperature, the reaction mixture was passed through a short plug of Celite. The filtrate was concentrated under a vacuum to give a crude product, which was purified by flash column chromatography on silica gel (5% MeOH in CHCl3) to give D’ (13.6 mg, 71%) as a white solid. Rf = 0.16 (3% MeOH in CHCl3). −30.2 (c 1.38, CHCl3). 1H NMR (500 MHz, CDCl3) δ: 8.49 (br s, 1H), 7.27 (d, J = 7.3 Hz, 1H), 7.23 (d, J = 6.1 Hz, 1H), 7.09 (d, J = 7.8 Hz, 1H), 6.96 (s, 1H), 6.38 (br s, 1H), 4.48 (ddd, J = 10.0, 6.1, 3.4 Hz, 1H), 4.40–4.27 (m, 2H), 4.03–3.93 (m, 2H), 3.90 (dd, J = 9.8, 3.4 Hz, 1H), 3.77–3.68 (m, 1H), 3.68 (s, 3H), 3.54 (dt, J = 9.2, 4.5 Hz, 1H), 3.50–3.43 (m, 1H), 3.14–3.06 (m, 1H), 2.86–2.78 (m, 1H), 2.31–2.20 (m, 2H), 2.19–2.12 (m, 1H), 2.11–1.99 (m, 2H), 1.99–1.91 (m, 1H), 1.90–1.58 (m, 21H), 1.57–1.36 (m, 7H), 1.05–0.82 (m, 24H). 13C NMR (125 MHz, CDCl3) δ: 174.9, 174.2, 173.39, 173.38, 172.5, 172.4, 170.4, 69.0, 68.8, 65.8, 56.0, 54.5, 54.3, 53.5, 52.2, 52.0, 46.6, 40.3, 40.1, 39.68, 39.66, 38.7, 37.2, 36.9, 36.8, 28.2, 27.4, 26.4, 25.5, 25.2, 25.1, 24.9, 24.7, 24.40, 24.35 (2C), 24.2, 23.3 (2C), 23.0, 22.7, 21.8, 21.2, 21.1, 21.0. IR (KBr): 3296, 3109, 2953, 1641, 1530 cm−1. HRMS (ESI) m/z: [M + Na]+ calcd for C45H79N7O9Na, 884.5837; found, 884.5874.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

