
Molecular Sensing with Host Systems for Hyperpolarized 129Xe


Abstract

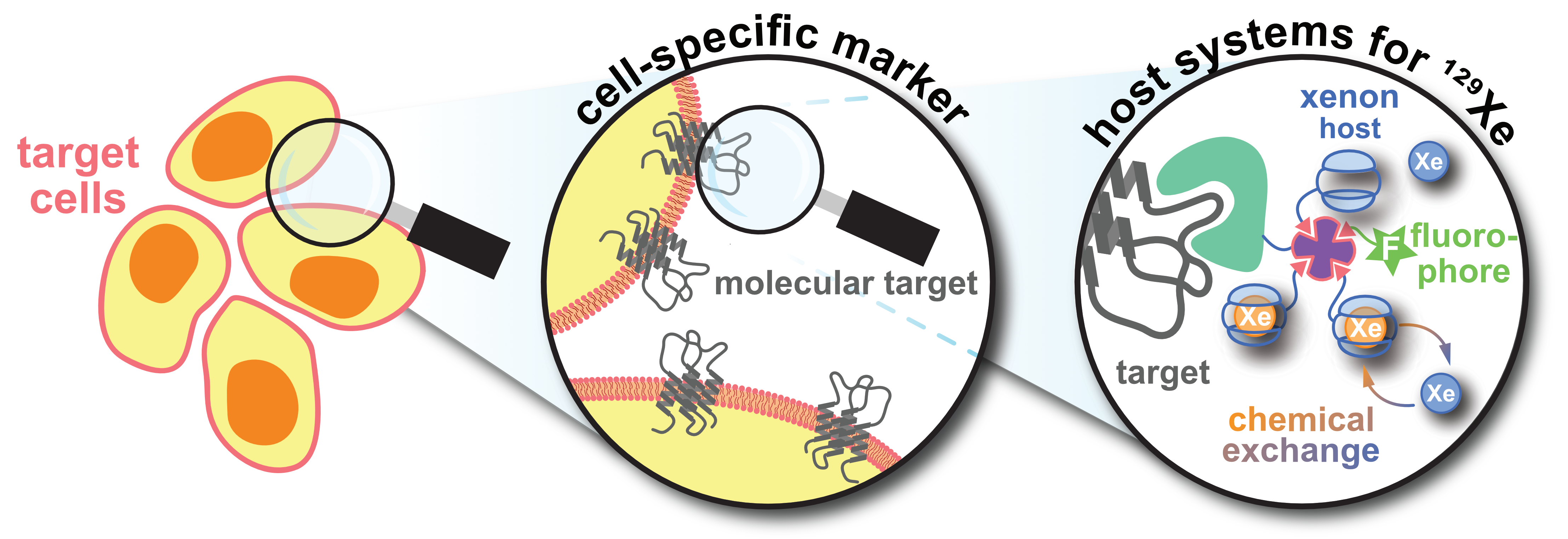
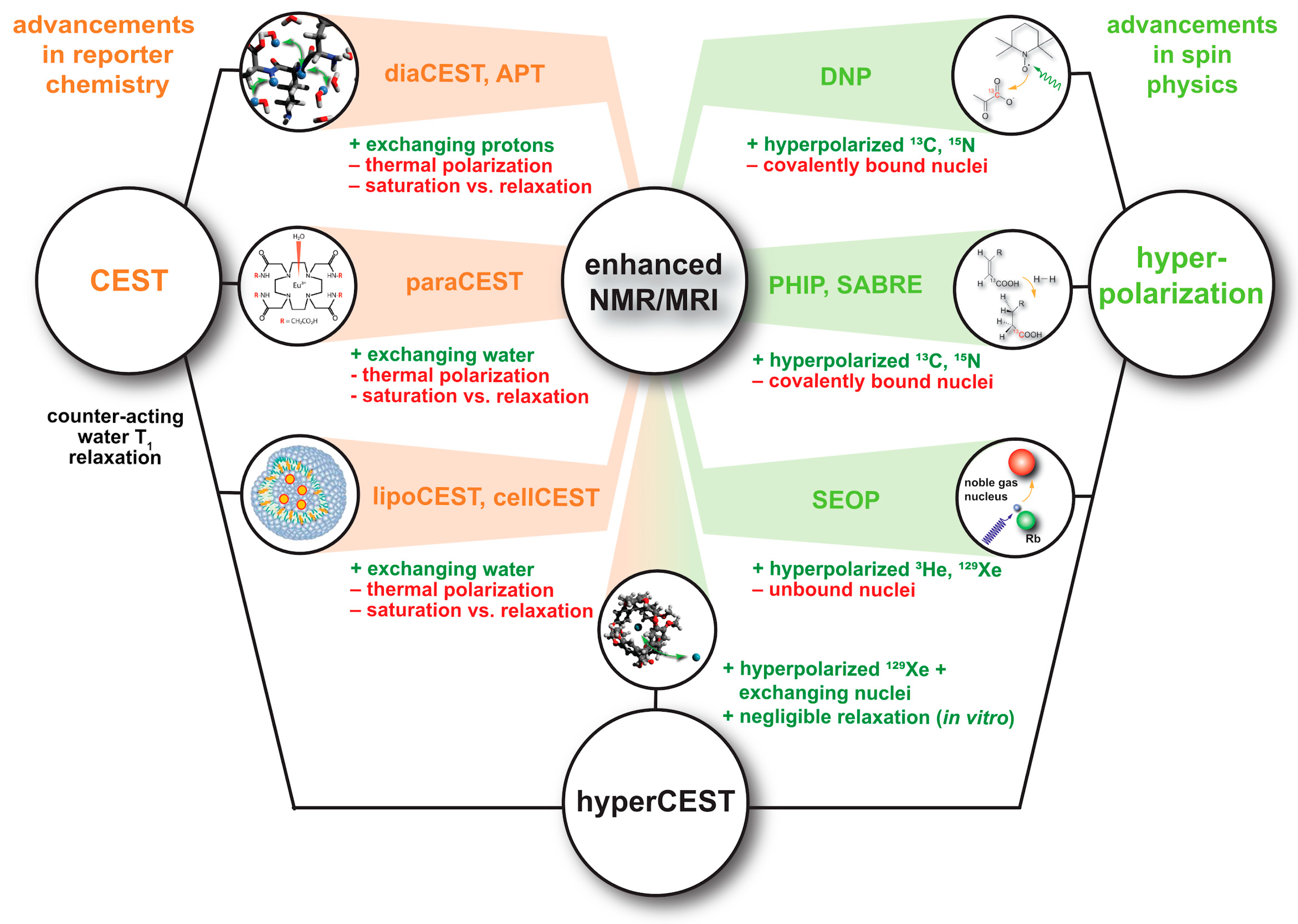
1. Introduction
2. Xenon-Host Interactions: The Basis for Functionalized 129Xe NMR
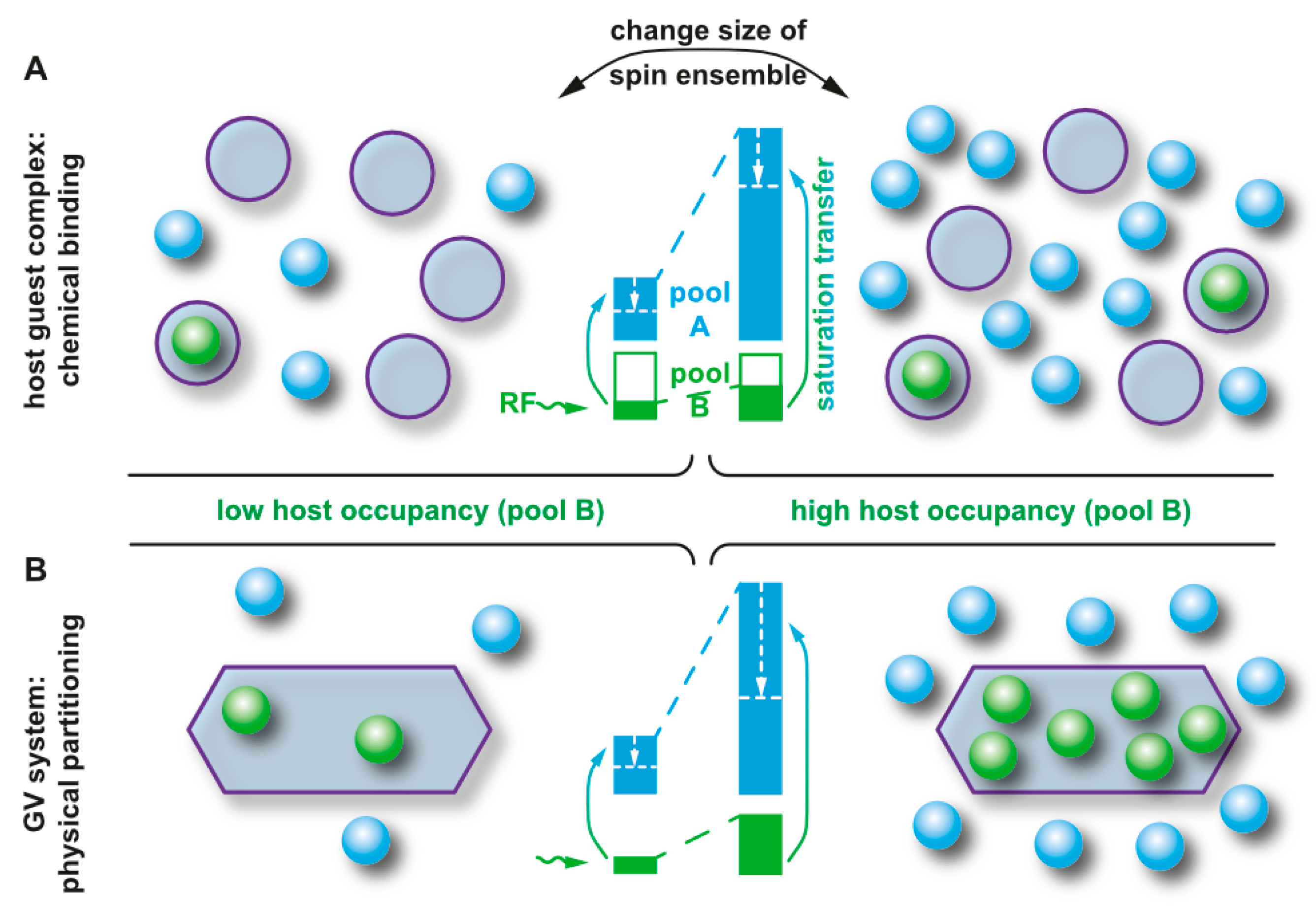
2.1. Unspecific Loose Interactions with Proteins
2.2. High-Affinity, but Reversible Binding in Synthetic Structures
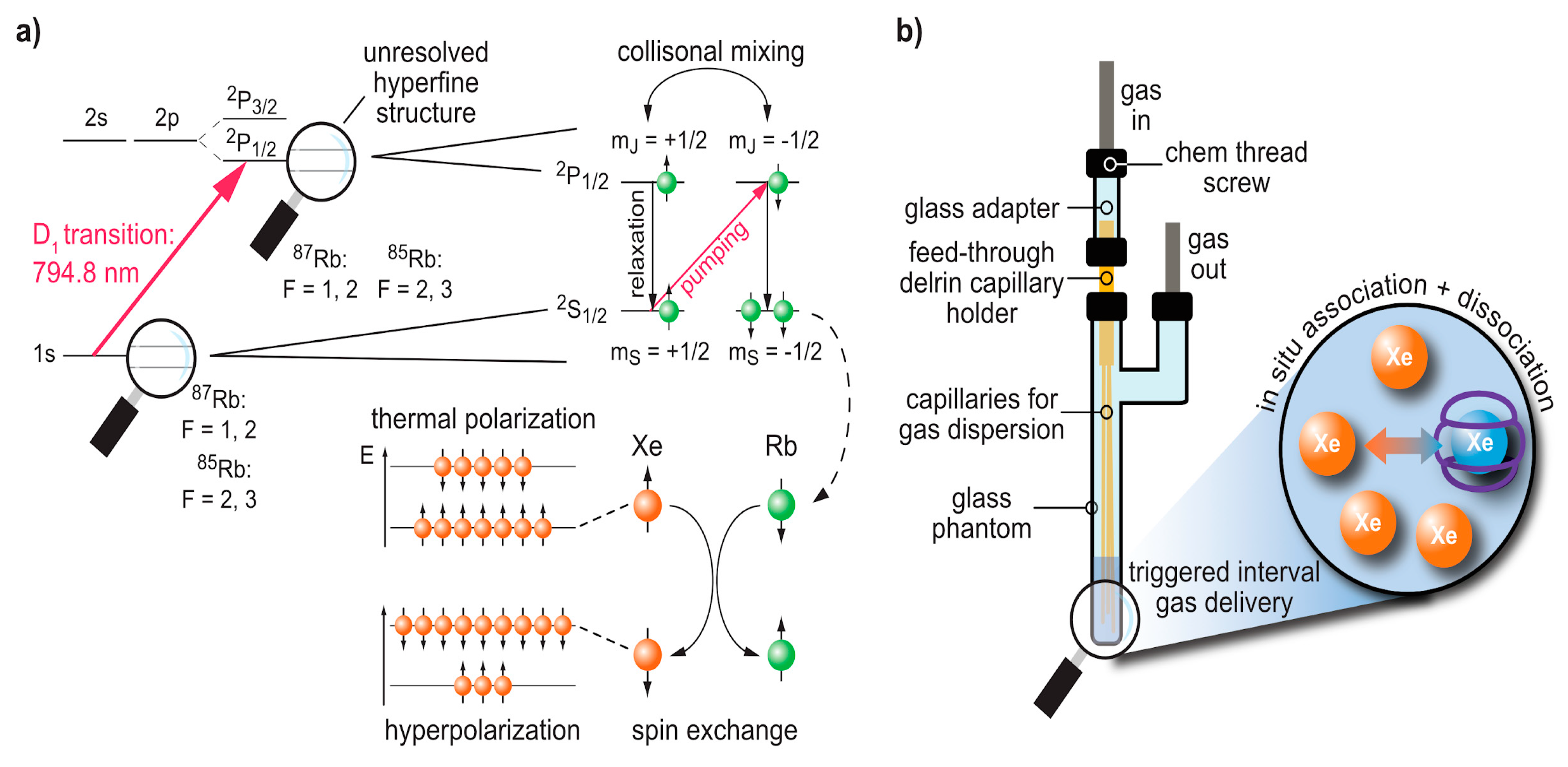
3. Production and In Situ Delivery of hp Xe
3.1. Spin Exchange Optical Pumping (SEOP)
3.2. Delivery and Handling of Xe Compared to Other hp Tracers
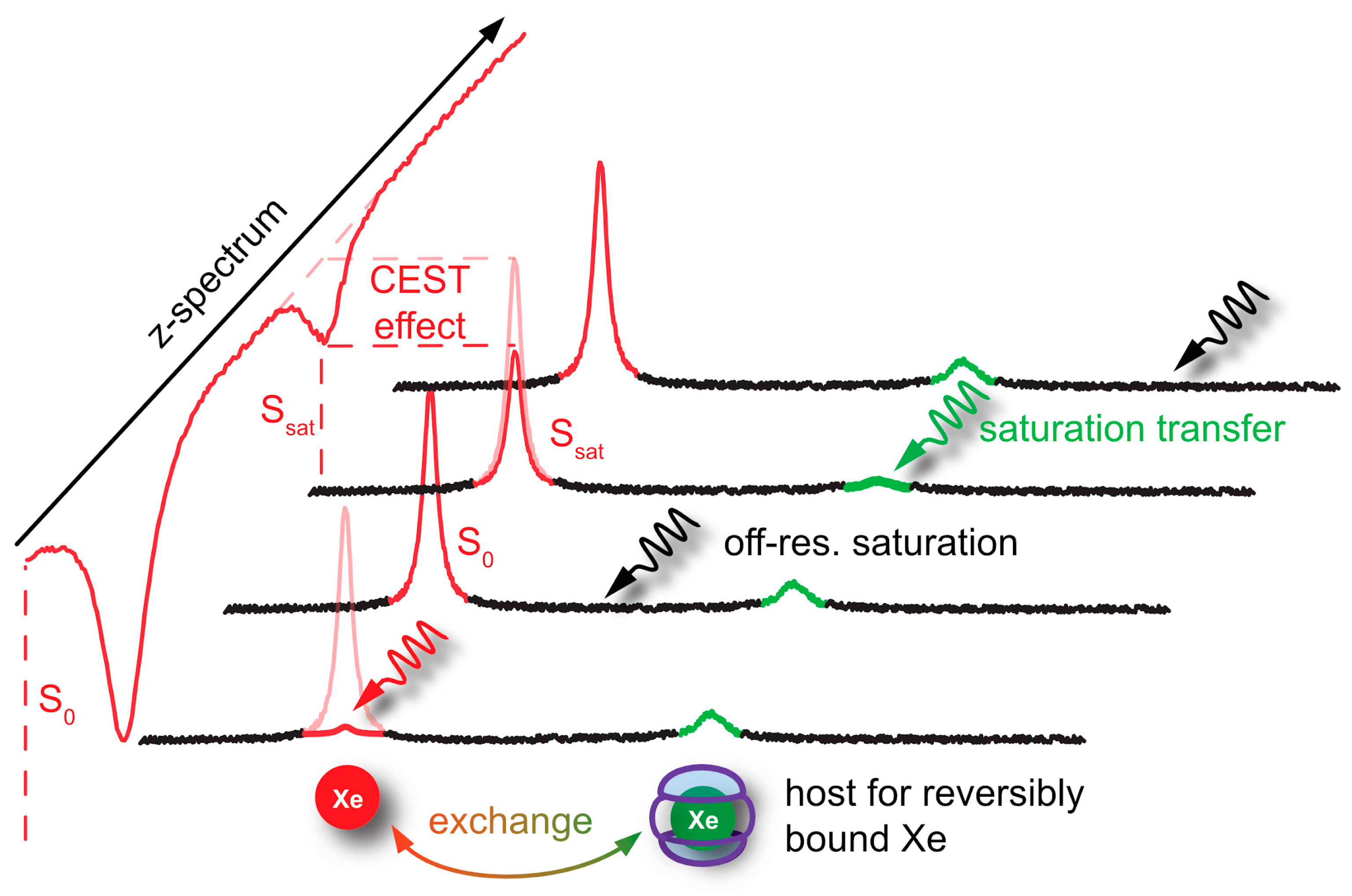
4. Signal Transfer Detection Concepts for Xe
4.1. Spin Polarization Induced Nuclear Overhauser Effect (SPINOE)
4.2. HyperCEST and MT Detection
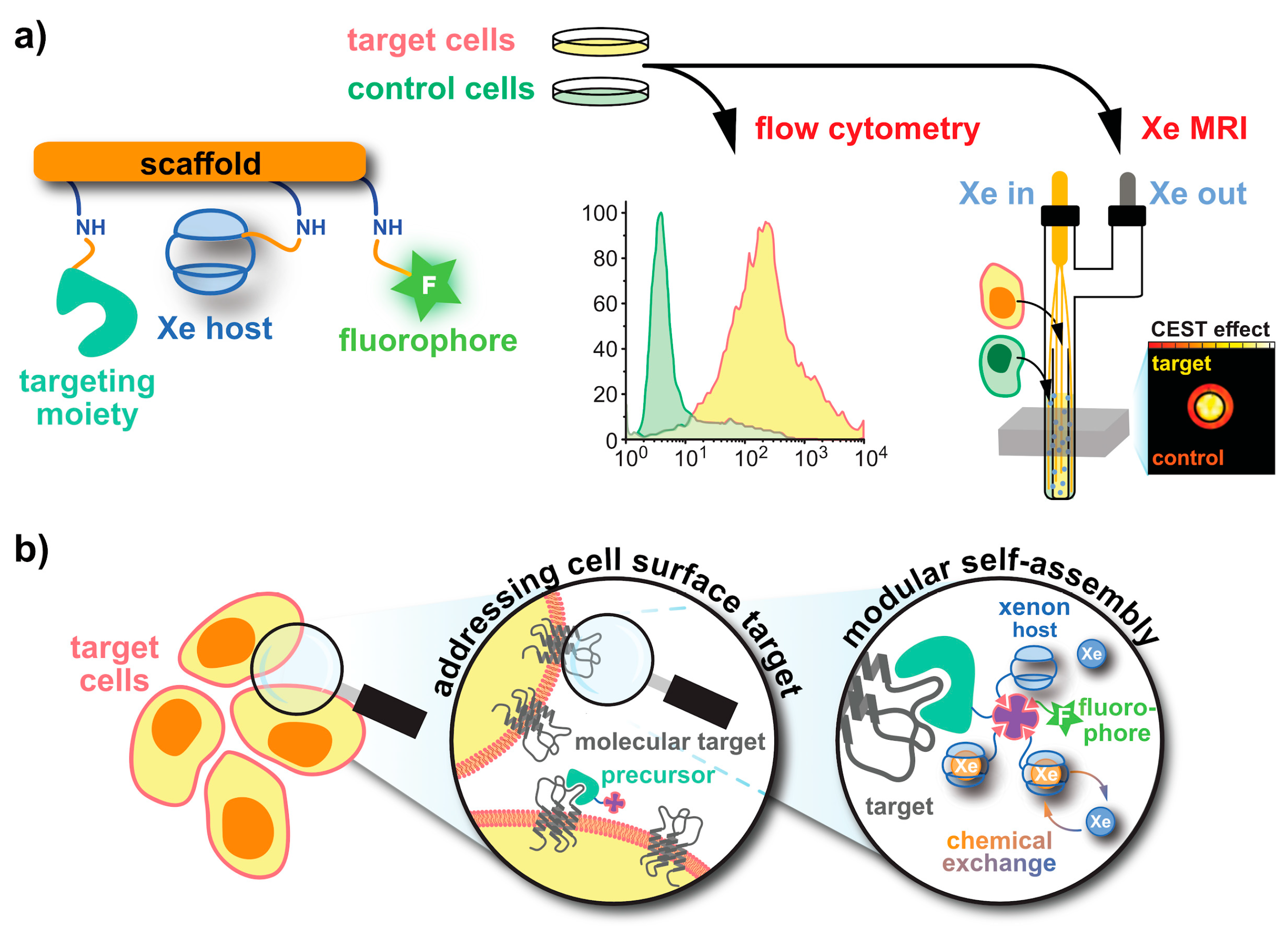
5. Aspects of 129Xe Biosensor Design
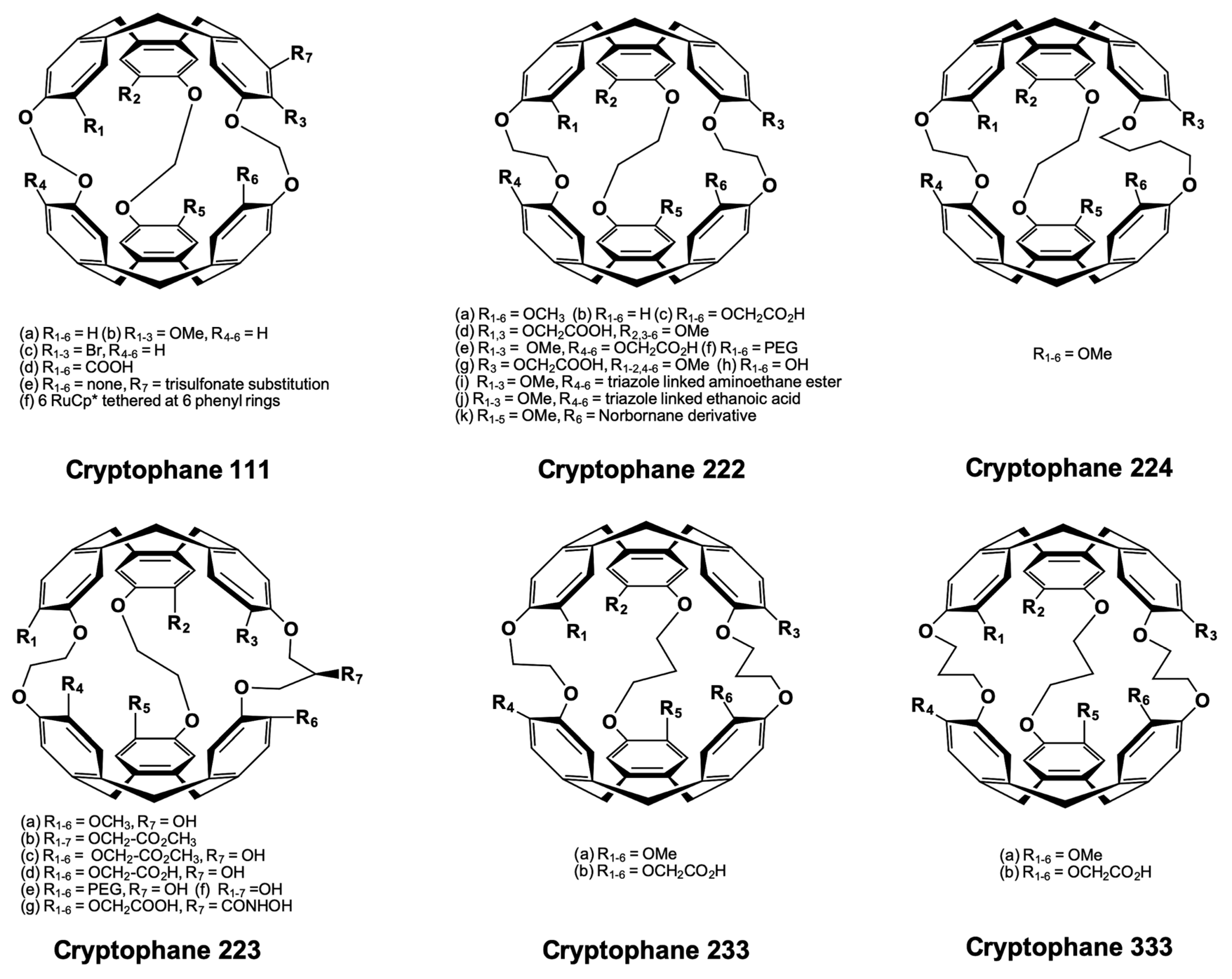
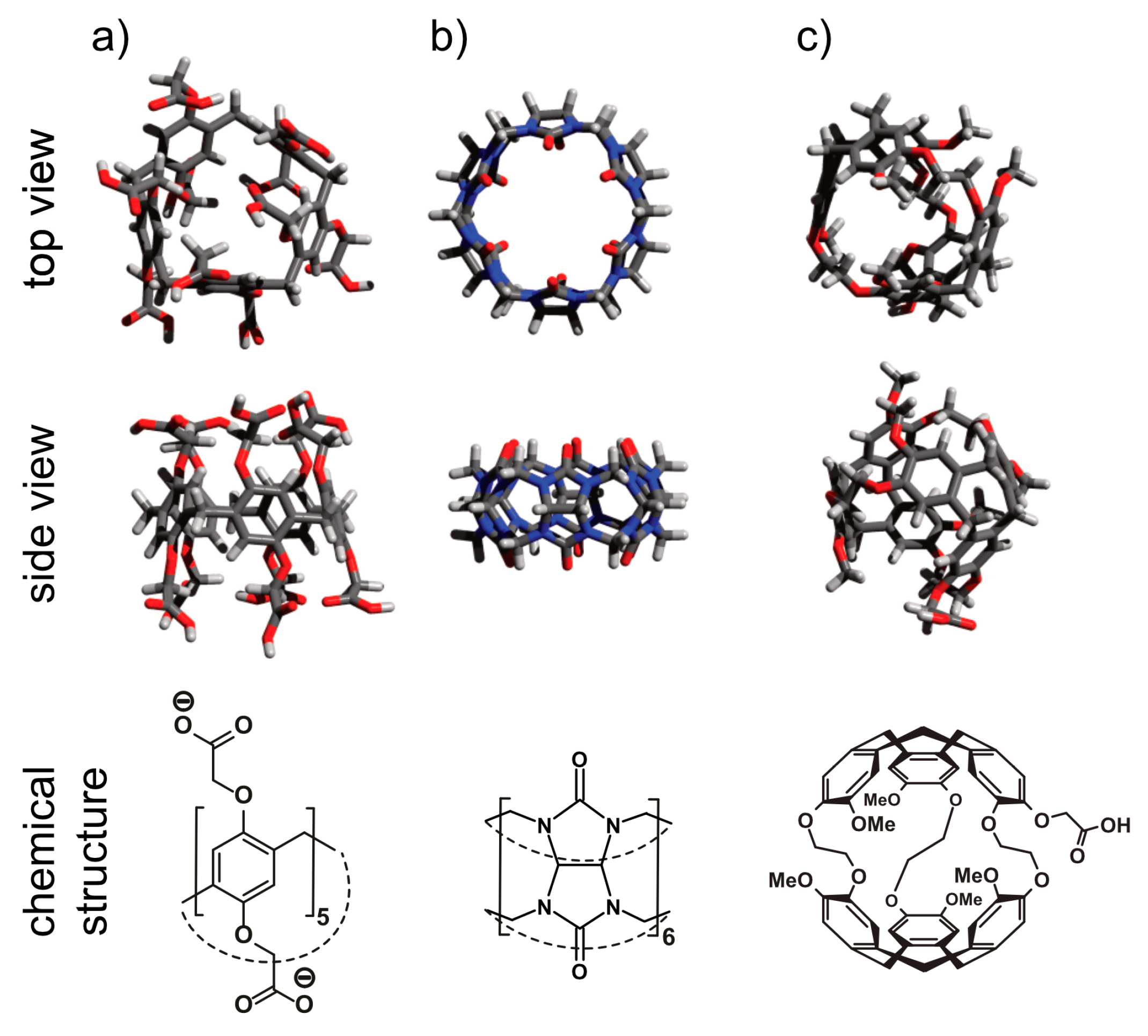
5.1. Cryptophanes as Xe Hosts
5.2. Peptide Chemistry
5.2.1. Avidin-Biotin Systems
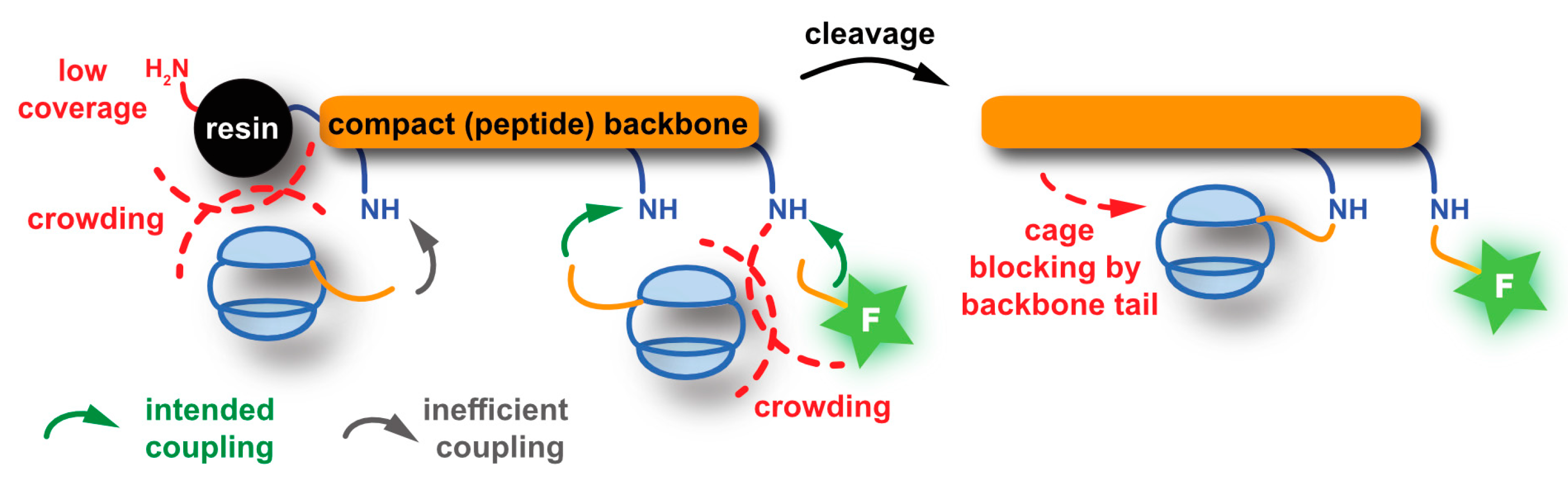
5.2.2. SPPS-Based Approaches
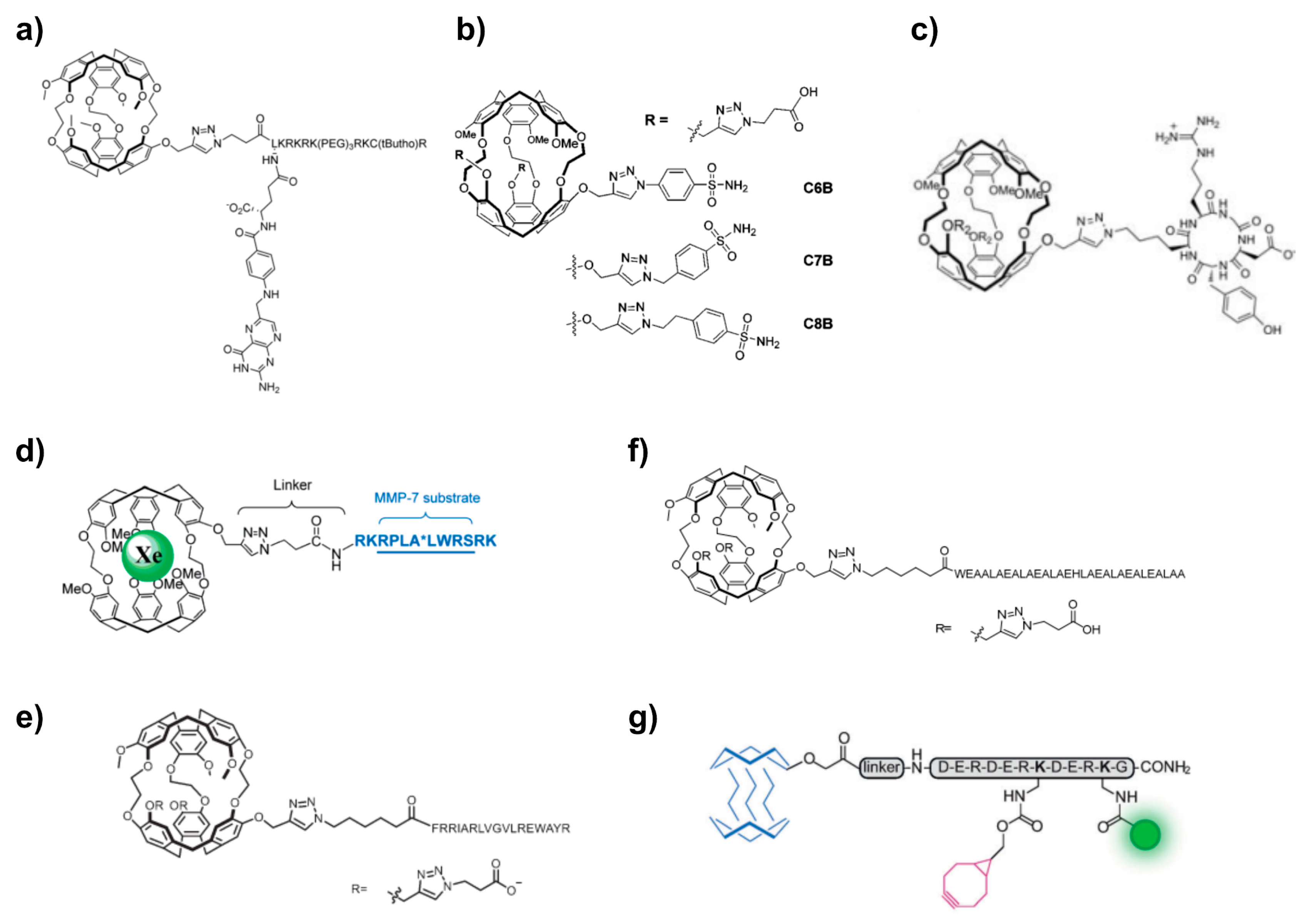
5.3. Click Chemistry Approaches
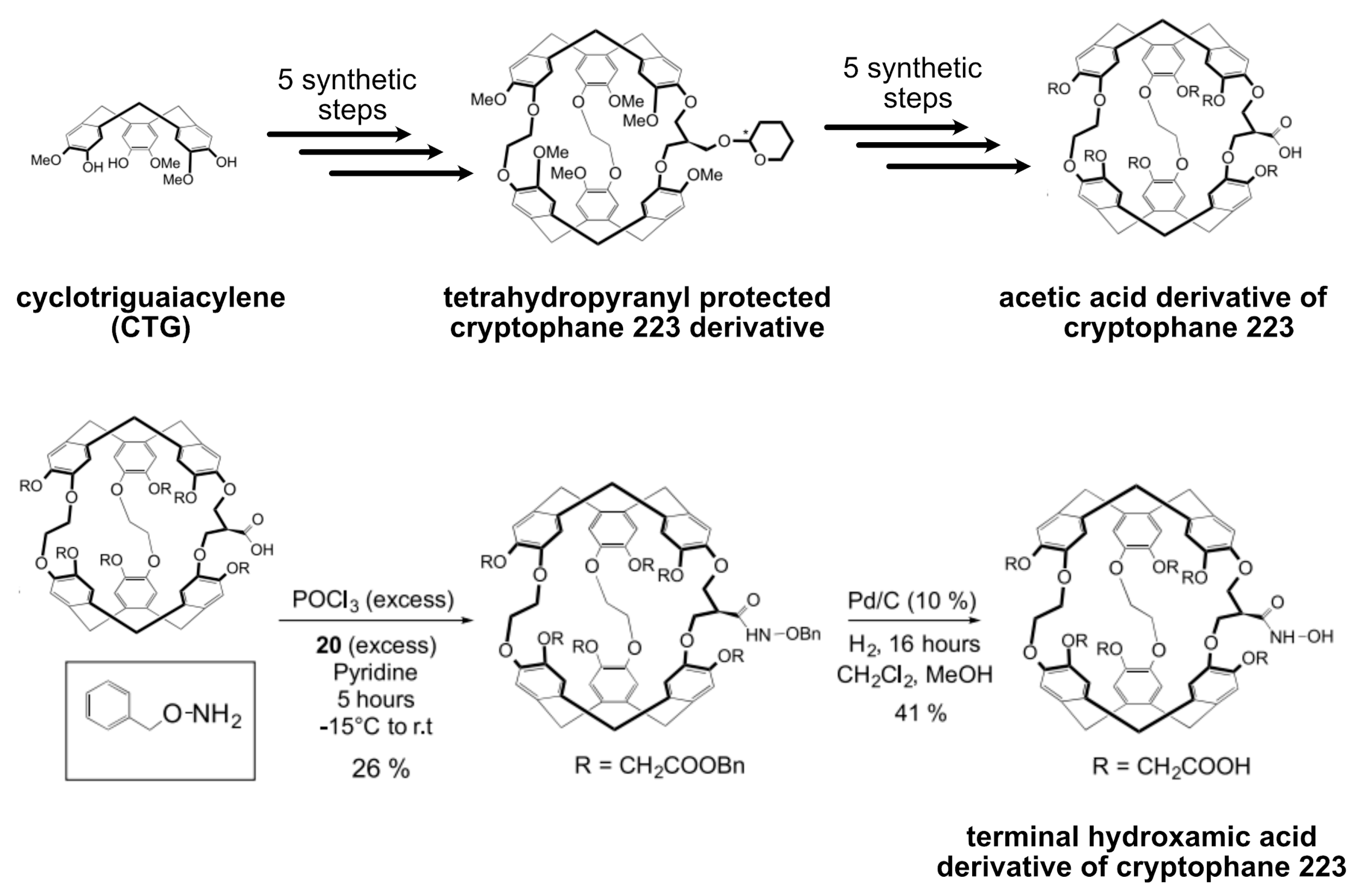
5.4. Xe Biosensor Generation via Single Arm Functionalization of Cryptophanes-223
5.5. Multiplexing Approaches Involving Cryptophanes
5.6. Fluorophore Coupling for Dual Readout
5.7. Cryptophane-Based Theranostic Probes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host | Pros | Cons |
|---|---|---|
| Cryptophanes |
|
|
| Cucurbit[n]urils (CB[n]s) |
|
|
| Pillar[n]arenes |
|
|
| Metal organic polyhedral (MOP) |
|
|
| Modular hosts (micelles & liposomes) |
|
|
| Biogenic scaffolds (viral capsids, bacteriophages) |
|
|
| Gas binding proteins |
|
|
| Superhosts (Nanoemulsions, gas vesicles) |
|
|
5.8. Xe Binding to Other Synthetic Hosts: Different Exchange Kinetics
5.8.1. Cucurbit[n]urils-CB[n]
5.8.2. Pillar[n]arenes
5.8.3. Metal-Organic Polyhedra—MOPs
| Xe Hosts | Solvent and Binding Guests | Chemical Shift (ppm) | Binding Constant (M−1) | Xe HyperCEST Effect (ppm) and HyperCEST MRI |
|---|---|---|---|---|
| Cryptophanes | ||||
| Cryptophane 111 | Xe in (CDCl2)2 | ~31 | 28,000 (278 K) | |
| (1). [(Cp*Ru)6(Cry111)]Cl6 | D2O | 308 (293 K) | ||
| Cryptophane 222 (CryA) | CH2Cl2 | 475 (298 K) | ||
| CHCl3 | 230 (298 K) | |||
| CH4 | 130 (298 K) | |||
| Xe in (CHCl2)2 | 3000 | |||
| (1). hexahydroxy substituted (MM)-CryA derivatives | Cs+ in H2O | 6 × 109 | ||
| (2). hexahydroxy substituted (PP)-CryA derivatives | Cs+ in H2O | 2 × 109 | ||
| (3). triacetic acid functionalized CryA derivative | Xe in H2O | 33,000 ± 2000 | ||
| (4). Trifunctionalized CryA | Xe in H2O | 4.2 × 104 | ||
| (5). CryA | Xe in (CDCl2)2 | ~62 (278 K) | 3.95 × 103 (278 K) | |
| (6). hexaacid functionalized CryA derivatives | Xe in D2O | 64 (293 K) | 6800 (293 K) | |
| (7). PEG-modified CryA | Xe in H2O | 77 | ||
| (8). CryA-linker-biotin (B1) | B1 (300 μM) and ~80 nmol avidin | 193 (Xe@aq), 70 (Xe@CryA), 2.3 ppm downfield to bound Xe (avidin-bound B1) | ||
| (9). cysteine-maleimide by a lysine in B1 (B2) | B2 (140 µM) and 29 µM avidin | RL and LL diastereomeric peaks at 65.1 and 64.5 ppm shifted to 67.9 and 66.4 ppm | ||
| (10). B2 with rigid linker (B3) | B3 (77 μM) and 20 μM avidin | four peaks with large line widths (45–55 Hz) | ||
| (11). B2 with short linker (B4) | increased both line width and chemical shift sensitivity | |||
| (12). Biosensor B3 | 250 μL avidin-agarose beads | 65.4 (Xe@B3), 193.6 (Xe@agarose), 192.5 (Xe@aq) | ||
| (13). CryA-based CD14 biosensor | cells | ~120 ppm only for RAW264.7 cells | ||
| (14). CryA-based claudin biosensor | cells | 59% CEST effect for transfected HEK cells compared to 11% in non-transfected HEKs. Xe@CryA in lipidic/cellular environment (~120 ppm) | ||
| (15). CryA-dicarboxylic acid derivative based biosensor | H2O + 1% DMSO | −131.2 ppm | ||
| (16). CryA-monocarboxylic acid | H2O + 1% DMSO | −132.4 ppm | ||
| (17). CryA-based MMP7 biosensor | D2O | 61.8 (RL) and 62.4 (LL) diastereomers. Two groups of two peaks separated by 0.6 and 0.8 ppm observed for mix of uncleaved and cleaved MMP7 | ||
| (18). CryA-based folate biosensor | acetate buffer (pH 5) | 64.8 (LRL) and 66.0 (LRR) diastereomers | ||
| (19). Trisubstituted CryA derivatives (subst. = tripropargyl, triallyl, tribenzyl, trihydroxy) | 63–65 ppm, 57 (trihydroxy substituted CryA) | |||
| (20). CryA-based pH biosensor | solution and cells | 64.2 (pH 7.5), 67.6 (pH 5.5) at room temperature. In HeLa cells: 78.4 ppm (pH 5.5), 65 ppm (pH 7.5) | ||
| (21). CryA-based integrin biosensor | Tris buffer (pH 7.2) | 65.8, two resonances at 67.1 (free CryA) and 71.2 (bound CryA) on treating αIIbβ3 (16 μM) with biosensor (50 μM). | ||
| (22). CryA-based carbonic anhydrase biosensor (triazole linkers with 6-, 7- and 8-bonds to benzenesulfonamide forms C6B, C7B and C8B ligands) | H2O | bound Xe peaks for C6B, C7B and C8B are at 63.5, 63.9 and 62.9, respectively. | ||
| (23). Tripropargyl CryA-based Calmodulin biosensor | buffer | biosensor + eq.molar CaM (halo form) showed two peaks for bound Xe (65.9, 67.6). | ||
| (24). PEGylated tripropargylated CryA derivative | H2O (20 mM solubility) | bound Xe at 77.4 ppm | ||
| (25). Zn2+ chelating hexacarboxylic acid CryA derivative | Sensor (260 μM) in PBS buffer | 65.6 (bound Xe), Zn2+ addition splits bound Xe peak into 65.75 and 67.2. | ||
| (26). (+)-MM-cryptophane-(L)-NTA | Sensor (970 μM) in HEPES buffer | 5% Zn2+, 5% Cd2+, 5% Pb2+ indicated downfield shift of bound Xe i.e., 1.5 (Zn2+), 0.3 (Cd2+), 4.5 (Pb2+) | ||
| (27). CryA-based porphyrin sensor | solution and cells | CEST peak at 72 ppm (between pH 3 and 5). pH increase from 5.1 until 9.3 showed another peak at 70 ppm. A549 cells incubation with sensor indicated CEST peak at 74 ppm (pH 5.1 and 7.4). | ||
| Cryptophane 223 (Cry223) | Xe in (CDCl2)2 | 52 | 2810 (278 K) | |
| (1). hexaacid derivative of Cry223 | Xe in D2O | 42 | 2200 | |
| (2). Cry223 (R11−6 = CH3, R2 = OH) | 64 | |||
| (3). Cry223 (R11−6 = R2 = CH2CO2CH3) | (CDCl2)2 | 82 | ||
| (4). Cry223 (R11−6 = CH2CO2CH3, R2 = OH) | 58 | |||
| (5). Cry223 (R11−6 = CH2CO2H, R2 = OH) | 50 | |||
| (6). Cry223 (R1 = R2 = OH) | 63 | |||
| (7). Cry223 (R11−6 = PEG, R2 = OH) | 67 | |||
| (8). Cry223 (R11−6 = CH2COOH, R2 = CONHOH) | 58.4 (pH 9.9), 59.4 (pH 8.2), 61.3 (pH 3.3) | |||
| Cryptophane 333 | solution | 35 | ||
| Cucurbit[n]urils | ||||
| Cucurbit[6]uril (CB[6]) | ||||
| (1). CB[6]-Na+ coordination | THF encapsulation | 5.1 ± 0.5 × 102 at room temperature | ||
| (2). CB[6]-Cs+ coordination | THF encapsulation | 1.1 ± 0.1 × 103 at room temperature | ||
| (3). CB[6] | H2O, acidic or saline solution | 122 ± 0.5 (bound Xe signal), 199 ± 0.5 (free Xe signal) | ||
| (4). Cy6CB[6] (Cy = cyclohexane) | H2O | 93 (bound Xe peak), T1 ~40 s | 1.3 ± 0.1 × 103 | |
| (5). CB[6]-based rotaxanes | solution | rotaxane (100 μM): no Xe@CB[6] signal. Base-catalyzed hydrolysis led to semi-rotaxanes and appearance of Xe@CB[6] signal. | ||
| (6). CB[6]-based MMP2 sensor | rotaxane (5 μM), MMP2 (5 nM) | cleavage of rotaxane led to CB[6] release from former’s axle (15% CEST effect) | ||
| (7). CB[6]-based H2O2 sensor | rotaxane (25 μM), H2O2 (50 μM) | cleavage of rotaxane led to an apparent maximum saturation of about 25% | ||
| Cucurbit[7]uril (CB[7]) | ||||
| (1). CB[7]-based biotin biosensor | −68 ppm Xe@btCB[7] similar to free CB[7]. Avidin to btCB[7] (50 μM) led to four peaks at −68 ppm (Xe@ibtCB[7]), 0 ppm (Xeaq), 100 ppm (Xe@avidin) and −40 ppm (Xe@btCB[7]-avidin). CB[7] interaction with avidin estimated to generate 50% CEST difference. | |||
| Pillar[n]arenes | ||||
| Pillar[5]arenes | cationic viologen salt | 8 ± 2 × 104 | ||
| (1). water soluble pillar[5]arenes decorated with carboxylate groups | amino acids e.g., L-arginine, L-lysine, L-histidine | ca. 103 | ||
| (2). pillar[5]arenes with 10 trimethyl ammonium groups | sodium alkyl sulfonates e.g., 1-octane sulfonate | 1.3 ± 0.9 × 104 | ||
| (3). Pillar[5]arene | hexane as thread | more than 75 ppm observed compared to external reference (Xe solubilized in CDCl3) 10 ppm downfield for Xe compared to Xe@pillar[5]arene. | ||
| Metal organic polyhedra (MOP) | ||||
| Fe-MOP | Fe-MOP (8.9 mM), Xe (20 mM) in D2O | Xe@Fe-MOP appeared at 16.6 ppm downfield than free Xe in D2O | 16 | Xe exchange rate in Fe-MOP is 10 Hz. |
| Supercage | ||||
| Cry 111 inside FeII4L4 MOP | Cs+ binding to Cry⊂MOP indicated bound resonance (316), free resonance (32, 133Cs NMR) | MOP for Cry binding constant is 9.5 ± 0.4 × 106 Cs+ binding to Cry⊂MOP revealed 34 ± 3 | Cry⊂MOP showed 179 ppm (free Xe), 47 ppm ([Xe@Cry]⊂MOP]) and an exchange rate of 11 ± 2 Hz. | |
| Liposomes | ||||
| (1). CryA-dicarboxylic acid derivative (CryA-da) | CryA-da (15 ± 5) μM in lipid emulsions | 62 (Xe@cage), 189 (Xeaq) Increasing lipid conc. between 1 and 5 % led to the appearance of two resonances in aq. solution and third one at 73 ppm. Chemical shift difference between Xe signals in aqueous and lipid environments is larger inside CryA-da (~10 ppm) than unbound Xe (~1 ppm). | ||
| (2). Targeted liposomes decorated with lipopeptide | cells | HBMEC cells (target cells) + liposomes resulted in two peaks at 71 ppm (Xe@CryA@cells) and 62 ppm (Xe@CryA@aq) compared to reduced signal in control HAoEC cells. 129Xe HyperCEST intensity of (68 ± 4)% in HBMECs compared to (23 ± 5)% for control cells | ||
| (3). CryA-monocarboxylic acid (CryA-ma) in liposomes | CryA-ma (5 μM) + POPC or DPPC (200 μM) at 300 K | 64 ppm (Xe@CryA-ma@aq), 74 ppm (Xe@CryA-ma@lipid) | ||
| (4). POPC/cholesterol model membranes | CryA-ma loading into liposomes | 70 ppm (Xe@CryA-ma@aq), ~77 ppm (Xe@CryA-ma@lipid) | ||
| Biogenic scaffolds | ||||
| MS2 viral capsids | CryA (1 μM): 70% modification of MS2 proteins i.e., ~125 copies of CryA per capsid. | 190 ppm (Xeaq), 60 ppm (bound Xe peak) | ||
| CryA, fluorophore installed at capsid interior (~110 CryA per capsid) and exterior decorated with DNA aptamer TD05.1 | scaffold (167 nM in capsids) incubated with Ramos (positive cells) showed a broad Xe@CryA peak at 57 ppm and 50% higher image contrast compared to control Jurkats. | |||
| M13 bacteriophage | p8 proteins modified with PEG5k (approx. 28% i.e., 760 copies) and CryA (approx. 39% i.e., 1050 copies) | sensor (2.3 nM, 293 K) revealed peaks at 192 ppm (Xeaq), 61.8 ppm (Xe@CryA@M13), 59.4 ppm (Xe@CryA). At 310 K led to changes in the observed peaks i.e., 192.4 ppm (Xeaq), bound Xe 64.6 ppm (Xe@CryA@M13), 62.2 ppm (Xe@CryA). | ||
| fd bacteriophage (EGFR targeted biosensor) | p8 proteins modified by 6–8% (~330 cages per phage) | Target cells (MDA-MB-231) incubated with fd-CryA sensor (0.7 nM) exhibited a bound peak at 70 ppm and 16.0 ± 9.4% contrast compared to control Jurkats. | ||
| Gas binding protein structures | ||||
| (1). TEM-1-beta-lactamase (bla, 29 kDa) | 40 | CEST of recombinant bla (80 μM) showed two peaks at 195 ppm (Xeaq) and 255 ppm (Xe@bla) | ||
| (2). Maltose binding protein (MBP) | HyperCEST response for bound Xe appeared at 95 ppm (Xe@MBP) after treating MBP (80 μM) with 1 mM maltose. | |||
| Superhosts | ||||
| Perfluorooctyl bromide (PFOB) nanoemulsions | PFOB (130 nm) | Two CEST peaks found at 111 ± 9 ppm (Xe@PFOB) and ~192 ppm (Xeaq) | ||
| Targeted PFOB | 106 ppm (PFOB-dissolved Xe) if particle size larger than 250 nm and conc. of 5 pM. Target cells (A549) incubation with RGD-tagged PFOB led to CEST peak at 106 ppm and 54% HyperCEST effect compared to no uptake by control cells (WI-38). | |||
| Gas vesicles (GVs) | 145 nm diameter, 250–1000 nm length | CEST peak at 31.2 ppm (Xe@GVs), 195 ppm (Xeaq) and saturation contrast of about (33 ± 2)%. GVs engineered from Halobacteria sp. NRC-1, Microcystis SP and E. Coli exhibited following CEST responses at 14.4, 30.6 and 51.4 ppm. |
5.9. Nanoscopic Formulations as Multivalent Xe Hosts
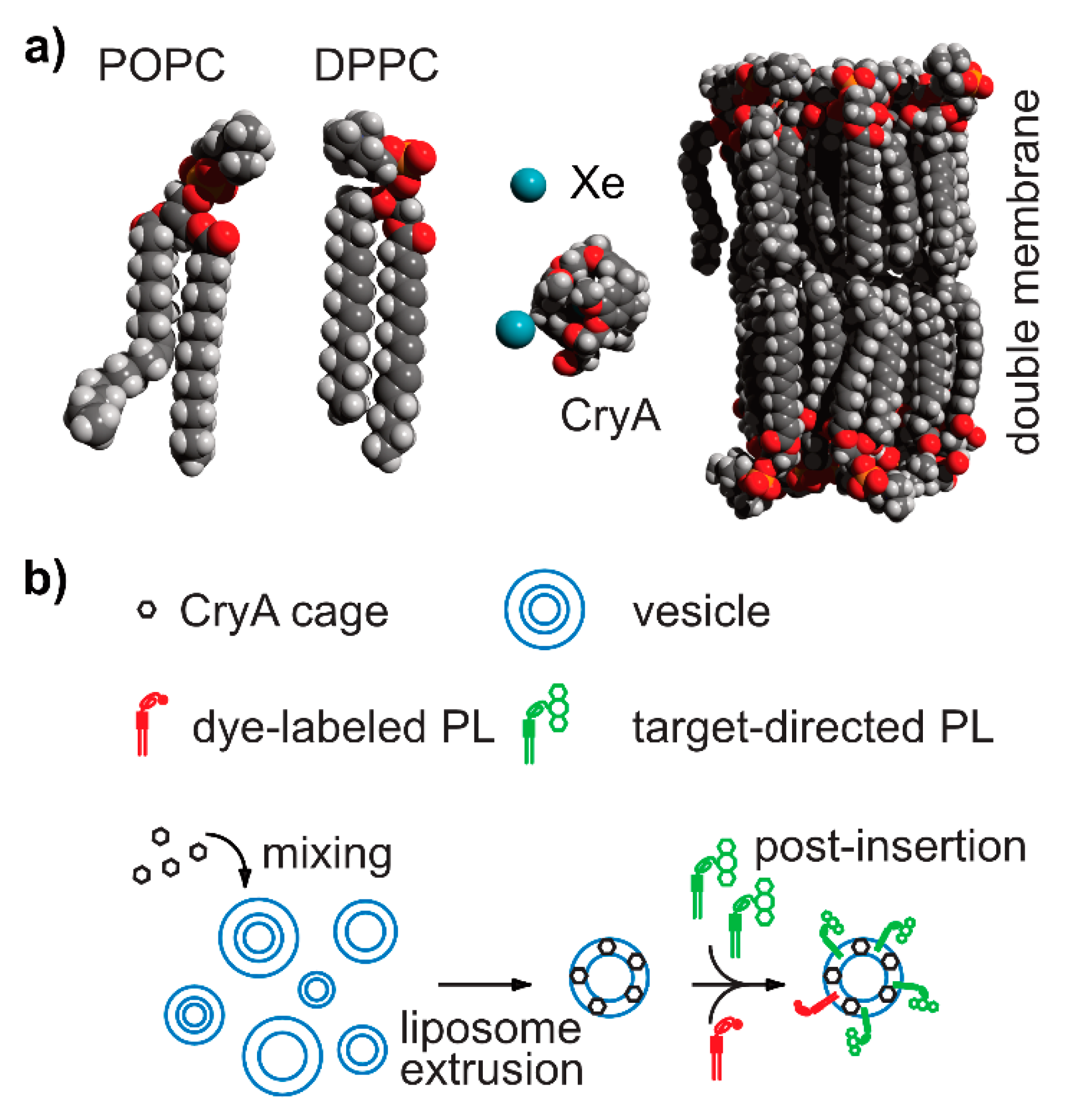
5.9.1. Liposomes and Micelles as Modular Platform
5.9.2. Biomembrane Fluidity Studies
5.10. Biogenic Scaffolds and Gas-Binding Proteins
5.10.1. Capsid-Based Scaffolds
5.10.2. Bacteriophage-Based Scaffolds
5.10.3. Gas-Binding Protein Structures as Xe Hosts
5.11. Super Hosts
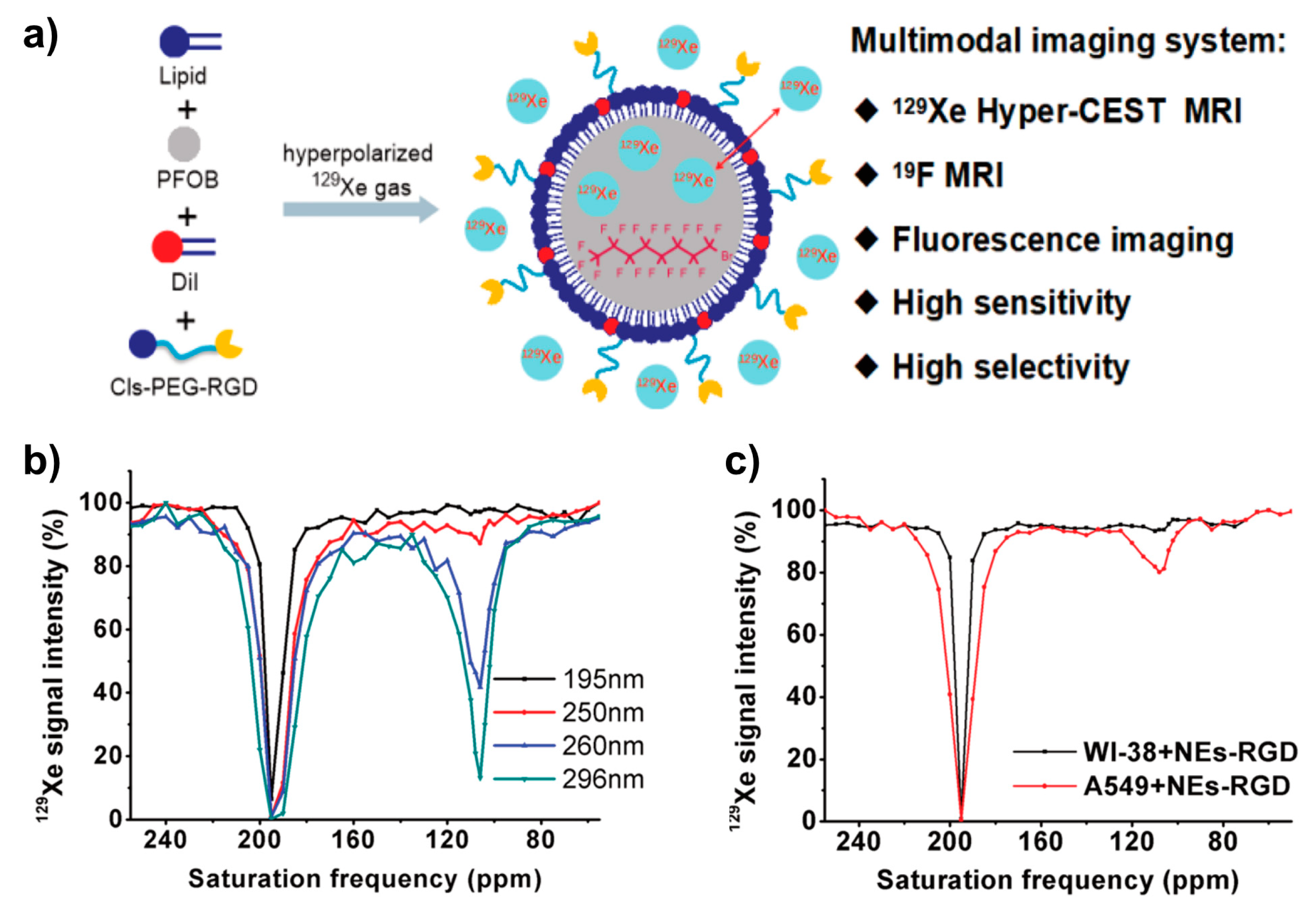
5.11.1. Nanodroplets/Nanoemulsions
5.11.2. Bacterial Gas Vesicle
- creating theranostic GVs for both US-imaging and photodynamic therapy (PDT) agent addressing human breast carcinoma (MCF-7) [451];
- obtaining a genetically engineered variant of Ana GvpC-containing N- or C-terminal hexahistidine sequences in E. coli for protein fusion with lysine-rich protein (LRP). The latter introduces 100 positive charges that modulate the GVs behavior in solution and in vivo [452];
- expressing GvpCRGD on their surface leading to specific targeting of integrin-overexpressing U87 human glioblastoma cell line in vitro compared to no uptake in the case of scrambled GvpCRDG, respectively [453];
- uptake by phagocytic cells through polycationic polyarginine (R8) peptide fusion [454];
- a modular approach by fusing GvpC with SpyTag (ST; a 13-residue peptide that forms a covalent amide bond with a partner SpyCatcher protein under physiological conditions [455] for demonstrating such the capability of such GVs (SpyTag-SpyCatcher) as multimodal probes (e.g., acoustic and fluorescence imaging) [442].
6. Quantitative Analysis of 129Xe Host Systems
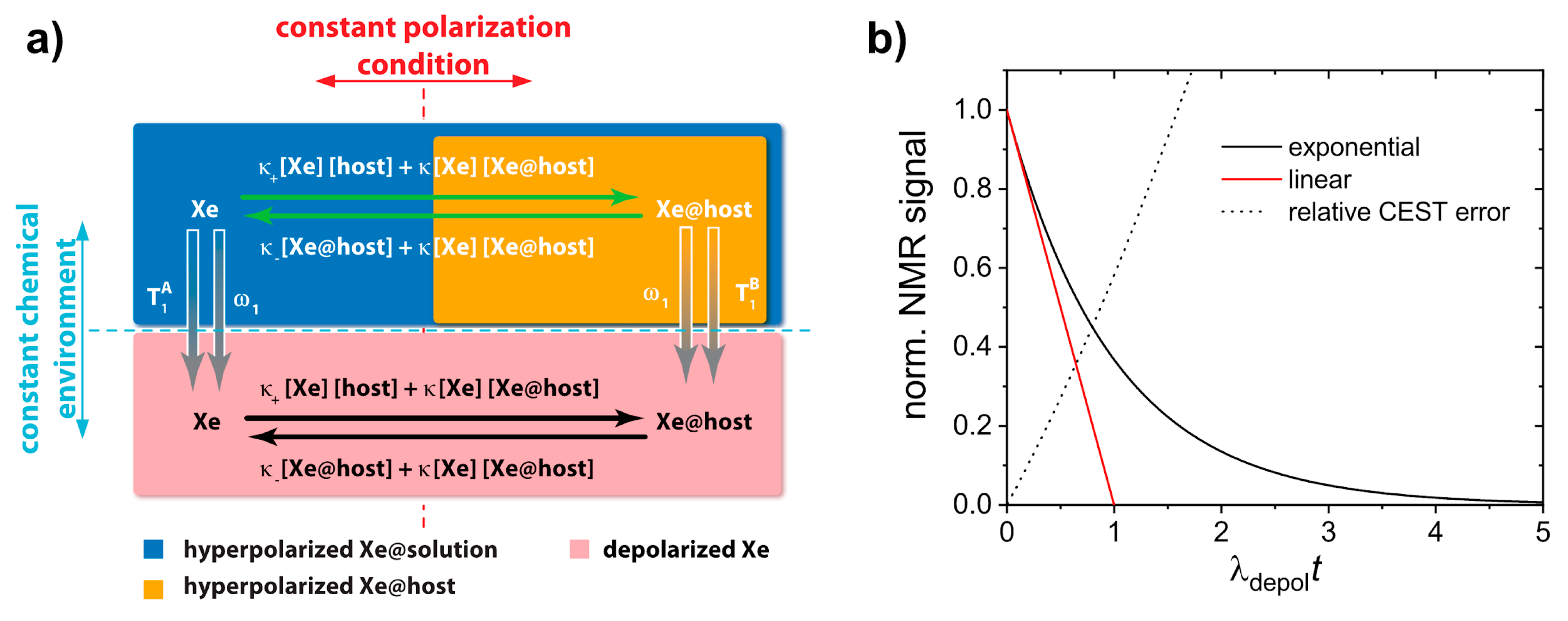
6.1. Quantitative hp Xe Saturation Transfer Analysis (qHyperCEST) for Host Characterization
6.2. Quantifying HyperCEST Changes in Displacement Assays
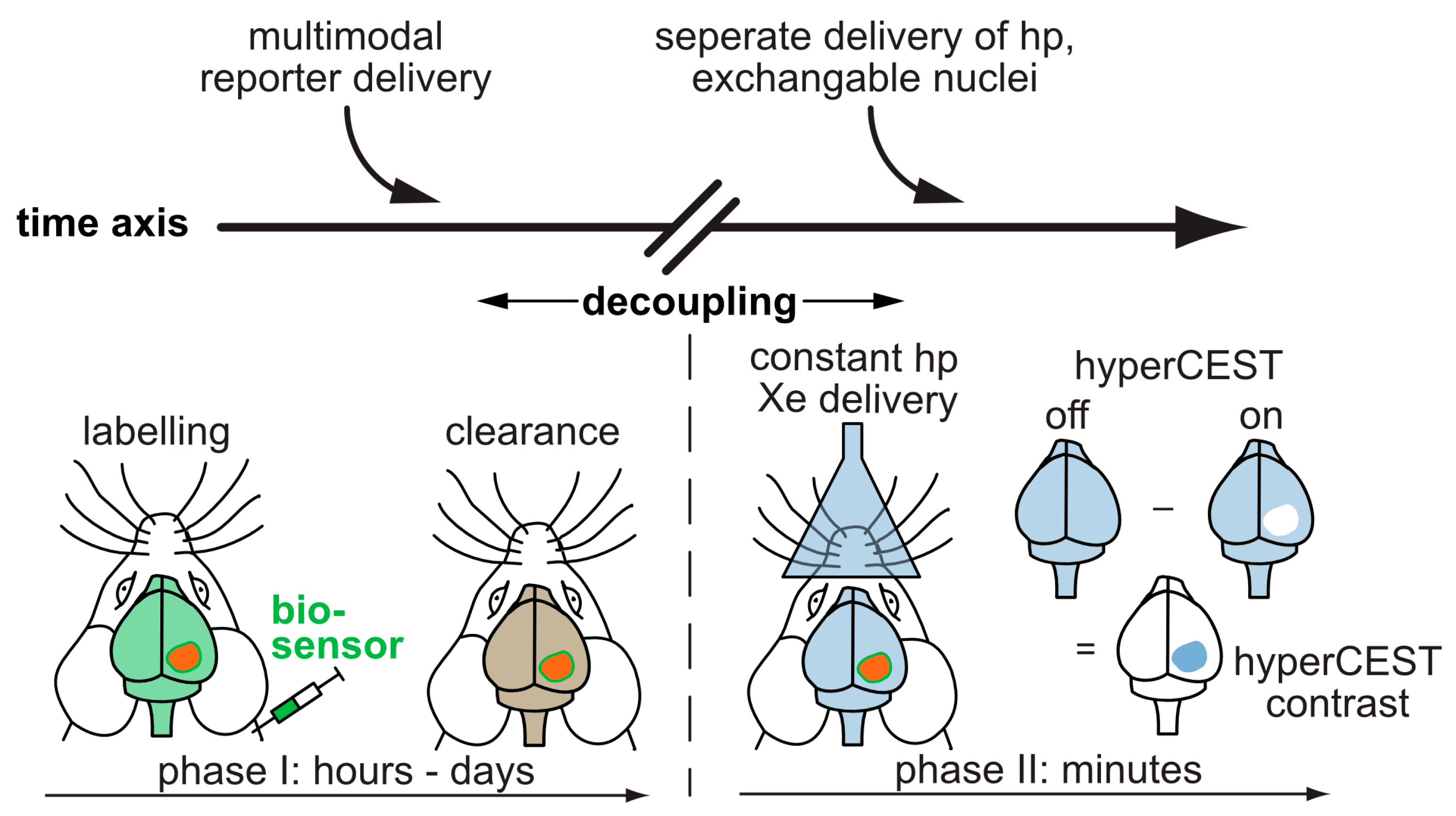
7. Translation Potential
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Kurhanewicz, J.; Vigneron, D.B.; Ardenkjaer-Larsen, J.H.; Bankson, J.A.; Brindle, K.; Cunningham, C.H.; Gallagher, F.A.; Keshari, K.R.; Kjaer, A.; Laustsen, C.; et al. Hyperpolarized C MRI: Path to Clinical Translation in Oncology. Neoplasia 2019, 21, 1–16. [Google Scholar] [CrossRef]
- Adamson, E.B.; Ludwig, K.D.; Mummy, D.G.; Fain, S.B. Magnetic resonance imaging with hyperpolarized agents: Methods and applications. Phys. Med. Biol. 2017, 62, R81–R123. [Google Scholar] [CrossRef]
- Ruiz-Cabello, J.; Barnett, B.P.; Bottomley, P.A.; Bulte, J.W.M. Fluorine 19F MRS and MRI in biomedicine. NMR Biomed. 2011, 24, 114–129. [Google Scholar] [CrossRef]
- Kurhanewicz, J.; Vigneron, D.B.; Brindle, K.; Chekmenev, E.Y.; Comment, A.; Cunningham, C.H.; DeBerardinis, R.J.; Green, G.G.; Leach, M.O.; Rajan, S.S.; et al. Analysis of Cancer Metabolism by Imaging Hyperpolarized Nuclei: Prospects for Translation to Clinical Research. Neoplasia 2011, 13, 81–97. [Google Scholar] [CrossRef]
- Witte, C.; Schröder, L. NMR of hyperpolarised probes. NMR Biomed. 2013, 26, 788–802. [Google Scholar] [CrossRef]
- Ball, D.R.; Cruickshank, R.; Carr, C.A.; Stuckey, D.J.; Lee, P.; Clarke, K.; Tyler, D.J. Metabolic imaging of acute and chronic infarction in the perfused rat heart using hyperpolarised [1-13C]pyruvate. NMR Biomed. 2013, 26, 1441–1450. [Google Scholar] [CrossRef]
- Atherton, H.J.; Schroeder, M.A.; Dodd, M.S.; Heather, L.C.; Carter, E.E.; Cochlin, L.E.; Nagel, S.; Sibson, N.R.; Radda, G.K.; Clarke, K.; et al. Validation of the in vivo assessment of pyruvate dehydrogenase activity using hyperpolarised 13C MRS. NMR Biomed. 2011, 24, 201–208. [Google Scholar] [CrossRef]
- Rider, O.J.; Tyler, D.J. Clinical Implications of Cardiac Hyperpolarized Magnetic Resonance Imaging. J. Cardiovasc. Magn. Reson. 2013, 15, 556. [Google Scholar] [CrossRef]
- Walkup, L.L.; Woods, J.C. Translational applications of hyperpolarized 3He and 129Xe. NMR Biomed. 2014, 27, 1429–1438. [Google Scholar] [CrossRef]
- Tilton, R.F., Jr.; Kuntz, I.D., Jr. Nuclear magnetic resonance studies of xenon-129 with myoglobin and hemoglobin. Biochemistry 1982, 21, 6850–6857. [Google Scholar] [CrossRef]
- Hollenbach, J.; Anger, B.; Matysik, J. Chapter 9. Probing Exchange and Diffusion in Confined Systems by 129Xe NMR Spectroscopy. In New Developments in NMR; Royal Society of Chemistry: Cambridge, UK, 2016; pp. 294–317. [Google Scholar]
- Goodson, B.M. Nuclear magnetic resonance of laser-polarized noble gases in molecules, materials, and organisms. J. Magn. Reson. 2002, 155, 157–216. [Google Scholar] [CrossRef]
- Schröder, L. Xenon for NMR biosensing-inert but alert. Phys. Med. 2013, 29, 3–16. [Google Scholar] [CrossRef]
- Klippel, S.; Freund, C.; Schröder, L. Multichannel MRI labeling of mammalian cells by switchable nanocarriers for hyperpolarized xenon. Nano Lett. 2014, 14, 5721–5726. [Google Scholar] [CrossRef]
- Rose, H.M.; Witte, C.; Rossella, F.; Klippel, S.; Freund, C.; Schröder, L. Development of an antibody-based, modular biosensor for 129Xe NMR molecular imaging of cells at nanomolar concentrations. Proc. Natl. Acad. Sci. USA 2014, 111, 11697–11702. [Google Scholar] [CrossRef]
- Spence, M.M.; Rubin, S.M.; Dimitrov, I.E.; Ruiz, E.J.; Wemmer, D.E.; Pines, A.; Yao, S.Q.; Tian, F.; Schultz, P.G. Functionalized xenon as a biosensor. Proc. Natl. Acad. Sci. USA 2001, 98, 10654–10657. [Google Scholar] [CrossRef]
- Schroder, L.; Lowery, T.J.; Hilty, C.; Wemmer, D.E.; Pines, A. Molecular Imaging Using a Targeted Magnetic Resonance Hyperpolarized Biosensor. Science 2006, 314, 446–449. [Google Scholar] [CrossRef]
- Colloc’h, N.; Sopkova-de Oliveira Santos, J.; Retailleau, P.; Vivarès, D.; Bonneté, F.; Langlois d’Estainto, B.; Gallois, B.; Brisson, A.; Risso, J.-J.; Lemaire, M.; et al. Protein crystallography under xenon and nitrous oxide pressure: Comparison with in vivo pharmacology studies and implications for the mechanism of inhaled anesthetic action. Biophys. J. 2007, 92, 217–224. [Google Scholar] [CrossRef]
- Cohen, J.; Arkhipov, A.; Braun, R.; Schulten, K. Imaging the migration pathways for O2, CO, NO, and Xe inside myoglobin. Biophys. J. 2006, 91, 1844–1857. [Google Scholar] [CrossRef]
- Johnson, B.J.; Cohen, J.; Welford, R.W.; Pearson, A.R.; Schulten, K.; Klinman, J.P.; Wilmot, C.M. Exploring molecular oxygen pathways in Hansenula polymorpha copper-containing amine oxidase. J. Biol. Chem. 2007, 282, 17767–17776. [Google Scholar] [CrossRef]
- Miller, K.W.; Reo, N.V.; Schoot Uiterkamp, A.J.; Stengle, D.P.; Stengle, T.R.; Williamson, K.L. Xenon NMR: Chemical shifts of a general anesthetic in common solvents, proteins, and membranes. Proc. Natl. Acad. Sci. USA 1981, 78, 4946–4949. [Google Scholar] [CrossRef]
- Tilton, R.F., Jr.; Kuntz, I.D., Jr.; Petsko, G.A. Cavities in proteins: Structure of a metmyoglobin-xenon complex solved to 1.9 A. Biochemistry 1984, 23, 2849–2857. [Google Scholar] [CrossRef]
- Bowers, C.R.; Storhaug, V.; Webster, C.E.; Bharatam, J.; Cottone, A.; Gianna, R.; Betsey, A.K.; Gaffney, B.J. Exploring Surfaces and Cavities in Lipoxygenase and Other Proteins by Hyperpolarized Xenon-129 NMR. J. Am. Chem. Soc. 1999, 121, 9370–9377. [Google Scholar] [CrossRef][Green Version]
- Mura, A.; Anedda, R.; Pintus, F.; Casu, M.; Padiglia, A.; Floris, G.; Medda, R. An important lysine residue in copper/quinone-containing amine oxidases. FEBS J. 2007, 274, 2585–2595. [Google Scholar] [CrossRef]
- Anderson, M.A.; Xu, Y.; Grissom, C.B. Electron spin catalysis by xenon in an enzyme. J. Am. Chem. Soc. 2001, 123, 6720–6721. [Google Scholar] [CrossRef]
- Anedda, R.; Era, B.; Casu, M.; Fais, A.; Ceccarelli, M.; Corda, M.; Ruggerone, P. Evidences of xenon-induced structural changes in the active site of cyano-metmyoglobins: A 1H NMR study. J. Phys. Chem. B 2008, 112, 15856–15866. [Google Scholar] [CrossRef]
- Gröger, C.; Möglich, A.; Pons, M.; Koch, B.; Hengstenberg, W.; Kalbitzer, H.R.; Brunner, E. NMR-Spectroscopic Mapping of an Engineered Cavity in the I14A Mutant of HPr fromStaphylococcuscarnosusUsing Xenon. J. Am. Chem. Soc. 2003, 125, 8726–8727. [Google Scholar] [CrossRef]
- Shulman, R.G.; Peisach, J.; Wyluda, B.J. Effects of cyclopropane and xenon upon the high-resolution nuclear magnetic resonance spectrum of ferrimyoglobin cyanide. J. Mol. Biol. 1970, 48, 517–523. [Google Scholar] [CrossRef]
- Mayer, A.; Ogawa, S.; Shulman, R.G.; Yamane, T.; Cavaleiro, J.A.; Rocha Gonsalves, A.M.; Kenner, G.W.; Smith, K.M. Assignments of the paramagnetically shifted heme methyl nuclear magnetic resonance peaks of cyanometmyoglobin by selective deuteration. J. Mol. Biol. 1974, 86, 749–756. [Google Scholar] [CrossRef]
- Stengle, T.R.; Hosseini, S.M.; Basiri, H.G.; Williamson, K.L. NMR chemical shifts of xenon in aqueous solutions of amphiphiles: A new probe of the hydrophobic environment. J. Solut. Chem. 1984, 13, 779–787. [Google Scholar] [CrossRef]
- Locci, E.; Dehouck, Y.; Casu, M.; Saba, G.; Lai, A.; Luhmer, M.; Reisse, J.; Bartik, K. Probing Proteins in Solution by 129Xe NMR Spectroscopy. J. Magn. Reson. 2001, 150, 167–174. [Google Scholar] [CrossRef]
- Moudrakovski, I.; Ratcliffe, C.I.; Ripmeester, J.A. 129Xe NMR studies of the dynamics of xenon in siliceous ZSM-12 zeolite. Appl. Magn. Reson. 1996, 10, 559–574. [Google Scholar] [CrossRef]
- Jameson, C.J.; Jameson, A.K.; Gerald, R.E.; Lim, H.-M. Anisotropic Xe Chemical Shifts in Zeolites. The Role of Intra- and Intercrystallite Diffusion. J. Phys. Chem. B 1997, 101, 8418–8437. [Google Scholar] [CrossRef]
- Chen, Q.; Springuel-Huet, M.; Fraissard, J. 129Xe NMR of xenon adsorbed on the molecular sieves AlPO4-5, SAPO-5, MAPO-5, and SAPO-37. Chem. Phys. Lett. 1989, 159, 117–121. [Google Scholar] [CrossRef]
- Ratcliffe, C.I.; Ripmeester, J.A. 129Xe NMR: Dynamic Behavior of Xenon in NaY Zeolite at 77 K. J. Am. Chem. Soc. 1995, 117, 1445–1446. [Google Scholar] [CrossRef]
- Suzuki, T.; Miyauchi, M.; Yoshimizu, H.; Tsujita, Y. Characterization of Microvoids in Glassy Polymers by Means of 129Xe NMR Spectroscopy. Polym. J. 2001, 33, 934–938. [Google Scholar] [CrossRef]
- Wang, Y.; Inglefield, P.T.; Jones, A.A. Gas sorption environments in poly (2,6-dimethyl-1,4-phenylene oxide) by xenon-129 nuclear magnetic resonance: Effects of processing. J. Polym. Sci. Part B Polym. Phys. 2002, 40, 1965–1974. [Google Scholar] [CrossRef]
- Golemme, G.; Nagy, J.; Fonseca, A.; Algieri, C.; Yampolskii, Y. 129Xe-NMR study of free volume in amorphous perfluorinated polymers: Comparsion with other methods. Polymer 2003, 44, 5039–5045. [Google Scholar] [CrossRef]
- Branda, N.; Grotzfeld, R.M.; Valdes, C.; Rebek, J.J.; Valdés, C. Control of Self-Assembly and Reversible Encapsulation of Xenon in a Self-Assembling Dimer by Acid-Base Chemistry. J. Am. Chem. Soc. 1995, 117, 85–88. [Google Scholar] [CrossRef]
- Sozzani, P.; Comotti, A.; Simonutti, R.; Meersmann, T.; Logan, J.W.; Pines, A. A Porous Crystalline Molecular Solid Explored by Hyperpolarized Xenon. Angew. Chem. Int. Ed. 2000, 39, 2695–2699. [Google Scholar] [CrossRef]
- Hoffmann, H.C.; Debowski, M.; Müller, P.; Paasch, S.; Senkovska, I.; Kaskel, S.; Brunner, E. Solid-State NMR Spectroscopy of Metal–Organic Framework Compounds (MOFs). Materials 2012, 5, 2537–2572. [Google Scholar] [CrossRef]
- Klein, N.; Herzog, C.; Sabo, M.; Senkovska, I.; Getzschmann, J.; Paasch, S.; Lohe, M.R.; Brunner, E.; Kaskel, S. Monitoring adsorption-induced switching by 129Xe NMR spectroscopy in a new metal–organic framework Ni2(2,6-ndc)2(dabco). Phys. Chem. Chem. Phys. 2010, 12, 11778–11784. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Kim, D.; Hopson, R.; Sailor, M.J.; Wang, L.-Q. Investigation of grafted mesoporous silicon sponge using hyperpolarized 129Xe NMR spectroscopy. J. Mater. Res. 2018, 33, 2637–2645. [Google Scholar] [CrossRef]
- Pawsey, S.; Moudrakovski, I.; Ripmeester, J.; Wang, L.-Q.; Exarhos, G.J.; Rowsell, J.L.C.; Yaghi, O.M. Hyperpolarized 129Xe Nuclear Magnetic Resonance Studies of Isoreticular Metal-Organic Frameworks. J. Phys. Chem. C 2007, 111, 6060–6067. [Google Scholar] [CrossRef]
- Böhlmann, W.; Pöppl, A.; Sabo, M.; Kaskel, S. Characterization of the Metal−Organic Framework Compound Cu3(benzene 1,3,5-tricarboxylate)2by Means of129Xe Nuclear Magnetic and Electron Paramagnetic Resonance Spectroscopy. J. Phys. Chem. B 2006, 110, 20177–20181. [Google Scholar] [CrossRef]
- Contributors to Wikimedia Projects Xenon. Available online: https://en.wikipedia.org/wiki/Xenon (accessed on 24 September 2020).
- Mecozzi, S.; Rebek, J. The 55 % Solution: A Formula for Molecular Recognition in the Liquid State. Chem. A Eur. J. 1998, 4, 1016–1022. [Google Scholar] [CrossRef]
- Garel, L.; Dutasta, J.-P. Complexation of Methane and Chlorofluorocarbons by Cryptophane-A in Organic Solution. Angew. Chem. Int. Ed. 1993, 32, 1169–1171. [Google Scholar] [CrossRef]
- Berthault, P.; Huber, J.G.; Desvaux, H. Biosensing using laser-polarized xenon NMR/MRI. Prog. Nucl. Magn. Reson. Spectrosc. 2009, 55, 35–60. [Google Scholar] [CrossRef]
- Barskiy, D.A.; Coffey, A.M.; Nikolaou, P.; Mikhaylov, D.M.; Goodson, B.M.; Branca, R.T.; Lu, G.J.; Shapiro, M.G.; Telkki, V.-V.A.; Zhivonitko, V.V.; et al. Frontispiece: NMR Hyperpolarization Techniques of Gases. Chem. A Eur. J. 2017, 23, 725–751. [Google Scholar] [CrossRef]
- Krjukov, E.V.; O’Neill, J.D.; Owers-Bradley, J. Brute Force Polarization of 129Xe. J. Low Temp. Phys. 2005, 140, 397–408. [Google Scholar] [CrossRef]
- Halse, M.E. Perspectives for hyperpolarisation in compact NMR. TrAC Trends Anal. Chem. 2016, 83, 76–83. [Google Scholar] [CrossRef]
- Hirsch, M.L.; Kalechofsky, N.; Belzer, A.; Rosay, M.; Kempf, J.G. Brute-Force Hyperpolarization for NMR and MRI. J. Am. Chem. Soc. 2015, 137, 8428–8434. [Google Scholar] [CrossRef]
- Walker, T.G.; Happer, W. Spin-exchange optical pumping of noble-gas nuclei. Rev. Mod. Phys. 1997, 69, 629–642. [Google Scholar] [CrossRef]
- Zeng, X.; Wu, Z.; Call, T.; Miron, E.; Schreiber, D.; Happer, W. Experimental determination of the rate constants for spin exchange between optically pumped K, Rb, and Cs atoms andXe129nuclei in alkali-metal–noble-gas van der Waals molecules. Phys. Rev. A 1985, 31, 260–278. [Google Scholar] [CrossRef]
- Nikolaou, P.; Coffey, A.M.; Walkup, L.L.; Gust, B.M.; Whiting, N.; Newton, H.; Barcus, S.; Muradyan, I.; Dabaghyan, M.; Moroz, G.D.; et al. Near-unity nuclear polarization with an open-source 129Xe hyperpolarizer for NMR and MRI. Proc. Natl. Acad. Sci. USA 2013, 110, 14150–14155. [Google Scholar] [CrossRef]
- Han, S.; Garcia, S.; Lowery, T.J.; Ruiz, E.J.; Seeley, J.A.; Chavez, L.; King, D.S.; Wemmer, D.E.; Pines, A. NMR-Based Biosensing with Optimized Delivery of Polarized129Xe to Solutions. Anal. Chem. 2005, 77, 4008–4012. [Google Scholar] [CrossRef]
- Witte, C.; Kunth, M.; Rossella, F.; Schröder, L. Observing and preventing rubidium runaway in a direct-infusion xenon-spin hyperpolarizer optimized for high-resolution hyper-CEST (chemical exchange saturation transfer using hyperpolarized nuclei) NMR. J. Chem. Phys. 2014, 140, 084203. [Google Scholar] [CrossRef]
- Lakshmanan, A.; Lu, G.J.; Farhadi, A.; Nety, S.P.; Kunth, M.; Lee-Gosselin, A.; Maresca, D.; Bourdeau, R.W.; Yin, M.; Yan, J.; et al. Preparation of biogenic gas vesicle nanostructures for use as contrast agents for ultrasound and MRI. Nat. Protoc. 2017, 12, 2050–2080. [Google Scholar] [CrossRef]
- Mazzanti, M.L.; Walvick, R.P.; Zhou, X.; Sun, Y.; Shah, N.; Mansour, J.; Gereige, J.; Albert, M.S. Distribution of hyperpolarized xenon in the brain following sensory stimulation: Preliminary MRI findings. PLoS ONE 2011, 6, e21607. [Google Scholar] [CrossRef]
- Norquay, G.; Leung, G.; Stewart, N.J.; Tozer, G.M.; Wolber, J.; Wild, J.M. Relaxation and exchange dynamics of hyperpolarized 129Xe in human blood. Magn. Reson. Med. 2015, 74, 303–311. [Google Scholar] [CrossRef]
- Zhou, X.; Mazzanti, M.L.; Chen, J.J.; Tzeng, Y.-S.; Mansour, J.K.; Gereige, J.D.; Venkatesh, A.K.; Sun, Y.; Mulkern, R.V.; Albert, M.S. Reinvestigating hyperpolarized 129Xe longitudinal relaxation time in the rat brain with noise considerations. NMR Biomed. 2008, 21, 217–225. [Google Scholar] [CrossRef]
- von Morze, C.; Merritt, M.E. Cancer in the crosshairs: Targeting cancer metabolism with hyperpolarized carbon-13 MRI technology. NMR Biomed. 2018, e3937. [Google Scholar]
- Gabellieri, C.; Reynolds, S.; Lavie, A.; Payne, G.S.; Leach, M.O.; Eykyn, T.R. Therapeutic target metabolism observed using hyperpolarized 15N choline. J. Am. Chem. Soc. 2008, 130, 4598–4599. [Google Scholar] [CrossRef] [PubMed]
- Lumata, L.; Ratnakar, S.J.; Jindal, A.; Merritt, M.; Comment, A.; Malloy, C.; Sherry, A.D.; Kovacs, Z. BDPA: An efficient polarizing agent for fast dissolution dynamic nuclear polarization NMR spectroscopy. Chemistry 2011, 17, 10825–10827. [Google Scholar] [CrossRef]
- Jiang, W.; Lumata, L.; Chen, W.; Zhang, S.; Kovacs, Z.; Sherry, A.D.; Khemtong, C. Hyperpolarized 15N-pyridine derivatives as pH-sensitive MRI agents. Sci. Rep. 2015, 5, 9104. [Google Scholar] [CrossRef] [PubMed]
- Goodson, B.M.; Song, Y.-Q.; Taylor, R.E.; Schepkin, V.D.; Brennan, K.M.; Chingas, G.C.; Budinger, T.F.; Navon, G.; Pines, A. In vivo NMR and MRI using injection delivery of laser-polarized xenon. Proc. Natl. Acad. Sci. USA 1997, 94, 14725–14729. [Google Scholar] [CrossRef]
- Cross, A.R.; McPhee, D.; Stevens, D.; McDonald, M.; Santyr, G.E. Hyperpolarized xenon relaxation times in perfluorocarbon emulsion and plasma mixtures. In Proceedings of the 22nd Annual International Conference of the IEEE Engineering in Medicine and Biology Society (Cat. No.00CH37143), Chicago, IL, USA, 23–28 July 2000; Volume 3, pp. 2171–2172. [Google Scholar]
- Rao, M.R.; Stewart, N.J.; Griffiths, P.D.; Norquay, G.; Wild, J.M. Imaging Human Brain Perfusion with Inhaled Hyperpolarized Xe MR Imaging. Radiology 2018, 286, 659–665. [Google Scholar] [CrossRef]
- Chacon-Caldera, J.; Maunder, A.; Rao, M.; Norquay, G.; Rodgers, O.I.; Clemence, M.; Puddu, C.; Schad, L.R.; Wild, J.M. Dissolved hyperpolarized xenon-129 MRI in human kidneys. Magn. Reson. Med. 2019. [Google Scholar] [CrossRef]
- Anger, B.C.; Schrank, G.; Schoeck, A.; Butler, K.A.; Solum, M.S.; Pugmire, R.J.; Saam, B. Gas-phase spin relaxation of 129Xe. Phys. Rev. A 2008, 78, 191. [Google Scholar] [CrossRef]
- Nelson, S.J.; Kurhanewicz, J.; Vigneron, D.B.; Larson, P.E.Z.; Harzstark, A.L.; Ferrone, M.; van Criekinge, M.; Chang, J.W.; Bok, R.; Park, I.; et al. Metabolic imaging of patients with prostate cancer using hyperpolarized [1-13C]pyruvate. Sci. Transl. Med. 2013, 5, 198ra108. [Google Scholar] [CrossRef]
- Bifone, A.; Song, Y.-Q.; Seydoux, R.; Taylor, R.E.; Goodson, B.M.; Pietrass, T.; Budinger, T.F.; Navon, G.; Pines, A. NMR of laser-polarized xenon in human blood. Proc. Natl. Acad. Sci. USA 1996, 93, 12932–12936. [Google Scholar] [CrossRef]
- Witte, C.; Kunth, M.; Döpfert, J.; Rossella, F.; Schröder, L. Hyperpolarized xenon for NMR and MRI applications. J. Vis. Exp. 2012. [Google Scholar] [CrossRef] [PubMed]
- Amor, N.; Zänker, P.P.; Blümler, P.; Meise, F.M.; Schreiber, L.M.; Scholz, A.; Schmiedeskamp, J.; Spiess, H.W.; Münnemann, K. Magnetic resonance imaging of dissolved hyperpolarized 129Xe using a membrane-based continuous flow system. J. Magn. Reson. 2009, 201, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Kunth, M.; Witte, C.; Schröder, L. Quantitative chemical exchange saturation transfer with hyperpolarized nuclei (qHyper-CEST): Sensing xenon-host exchange dynamics and binding affinities by NMR. J. Chem. Phys. 2014, 141, 194202. [Google Scholar] [CrossRef]
- Miao, Y.-F.; Peng, T.; Moody, M.R.; Klegerman, M.E.; Aronowski, J.; Grotta, J.; McPherson, D.D.; Kim, H.; Huang, S.-L. Delivery of xenon-containing echogenic liposomes inhibits early brain injury following subarachnoid hemorrhage. Sci. Rep. 2018, 8, 450. [Google Scholar] [CrossRef]
- Bartik, K.; Luhmer, M.; Heyes, S.J.; Ottinger, R.; Reisse, J. Probing Molecular Cavities in α-Cyclodextrin Solutions by Xenon NMR. J. Magn. Reson. B 1995, 109, 164–168. [Google Scholar] [CrossRef]
- Xu, Y.; Tang, P. Amphiphilic sites for general anesthetic action? Evidence from 129Xe-{1H} intermolecular nuclear Overhauser effects. Biochim. Biophys. Acta BBA Biomembr. 1997, 1323, 154–162. [Google Scholar] [CrossRef][Green Version]
- Navon, G.; Song, Y.-Q.; Rõõm, T.; Appelt, S.; Taylor, R.E.; Pines, A. Enhancement of Solution NMR and MRI with Laser-Polarized Xenon. Science 1996, 271, 1848–1851. [Google Scholar] [CrossRef]
- Song, Y.-Q.; Goodson, B.M.; Taylor, R.E.; Laws, D.D.; Navon, G.; Pines, A. Selective Enhancement of NMR Signals forα-Cyclodextrin with Laser-Polarized Xenon. Angew. Chem. Int. Ed. 1997, 36, 2368–2370. [Google Scholar] [CrossRef]
- Fitzgerald, R.; Sauer, K.; Happer, W. Cross-relaxation in laser-polarized liquid xenon. Chem. Phys. Lett. 1998, 284, 87–92. [Google Scholar] [CrossRef]
- Raftery, D.; MacNamara, E.; Fisher, G.; Rice, C.V.; Smith, J. Optical Pumping and Magic Angle Spinning: Sensitivity and Resolution Enhancement for Surface NMR Obtained with Laser-Polarized Xenon. J. Am. Chem. Soc. 1997, 119, 8746–8747. [Google Scholar] [CrossRef]
- Haake, M.; Pines, A.; Reimer, J.A.; Seydoux, R. Surface-Enhanced NMR Using Continuous-Flow Laser-Polarized Xenon. J. Am. Chem. Soc. 1997, 119, 11711–11712. [Google Scholar] [CrossRef]
- Brunner, E.; Seydoux, R.; Haake, M.; Pines, A.; Reimer, J.A. Surface NMR Using Laser-Polarized129Xe under Magic Angle Spinning Conditions. J. Magn. Reson. 1998, 130, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Brunner, E.; Haake, M.; Pines, A.; Reimer, J.; Seydoux, R. Enhancement of 13C NMR signals in solid C60 and C70 using laser-polarized xenon. Chem. Phys. Lett. 1998, 290, 112–116. [Google Scholar] [CrossRef]
- Bartik, K.; Luhmer, M.; Dutasta, J.-P.; Collet, A.; Reisse, J. 129Xe and1H NMR Study of the Reversible Trapping of Xenon by Cryptophane-A in Organic Solution. J. Am. Chem. Soc. 1998, 120, 784–791. [Google Scholar] [CrossRef]
- Luhmer, M.; Goodson, B.M.; Song, Y.-Q.; Laws, D.D.; Kaiser, L.; Cyrier, M.C.; Pines, A. Study of Xenon Binding in Cryptophane-A Using Laser-Induced NMR Polarization Enhancement. J. Am. Chem. Soc. 1999, 121, 3502–3512. [Google Scholar] [CrossRef]
- Prangé, T.; Schiltz, M.; Pernot, L.; Colloc’h, N.; Longhi, S.; Bourguet, W.; Fourme, R. Exploring hydrophobic sites in proteins with xenon or krypton. Prot. Struct. Funct. Gen. 1998, 30, 61–73. [Google Scholar] [CrossRef]
- Quillin, M.L.; Breyer, W.A.; Griswold, I.J.; Matthews, B.W. Size versus polarizability in protein-ligand interactions: Binding of noble gases within engineered cavities in phage T4 lysozyme. J. Mol. Boil. 2000, 302, 955–977. [Google Scholar] [CrossRef]
- Otting, G.; Liepinsh, E.; Wüthrich, K. Protein hydration in aqueous solution. Science 1991, 254, 974–980. [Google Scholar] [CrossRef]
- Inoue, K.; Yamada, H.; Imoto, T.; Akasaka, K. High pressure NMR study of a small protein, gurmarin. J. Biomol. NMR 1998, 12, 535–541. [Google Scholar] [CrossRef]
- Desvaux, H.; Gautier, T.; Le Goff, G.; Petro, M.; Berthault, P. Direct evidence of a magnetization transfer between laser-polarized xenon and protons of a cage-molecule in water. Eur. Phys. J. D 2000, 12, 289–296. [Google Scholar] [CrossRef]
- Kader, J.-C. Lipid-transfer proteins in plants. Annu. Rev. Plant Boil. 1996, 47, 627–654. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, B.; Andersen, K.V.; Nielsen, P.R.; Bech, L.M.; Poulsen, F.M. Structure in solution of a four-helix lipid binding protein. Protein Sci. 1996, 5, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Min, K.; Cha, H.; Shin, D.H.; Hwang, K.Y.; Suh, S.W. Rice non-specific lipid transfer protein: The 1.6 a crystal structure in the unliganded state reveals a small hydrophobic cavity 1 1Edited by D. Rees. J. Mol. Boil. 1998, 276, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Poznanski, J.; Sodano, P.; Suh, S.W.; Lee, J.Y.; Ptak, M.; Vovelle, F. Solution structure of a lipid transfer protein extracted from rice seeds. Comparison with homologous proteins. JBIC J. Boil. Inorg. Chem. 1999, 259, 692–708. [Google Scholar]
- Gincel, E.; Simorre, J.-P.; Caille, A.; Marion, D.; Ptak, M.; Vovelle, F. Three-Dimensional Structure in Solution of a Wheat Lipid-Transfer Protein from Multidimensional 1H-NMR Data. A New Folding for Lipid Carriers. JBIC J. Boil. Inorg. Chem. 1994, 226, 413–422. [Google Scholar] [CrossRef]
- Tassin-Moindrot, S.; Caille, A.; Douliez, J.P.; Marion, D.; Vovelle, F. The wide binding properties of a wheat nonspecific lipid transfer protein. Solution structure of a complex with prostaglandin B2. JBIC J. Boil. Inorg. Chem. 2000, 267, 1117–1124. [Google Scholar]
- Charvolin, D.; Douliez, J.-P.; Marion, D.; Cohen-Addad, C.; Pebay-Peyroula, E. The crystal structure of a wheat nonspecific lipid transfer protein (ns-LTP1) complexed with two molecules of phospholipid at 2.1 A resolution. JBIC J. Boil. Inorg. Chem. 1999, 264, 562–568. [Google Scholar] [CrossRef]
- Lerche, M.H.; Poulsen, F.M. Solution structure of barley lipid transfer protein complexed with palmitate. Two different binding modes of palmitate in the homologous maize and barley nonspecific lipid transfer proteins. Protein Sci. 1998, 7, 2490–2498. [Google Scholar] [CrossRef]
- Sodano, P.; Caille, A.; Sy, D.; de Person, G.; Marion, D.; Ptak, M. 1 H NMR and fluorescence studies of the complexation of DMPG by wheat non-specific lipid transfer protein. Global fold of the complex. FEBS Lett. 1997, 416, 130–134. [Google Scholar] [CrossRef]
- Liepinsh, E.; Sodano, P.; Tassin, S.; Marion, D.; Vovelle, F.; Otting, G. Solvation study of the non-specific lipid transfer protein from wheat by intermolecular NOEs with water and small organic molecules. J. Biomol. NMR 1999, 15, 213–225. [Google Scholar] [CrossRef]
- Mulder, F.A.A.; Hon, B.; Muhandiram, D.R.; Dahlquist, F.W.; Kay, L.E. Flexibility and ligand exchange in a buried cavity mutant of T4 lysozyme studied by multinuclear NMR. Biochemistry 2000, 39, 12614–12622. [Google Scholar] [CrossRef]
- Landon, C.; Berthault, P.; Vovelle, F.; Desvaux, H. Magnetization transfer from laser-polarized xenon to protons located in the hydrophobic cavity of the wheat nonspecific lipid transfer protein. Protein Sci. 2001, 10, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Marco-Rius, I.; Bohndiek, S.E.; Kettunen, M.I.; Larkin, T.J.; Basharat, M.; Seeley, C.; Brindle, K.M. Quantitation of a spin polarization-induced nuclear Overhauser effect (SPINOE) between a hyperpolarized 13C-labeled cell metabolite and water protons. Contrast Media Mol. Imaging 2014, 9, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Dubois, L.; Berthault, P.; Huber, J.G.; Desvaux, H. Mapping hydrophobic molecular regions using dissolved laser-polarized xenon NMR. Comptes Rendus Phys. 2004, 5, 305–313. [Google Scholar] [CrossRef]
- Desvaux, H.; Huber, J.G.; Brotin, T.; Dutasta, J.-P.; Berthault, P. Magnetization Transfer from Laser-Polarized Xenon to Protons with Spin-Diffusion Quenching. ChemPhysChem 2003, 4, 384–387. [Google Scholar] [CrossRef]
- Garcia, S.; Chavez, L.; Lowery, T.J.; Han, S.-I.; Wemmer, D.E.; Pines, A. Sensitivity enhancement by exchange mediated magnetization transfer of the xenon biosensor signal. J. Magn. Reson. 2007, 184, 72–77. [Google Scholar] [CrossRef][Green Version]
- Forsén, S.; Hoffman, R.A. Study of Moderately Rapid Chemical Exchange Reactions by Means of Nuclear Magnetic Double Resonance. J. Chem. Phys. 1963, 39, 2892–2901. [Google Scholar] [CrossRef]
- Forsén, S.; Hoffman, R.A. Exchange Rates by Nuclear Magnetic Multiple Resonance. III. Exchange Reactions in Systems with Several Nonequivalent Sites. J. Chem. Phys. 1964, 40, 1189. [Google Scholar] [CrossRef]
- Wolff, S.D.; Balaban, R.S. NMR imaging of labile proton exchange. J. Magn. Reson. 1990, 86, 164–169. [Google Scholar] [CrossRef]
- Kunth, M.; Schröder, L. CEST MRI. In Quantification of Biophysical Parameters in Medical Imaging; Springer Science and Business Media LLC: Cham, Switzerland, 2017; pp. 213–253. [Google Scholar]
- Wu, B.; Warnock, G.; Zaiss, M.; Lin, C.-Y.; Chen, M.; Zhou, Z.; Mu, L.; Nanz, D.; Tuura, R.O.; Delso, G. An overview of CEST MRI for non-MR physicists. EJNMMI Phys. 2016, 3, 19. [Google Scholar] [CrossRef]
- Zaiss, M.; Xu, J.; Goerke, S.; Khan, I.S.; Singer, R.J.; Gore, J.C.; Gochberg, D.F.; Bachert, P. Inverse Z -spectrum analysis for spillover-, MT-, and T 1 -corrected steady-state pulsed CEST-MRI—Application to pH-weighted MRI of acute stroke: SIMPLE SPILLOVER-, MT-, AND T 1—CORRECTED CEST-MRI. NMR Biomed. 2014, 27, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Windschuh, J.; Zaiss, M.; Meissner, J.-E.; Paech, D.; Radbruch, A.; Ladd, M.E.; Bachert, P. Correction of B 1-inhomogeneities for relaxation-compensated CEST imaging at 7 T. NMR Biomed. 2015, 28, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Soesbe, T.C.; Wu, Y.; Sherry, A.D. Advantages of paramagnetic chemical exchange saturation transfer (CEST) complexes having slow to intermediate water exchange properties as responsive MRI agents. NMR Biomed. 2012, 26, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Tóth, É.; Bonnet, C.S. Responsive ParaCEST Contrast Agents. Inorganics 2019, 7, 68. [Google Scholar] [CrossRef]
- Sherry, A.D.; Woods, M. Chemical Exchange Saturation Transfer Contrast Agents for Magnetic Resonance Imaging. Annu. Rev. Biomed. Eng. 2008, 10, 391–411. [Google Scholar] [CrossRef]
- Ladd, M.E.; Bachert, P.; Meyerspeer, M.; Emoser, E.; Nagel, A.M.; Norris, D.G.; Schmitter, S.; Speck, O.; Straub, S.; Zaiss, M. Pros and cons of ultra-high-field MRI/MRS for human application. Prog. Nucl. Magn. Reson. Spectrosc. 2018, 109, 1–50. [Google Scholar] [CrossRef]
- Harel, E.; Schröder, L.; Xu, S. Novel Detection Schemes of Nuclear Magnetic Resonance and Magnetic Resonance Imaging: Applications from Analytical Chemistry to Molecular Sensors. Annu. Rev. Anal. Chem. 2008, 1, 133–163. [Google Scholar] [CrossRef]
- Kunth, M.; Witte, C.; Schröder, L. Continuous-wave saturation considerations for efficient xenon depolarization. NMR Biomed. 2015, 28, 601–606. [Google Scholar] [CrossRef]
- Kunth, M.; Witte, C.; Hennig, A.; Schröder, L. Identification, classification, and signal amplification capabilities of high-turnover gas binding hosts in ultra-sensitive NMR. Chem. Sci. 2015, 6, 6069–6075. [Google Scholar] [CrossRef]
- Schnurr, M.; Sloniec-Myszk, J.; Döpfert, J.; Schröder, L.; Hennig, A. Supramolecular Assays for Mapping Enzyme Activity by Displacement-Triggered Change in Hyperpolarized 129Xe Magnetization Transfer NMR Spectroscopy. Angew. Chem. Int. Ed. Engl. 2015, 54, 13444–13447. [Google Scholar] [CrossRef]
- Schnurr, M.; Joseph, R.; Naugolny-Keisar, A.; Kaizerman-Kane, D.; Bogdanoff, N.; Schuenke, P.; Cohen, Y.; Schröder, L. High Exchange Rate Complexes of Xe with Water-Soluble Pillar[5]arenes for Adjustable Magnetization Transfer MRI. ChemPhysChem 2019, 20, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Turati, L.; Moscatelli, M.; Mastropietro, A.; Dowell, N.G.; Zucca, I.; Erbetta, A.; Cordiglieri, C.; Brenna, G.; Bianchi, B.; Mantegazza, R.; et al. In vivo quantitative magnetization transfer imaging correlates with histology during de- and remyelination in cuprizone-treated mice. NMR Biomed. 2015, 28, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Seward, G.K.; Bai, Y.; Khan, N.S.; Dmochowski, I.J. Cell-compatible, integrin-targeted cryptophane-Xe NMR biosensors. Chem. Sci. 2011, 2, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Witte, C.; Martos, V.; Rose, H.M.; Reinke, S.; Klippel, S.; Schröder, L.; Hackenberger, C.P.R. Live-cell MRI with xenon hyper-CEST biosensors targeted to metabolically labeled cell-surface glycans. Angew. Chem. Int. Ed. Engl. 2015, 54, 2806–2810. [Google Scholar] [CrossRef] [PubMed]
- Boutin, C.; Stopin, A.; Lenda, F.; Brotin, T.; Dutasta, J.-P.; Jamin, N.; Sanson, A.; Boulard, Y.; Leteurtre, F.; Huber, G.; et al. Cell uptake of a biosensor detected by hyperpolarized 129Xe NMR: The transferrin case. Bioorg. Med. Chem. 2011, 19, 4135–4143. [Google Scholar] [CrossRef] [PubMed]
- Milanole, G.; Gao, B.; Paoletti, A.; Pieters, G.; Dugave, C.; Deutsch, E.; Rivera, S.; Law, F.; Perfettini, J.-L.; Mari, E.; et al. Bimodal fluorescence/129Xe NMR probe for molecular imaging and biological inhibition of EGFR in Non-Small Cell Lung Cancer. Bioorg. Med. Chem. 2017, 25, 6653–6660. [Google Scholar] [CrossRef] [PubMed]
- Schnurr, M.; Sydow, K.; Rose, H.M.; Dathe, M.; Schröder, L. Brain endothelial cell targeting via a peptide-functionalized liposomal carrier for xenon hyper-CEST MRI. Adv. Healthc. Mater. 2015, 4, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Farhadi, A.; Ho, G.; Kunth, M.; Ling, B.; Lakshmanan, A.; Lu, G.; Bourdeau, R.W.; Schröder, L.; Shapiro, M.G. Recombinantly Expressed Gas Vesicles as Nanoscale Contrast Agents for Ultrasound and Hyperpolarized MRI. AIChE J. 2018, 64, 2927–2933. [Google Scholar] [CrossRef]
- Kunth, M.; Lu, G.J.; Witte, C.; Shapiro, M.G.; Schröder, L. Protein Nanostructures Produce Self-Adjusting Hyperpolarized Magnetic Resonance Imaging Contrast through Physical Gas Partitioning. ACS Nano 2018, 12, 10939–10948. [Google Scholar] [CrossRef]
- Gabard, J. Synthesis of a (D 3)-bis(cyclotriveratrylenyl) macrocage by stereospecific replication of a (C 3)-subunit. J. Chem. Soc. Chem. Commun. 1981, 1137–1139. [Google Scholar] [CrossRef]
- Canceill, J.; Guilhem, J.; Pascard, C. A new bis-cyclotribenzyl cavitand capable of selective inclusion of neutral molecules in solution. Crystal structure of its CH2Cl2 cavitate. J. Chem. Soc. Chem. Commun. 1985, 361–363. [Google Scholar] [CrossRef]
- Canceill, J.; Lacombe, L.; Collet, A. A new cryptophane forming unusually stable inclusion complexes with neutral guests in a lipophilic solvent. J. Am. Chem. Soc. 1986, 108, 4230–4232. [Google Scholar] [CrossRef]
- Brotin, T.; Daugey, N.; Vanthuyne, N.; Jeanneau, E.; Ducasse, L.; Buffeteau, T. Chiroptical Properties of Cryptophane-223 and -233 Investigated by ECD, VCD, and ROA Spectroscopy. J. Phys. Chem. B 2015, 119, 8631–8639. [Google Scholar] [CrossRef] [PubMed]
- Collet, A.; Gabard, J. Optically active (C3)-cyclotriveratrylene-d9. Energy barrier for the “crown to crown” conformational interconversion of its nine-membered ring system. J. Org. Chem. 1980, 45, 5400–5401. [Google Scholar] [CrossRef]
- Zimmermann, H.; Tolstoy, P.M.; Limbach, H.H.; Poupko, R.; Luz, Z. The Saddle Form of Cyclotriveratrylene. J. Phys. Chem. B 2004, 108, 18772–18778. [Google Scholar] [CrossRef]
- Brotin, T.; Jeanneau, E.; Berthault, P.; Léonce, E.; Pitrat, D.; Mulatier, J.-C. Synthesis of Cryptophane-B: Crystal Structure and Study of Its Complex with Xenon. J. Org. Chem. 2018, 83, 14465–14471. [Google Scholar] [CrossRef]
- Canceill, J.; Guilhem, J.; Riche, C.; Pascard, C. Selective recognition of neutral molecules: 1H n.m.r. Study of the complexation of CH2Cl2 and CH2Br2 by cryptophane-D in solution and crystal structure of its CH2Cl2cavitate. J. Chem. Soc. Chem. Commun. 1986, 339–341. [Google Scholar] [CrossRef]
- Gabard, J.; Canceill, J.; Collet, A. New D3 and C3h cryptophanes with ethylenic bridges. Tetrahedron 1987, 43, 4531–4538. [Google Scholar] [CrossRef]
- Little, M.A.; Donkin, J.; Fisher, J.; Halcrow, M.A.; Loder, J.; Hardie, M.J. Synthesis and methane-binding properties of disulfide-linked cryptophane-0.0.0. Angew. Chem. Int. Ed. Engl. 2012, 51, 764–766. [Google Scholar] [CrossRef]
- Fogarty, H.A.; Berthault, P.; Brotin, T.; Huber, G.; Desvaux, H.; Dutasta, J.-P. A cryptophane core optimized for xenon encapsulation. J. Am. Chem. Soc. 2007, 129, 10332–10333. [Google Scholar] [CrossRef]
- Darzac, M.; Brotin, T.; Bouchu, D.; Dutasta, J.-P. Cryptophanols, new versatile compounds for the synthesis of functionalized cryptophanes and polycryptophanes. Chem. Commun. 2001, 48–49. [Google Scholar] [CrossRef]
- Darzac, M.; Brotin, T.; Rousset-Arzel, L.; Bouchu, D.; Dutasta, J.-P. Synthesis and application of cryptophanol hosts: 129Xe NMR spectroscopy of a deuterium-labeled (Xe)2@bis-cryptophane complex. New J. Chem. 2004, 28, 502–512. [Google Scholar] [CrossRef]
- Taratula, O.; Hill, P.A.; Bai, Y.; Khan, N.S.; Dmochowski, I.J. Shorter Synthesis of Trifunctionalized Cryptophane-A Derivatives. Org. Lett. 2011, 13, 1414–1417. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wei, Q.; Seward, G.K.; Hill, P.A.; Patton, B.; Dimitrov, I.E.; Kuzma, N.N.; Dmochowski, I.J. Designing129Xe NMR Biosensors for Matrix Metalloproteinase Detection. J. Am. Chem. Soc. 2006, 128, 13274–13283. [Google Scholar] [CrossRef]
- Hill, P.A.; Wei, Q.; Eckenhoff, R.G.; Dmochowski, I.J. Thermodynamics of Xenon Binding to Cryptophane in Water and Human Plasma. J. Am. Chem. Soc. 2007, 129, 9262–9263. [Google Scholar] [CrossRef]
- Delacour, L.; Kotera, N.; Traoré, T.; Puente, C.; Leteurtre, F.; Tassali, N.; Boutin, C.; Léonce, E.; Boulard, Y.; Garcia-Argote, S.; et al. “Clickable” Hydrosoluble PEGylated Cryptophane as a Universal Platform for 129Xe Magnetic Resonance Imaging Biosensors. Chemistry 2013, 19, 6089–6093. [Google Scholar] [CrossRef]
- Coker, H. Empirical free-ion polarizabilities of the alkali metal, alkaline earth metal, and halide ions. J. Phys. Chem. 1976, 80, 2078–2084. [Google Scholar] [CrossRef]
- Coker, H. Polarizability changes on ion hydration. J. Phys. Chem. 1976, 80, 2084–2091. [Google Scholar] [CrossRef]
- Brotin, T.; Montserret, R.; Bouchet, A.; Cavagnat, D.; Linares, M.; Buffeteau, T. High Affinity of Water-Soluble Cryptophanes for Cesium Cations. J. Org. Chem. 2012, 77, 1198–1201. [Google Scholar] [CrossRef]
- Hill, P.A.; Wei, Q.; Troxler, T.; Dmochowski, I.J. Substituent Effects on Xenon Binding Affinity and Solution Behavior of Water-Soluble Cryptophanes. J. Am. Chem. Soc. 2009, 131, 3069–3077. [Google Scholar] [CrossRef][Green Version]
- Jacobson, D.R.; Khan, N.S.; Collé, R.; Fitzgerald, R.; Laureano-Pérez, L.; Bai, Y.; Dmochowski, I.J. Measurement of radon and xenon binding to a cryptophane molecular host. Proc. Natl. Acad. Sci. USA 2011, 108, 10969–10973. [Google Scholar] [CrossRef] [PubMed]
- Bondi, A. van der Waals Volumes and Radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Taratula, O.; Hill, P.A.; Khan, N.S.; Carroll, P.J.; Dmochowski, I.J. Crystallographic observation of ‘induced fit’ in a cryptophane host–guest model system. Nat. Commun. 2010, 1, 148. [Google Scholar] [CrossRef] [PubMed]
- Huber, J.G.; Beguin, L.; Desvaux, H.; Brotin, T.; Fogarty, H.A.; Dutasta, J.-P.; Berthault, P. Cryptophane-Xenon Complexes in Organic Solvents Observed through NMR Spectroscopy. J. Phys. Chem. A 2008, 112, 11363–11372. [Google Scholar] [CrossRef]
- Drake, S.D. Capsules and Cavitands: Concave Molecules Built on the Cyclotribenzylene (CTB) Scaffold. Ph.D. Thesis, Georgetown University, Washington, DC, USA, 2011. [Google Scholar]
- Berthault, P.; Boutin, C.; Léonce, E.; Jeanneau, E.; Brotin, T. Role of the Methoxy Groups in Cryptophanes for Complexation of Xenon: Conformational Selection Evidence from 129 Xe-1 H NMR SPINOE Experiments. ChemPhysChem 2017, 18, 1561–1568. [Google Scholar] [CrossRef] [PubMed]
- Huber, J.G.; Brotin, T.; Dubois, L.; Desvaux, H.; Dutasta, J.-P.; Berthault, P. Water Soluble Cryptophanes Showing Unprecedented Affinity for Xenon: Candidates as NMR-Based Biosensors. J. Am. Chem. Soc. 2006, 128, 6239–6246. [Google Scholar] [CrossRef]
- Lowery, T.J.; Garcia, S.; Chavez, L.; Ruiz, E.J.; Wu, T.; Brotin, T.; Dutasta, J.-P.; King, D.S.; Schultz, P.G.; Pines, A.; et al. Optimization of Xenon Biosensors for Detection of Protein Interactions. ChemBioChem 2005, 7, 65–73. [Google Scholar] [CrossRef]
- Hilty, C.; Lowery, T.J.; Wemmer, D.E.; Pines, A. Spectrally Resolved Magnetic Resonance Imaging of a Xenon Biosensor. Angew. Chem. Int. Ed. 2006, 45, 70–73. [Google Scholar] [CrossRef]
- Bagno, A.; Saielli, G. Understanding the Extraordinary Deshielding of129Xe in a Permetallated Cryptophane by Relativistic DFT. Chemistry 2012, 18, 7341–7345. [Google Scholar] [CrossRef]
- Fairchild, R.M.; Joseph, A.I.; Holman, K.T.; Fogarty, H.A.; Brotin, T.; Dutasta, J.-P.; Boutin, C.; Huber, J.G.; Berthault, P. A Water-Soluble Xe@cryptophane-111 Complex Exhibits Very High Thermodynamic Stability and a Peculiar129Xe NMR Chemical Shift. J. Am. Chem. Soc. 2010, 132, 15505–15507. [Google Scholar] [CrossRef]
- Lerouge, F.; Melnyk, O.; Durand, J.-O.; Raehm, L.; Berthault, P.; Huber, J.G.; Desvaux, H.; Constantinesco, A.; Choquet, P.; Detour, J.; et al. Towards thrombosis-targeted zeolitenanoparticles for laser-polarized 129 Xe MRI. J. Mater. Chem. 2009, 19, 379–386. [Google Scholar] [CrossRef]
- Cram, D.J.; Cram, J.M. Monographs in Supramolecular Chemistry; Stoddart, J.F., Ed.; Royal Society of Chemistry: Cambridge, UK, 1994; Volume 4. [Google Scholar]
- Houk, K.N.; Nakamura, K.; Sheu, C.; Keating, A.E. Gating as a Control Element in Constrictive Binding and Guest Release by Hemicarcerands. Science 1996, 273, 627–629. [Google Scholar] [CrossRef] [PubMed]
- Brotin, T.; Dutasta, J.-P. Xe@cryptophane Complexes with C2 Symmetry: Synthesis and Investigations by 129Xe NMR of the Consequences of the Size of the Host Cavity for Xenon Encapsulation. Eur. J. Org. Chem. 2003, 2003, 973–984. [Google Scholar] [CrossRef]
- Gomes, M.D.; Dao, P.; Jeong, K.; Slack, C.C.; Vassiliou, C.C.; Finbloom, J.A.; Francis, M.B.; Wemmer, D.E.; Pines, A. 129Xe NMR Relaxation-Based Macromolecular Sensing. J. Am. Chem. Soc. 2016, 138, 9747–9750. [Google Scholar] [CrossRef] [PubMed]
- El-Ayle, G.; Travis Holman, K. Cryptophanes. In Comprehensive Supramolecular Chemistry II; Elsevier: Amsterdam, The Netherlands, 2017; pp. 199–249. [Google Scholar]
- Traoré, T.; Clave, G.; Delacour, L.; Kotera, N.; Renard, P.-Y.; Romieu, A.; Berthault, P.; Boutin, C.; Tassali, N.; Rousseau, B. The first metal-free water-soluble cryptophane-111. Chem. Commun. 2011, 47, 9702. [Google Scholar] [CrossRef] [PubMed]
- Dubost, E.; Kotera, N.; Garcia-Argote, S.; Boulard, Y.; Léonce, E.; Boutin, C.; Berthault, P.; Dugave, C.; Rousseau, B. Synthesis of a Functionalizable Water-Soluble Cryptophane-111. Org. Lett. 2013, 15, 2866–2868. [Google Scholar] [CrossRef] [PubMed]
- Schilling, F.; Schröder, L.; Palaniappan, K.K.; Zapf, S.; Wemmer, D.E.; Pines, A. MRI Thermometry Based on Encapsulated Hyperpolarized Xenon. ChemPhysChem 2010, 11, 3529–3533. [Google Scholar] [CrossRef]
- Schilling, F.; Schröder, L.; Palaniappan, K.K.; Zapf, S.; Wemmer, D.E.; Pines, A. Cover Picture: MRI Thermometry Based on Encapsulated Hyperpolarized Xenon (ChemPhysChem 16/2010). Chem. Eur. J. Chem. Phys. 2010, 11, 3369. [Google Scholar] [CrossRef]
- Khan, N.S.; Riggle, B.A.; Seward, G.K.; Bai, Y.; Dmochowski, I.J. Cryptophane-Folate Biosensor for 129Xe NMR. Bioconjug. Chem. 2014, 26, 101–109. [Google Scholar] [CrossRef]
- Chambers, J.M.; Hill, P.A.; Aaron, J.A.; Han, Z.; Christianson, D.W.; Kuzma, N.N.; Dmochowski, I.J. Cryptophane Xenon-129 Nuclear Magnetic Resonance Biosensors Targeting Human Carbonic Anhydrase. J. Am. Chem. Soc. 2009, 131, 563–569. [Google Scholar] [CrossRef]
- Riggle, B.A.; Greenberg, M.L.; Wang, Y.; Wissner, R.F.; Zemerov, S.D.; Petersson, E.J.; Dmochowski, I.J. A cryptophane-based “turn-on” 129Xe NMR biosensor for monitoring calmodulin. Org. Biomol. Chem. 2017, 15, 8883–8887. [Google Scholar] [CrossRef] [PubMed]
- Riggle, B.A.; Wang, Y.; Dmochowski, I.J. A “Smart” 129Xe NMR Biosensor for pH-Dependent Cell Labeling. J. Am. Chem. Soc. 2015, 137, 5542–5548. [Google Scholar] [CrossRef] [PubMed]
- González, M.; Argaraña, C.E.; Fidelio, G.D. Extremely high thermal stability of streptavidin and avidin upon biotin binding. Biomol. Eng. 1999, 16, 67–72. [Google Scholar] [CrossRef]
- Rybak, J.-N.; Scheurer, S.B.; Neri, D.; Elia, G. Purification of biotinylated proteins on streptavidin resin: A protocol for quantitative elution. Proteomics 2004, 4, 2296–2299. [Google Scholar] [CrossRef]
- Ellison, D.; Hinton, J.; Hubbard, S.; Beynon, R.J. Limited proteolysis of native proteins: The interaction between avidin and proteinase K. Protein Sci. 1995, 4, 1337–1345. [Google Scholar] [CrossRef]
- Elia, G. Biotinylation reagents for the study of cell surface proteins. Proteomics 2008, 8, 4012–4024. [Google Scholar] [CrossRef]
- Jain, A.; Cheng, K. The principles and applications of avidin-based nanoparticles in drug delivery and diagnosis. J. Control. Release 2017, 245, 27–40. [Google Scholar] [CrossRef]
- Geninatti Crich, S.; Barge, A.; Battistini, E.; Cabella, C.; Coluccia, S.; Longo, D.; Mainero, V.; Tarone, G.; Aime, S. Magnetic resonance imaging visualization of targeted cells by the internalization of supramolecular adducts formed between avidin and biotinylated Gd3+ chelates. J. Biol. Inorg. Chem. 2005, 10, 78–86. [Google Scholar] [CrossRef]
- Wright, S.; Ramos, R.; Tobias, P.; Ulevitch, R.; Mathison, J. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 1990, 249, 1431–1433. [Google Scholar] [CrossRef]
- Wright, S.D. CD14 and innate recognition of bacteria. J. Immunol. 1995, 155, 6–8. [Google Scholar]
- Günzel, D.; Fromm, M. Claudins and other tight junction proteins. Compr. Physiol. 2012, 2, 1819–1852. [Google Scholar] [PubMed]
- Osanai, M.; Takasawa, A.; Murata, M.; Sawada, N. Claudins in cancer: Bench to bedside. Pflugers Arch. Eur. J. Physiol. 2017, 469, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Kokai-Kun, J.F.; McClane, B.A. Deletion analysis of the Clostridium perfringens enterotoxin. Infect. Immun. 1997, 65, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Kondoh, M.; Li, X.; Takahashi, A.; Matsuhisa, K.; Matsushita, K.; Kakamu, Y.; Yamane, S.; Kodaka, M.; Isoda, K.; et al. A toxicological evaluation of a claudin modulator, the C-terminal fragment of Clostridium perfringens enterotoxin, in mice. Die Pharm. 2011, 66, 543–546. [Google Scholar]
- McClane, B.A. The complex interactions between Clostridium perfringens enterotoxin and epithelial tight junctions. Toxicon 2001, 39, 1781–1791. [Google Scholar] [CrossRef]
- Takahashi, A.; Komiya, E.; Kakutani, H.; Yoshida, T.; Fujii, M.; Horiguchi, Y.; Mizuguchi, H.; Tsutsumi, Y.; Tsunoda, S.-I.; Koizumi, N.; et al. Domain mapping of a claudin-4 modulator, the C-terminal region of C-terminal fragment of Clostridium perfringens enterotoxin, by site-directed mutagenesis. Biochem. Pharmacol. 2008, 75, 1639–1648. [Google Scholar] [CrossRef]
- Piontek, A.; Witte, C.; May Rose, H.; Eichner, M.; Protze, J.; Krause, G.; Piontek, J.; Schröder, L. A cCPE-based xenon biosensor for magnetic resonance imaging of claudin-expressing cells. Ann. N. Y. Acad. Sci. 2017, 1397, 195–208. [Google Scholar] [CrossRef]
- Jayapaul, J.; Schröder, L. Complete Generation of a Xe Biosensor on the Solid Support by Systematic Backbone Assembly. Bioconjug. Chem. 2018, 29, 4004–4011. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Barry Sharpless, K. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Moses, J.E.; Moorhouse, A.D. The growing applications of click chemistry. Chem. Soc. Rev. 2007, 36, 1249–1262. [Google Scholar] [CrossRef]
- Wu, P.; Fokin, V.V. Catalytic Azide—Alkyne Cycloaddition: Reactivity and Applications. Chemin 2007, 38, 38. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. Engl. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Manetsch, R.; Krasiński, A.; Radić, Z.; Raushel, J.; Taylor, P.; Sharpless, K.B.; Kolb, H.C. In situ click chemistry: Enzyme inhibitors made to their own specifications. J. Am. Chem. Soc. 2004, 126, 12809–12818. [Google Scholar] [CrossRef] [PubMed]
- Speers, A.E.; Adam, G.C.; Cravatt, B.F. Activity-based protein profiling in vivo using a copper(i)-catalyzed azide-alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc. 2003, 125, 4686–4687. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.R.; Hilgraf, R.; Sharpless, K.B.; Fokin, V.V. Polytriazoles as copper(I)-stabilizing ligands in catalysis. Org. Lett. 2004, 6, 2853–2855. [Google Scholar] [CrossRef] [PubMed]
- Golas, P.L.; Matyjaszewski, K. Marrying click chemistry with polymerization: Expanding the scope of polymeric materials. Chem. Soc. Rev. 2010, 39, 1338–1354. [Google Scholar] [CrossRef] [PubMed]
- Hänni, K.D.; Leigh, D.A. The application of CuAAC “click” chemistry to catenane and rotaxane synthesis. Chem. Soc. Rev. 2010, 39, 1240–1251. [Google Scholar] [CrossRef]
- Taratula, O.; Kim, M.P.; Bai, Y.; Philbin, J.P.; Riggle, B.A.; Haase, D.N.; Dmochowski, I.J. Synthesis of Enantiopure, Trisubstituted Cryptophane-A Derivatives. Org. Lett. 2012, 14, 3580–3583. [Google Scholar] [CrossRef]
- Aaron, J.A.; Chambers, J.M.; Jude, K.M.; Di Costanzo, L.; Dmochowski, I.J.; Christianson, D.W. Structure of a 129Xe-Cryptophane Biosensor Complexed with Human Carbonic Anhydrase II. J. Am. Chem. Soc. 2008, 130, 6942–6943. [Google Scholar] [CrossRef]
- Taratula, O.; Bai, Y.; D’Antonio, E.L.; Dmochowski, I.J. Enantiopure Cryptophane-Xe Nuclear Magnetic Resonance Biosensors Targeting Carbonic Anhydrase. Supramol. Chem. 2015, 27, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Zemerov, S.D.; Roose, B.W.; Greenberg, M.L.; Wang, Y.; Dmochowski, I.J. Cryptophane Nanoscale Assemblies Expand Xe NMR Biosensing. Anal. Chem. 2018, 90, 7730–7738. [Google Scholar] [CrossRef] [PubMed]
- Dube, D.H.; Bertozzi, C.R. Glycans in cancer and inflammation—Potential for therapeutics and diagnostics. Nat. Rev. Drug Discov. 2005, 4, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Prescher, J.A.; Bertozzi, C.R. Chemistry in living systems. Nat. Chem. Biol. 2005, 1, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.T.; Sampathkumar, S.-G.; Yarema, K.J. Metabolic oligosaccharide engineering: Perspectives, applications, and future directions. Mol. Biosyst. 2007, 3, 187–194. [Google Scholar] [CrossRef]
- Laughlin, S.T.; Baskin, J.M.; Amacher, S.L.; Bertozzi, C.R. In vivo imaging of membrane-associated glycans in developing zebrafish. Science 2008, 320, 664–667. [Google Scholar] [CrossRef]
- Möller, H.; Böhrsch, V.; Bentrop, J.; Bender, J.; Hinderlich, S.; Hackenberger, C.P.R. Glycan-specific metabolic oligosaccharide engineering of C7-substituted sialic acids. Angew. Chem. Int. Ed. Engl. 2012, 51, 5986–5990. [Google Scholar] [CrossRef]
- Du, J.; Meledeo, M.A.; Wang, Z.; Khanna, H.S.; Paruchuri, V.D.P.; Yarema, K.J. Metabolic glycoengineering: Sialic acid and beyond. Glycobiology 2009, 19, 1382–1401. [Google Scholar] [CrossRef]
- Meldrum, T.; Seim, K.L.; Bajaj, V.S.; Palaniappan, K.K.; Wu, W.; Francis, M.B.; Wemmer, D.E.; Pines, A. A xenon-based molecular sensor assembled on an MS2 viral capsid scaffold. J. Am. Chem. Soc. 2010, 132, 5936–5937. [Google Scholar] [CrossRef]
- Bouchet, A.; Brotin, T.; Linares, M.; Cavagnat, D.; Buffeteau, T. Influence of the chemical structure of water-soluble cryptophanes on their overall chiroptical and binding properties. J. Org. Chem. 2011, 76, 7816–7825. [Google Scholar] [CrossRef]
- Kotera, N.; Tassali, N.; Léonce, E.; Boutin, C.; Berthault, P.; Brotin, T.; Dutasta, J.-P.; Delacour, L.; Traoré, T.; Buisson, D.-A.; et al. A sensitive zinc-activated 129Xe MRI probe. Angew. Chem. Int. Ed. Engl. 2012, 51, 4100–4103. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, R.; Witte, C.; Haag, R.; Schröder, L. Dendronized cryptophanes as water-soluble xenon hosts for 129Xe magnetic resonance imaging. Org. Lett. 2014, 16, 4436–4439. [Google Scholar] [CrossRef] [PubMed]
- Mynar, J.L.; Lowery, T.J.; Wemmer, D.E.; Pines, A.; Fréchet, J.M.J. Xenon biosensor amplification via dendrimer-cage supramolecular constructs. J. Am. Chem. Soc. 2006, 128, 6334–6335. [Google Scholar] [CrossRef] [PubMed]
- Chapellet, L.-L.; Cochrane, J.R.; Mari, E.; Boutin, C.; Berthault, P.; Brotin, T. Synthesis of Cryptophanes with Two Different Reaction Sites: Chemical Platforms for Xenon Biosensing. J. Org. Chem. 2015, 80, 6143–6151. [Google Scholar] [CrossRef] [PubMed]
- Brotin, T.; Roy, V.; Dutasta, J.-P. Improved synthesis of functional CTVs and cryptophanes using Sc(OTf)3 as catalyst. J. Org. Chem. 2005, 70, 6187–6195. [Google Scholar] [CrossRef] [PubMed]
- Haberhauer, G.; Woitschetzki, S.; Bandmann, H. Strongly underestimated dispersion energy in cryptophanes and their complexes. Nat. Commun. 2014, 5, 3542. [Google Scholar] [CrossRef]
- Berthault, P.; Desvaux, H.; Wendlinger, T.; Gyejacquot, M.; Stopin, A.; Brotin, T.; Dutasta, J.-P.; Boulard, Y. Effect of pH and counterions on the encapsulation properties of xenon in water-soluble cryptophanes. Chemistry 2010, 16, 12941–12946. [Google Scholar] [CrossRef]
- Baydoun, O.; De Rycke, N.; Léonce, E.; Boutin, C.; Berthault, P.; Jeanneau, E.; Brotin, T. Synthesis of Cryptophane-223-Type Derivatives with Dual Functionalization. J. Org. Chem. 2019, 84, 9127–9137. [Google Scholar] [CrossRef]
- Tassali, N.; Kotera, N.; Boutin, C.; Léonce, E.; Boulard, Y.; Rousseau, B.; Dubost, E.; Taran, F.; Brotin, T.; Dutasta, J.-P.; et al. Smart Detection of Toxic Metal Ions, Pb2 and Cd2, Using a 129Xe NMR-Based Sensor. Anal. Chem. 2014, 86, 1783–1788. [Google Scholar] [CrossRef]
- Berthault, P.; Bogaert-Buchmann, A.; Desvaux, H.; Huber, G.; Boulard, Y. Sensitivity and multiplexing capabilities of MRI based on polarized 129Xe biosensors. J. Am. Chem. Soc. 2008, 130, 16456–16457. [Google Scholar] [CrossRef]
- Seward, G.K.; Wei, Q.; Dmochowski, I.J. Peptide-mediated cellular uptake of cryptophane. Bioconjug. Chem. 2008, 19, 2129–2135. [Google Scholar] [CrossRef] [PubMed]
- Mari, E.; Bousmah, Y.; Boutin, C.; Léonce, E.; Milanole, G.; Brotin, T.; Berthault, P.; Erard, M. Bimodal Detection of Proteins by 129Xe NMR and Fluorescence Spectroscopy. ChemBioChem 2019, 20, 1450–1457. [Google Scholar] [CrossRef] [PubMed]
- Kotera, N.; Dubost, E.; Milanole, G.; Doris, E.; Gravel, E.; Arhel, N.; Brotin, T.; Dutasta, J.-P.; Cochrane, J.; Mari, E.; et al. A doubly responsive probe for the detection of Cys4-tagged proteins. Chem. Commun. 2015, 51, 11482–11484. [Google Scholar] [CrossRef] [PubMed]
- Witte, C.; Martos, V.; Rose, H.M.; Reinke, S.; Klippel, S.; Schröder, L.; Hackenberger, C.P.R. Inside Back Cover: Live-cell MRI with Xenon Hyper-CEST Biosensors Targeted to Metabolically Labeled Cell-Surface Glycans (Angew. Chem. Int. Ed. 9/2015). Angew. Chem. Int. Ed. 2015, 54, 2855. [Google Scholar] [CrossRef][Green Version]
- Klippel, S.; Döpfert, J.; Jayapaul, J.; Kunth, M.; Rossella, F.; Schnurr, M.; Witte, C.; Freund, C.; Schröder, L. Cell tracking with caged xenon: Using cryptophanes as MRI reporters upon cellular internalization. Angew. Chem. Int. Ed. Engl. 2014, 53, 493–496. [Google Scholar] [CrossRef]
- Sloniec, J.; Schnurr, M.; Witte, C.; Resch-Genger, U.; Schröder, L.; Hennig, A. Biomembrane interactions of functionalized cryptophane-A: Combined fluorescence and 129Xe NMR studies of a bimodal contrast agent. Chemistry 2013, 19, 3110–3118. [Google Scholar] [CrossRef]
- Rossella, F.; Rose, H.M.; Witte, C.; Jayapaul, J.; Schröder, L. Design and Characterization of Two Bifunctional Cryptophane A-Based Host Molecules for Xenon Magnetic Resonance Imaging Applications. ChemPlusChem 2014, 79, 1463–1471. [Google Scholar] [CrossRef]
- Zeng, Q.; Guo, Q.; Yuan, Y.; Yang, Y.; Zhang, B.; Ren, L.; Zhang, X.; Luo, Q.; Liu, M.; Bouchard, L.-S.; et al. Mitochondria Targeted and Intracellular Biothiol Triggered Hyperpolarized Xe Magnetofluorescent Biosensor. Anal. Chem. 2017, 89, 2288–2295. [Google Scholar] [CrossRef]
- Pottier, R.; Kennedy, J.C. New trends in photobiology. J. Photochem. Photobiol. B 1990, 8, 1–16. [Google Scholar] [CrossRef]
- Zeng, Q.; Guo, Q.; Yuan, Y.; Zhang, X.; Jiang, W.; Xiao, S.; Zhang, B.; Lou, X.; Ye, C.; Liu, M.; et al. A Small Molecular Multifunctional Tool for pH Detection, Fluorescence Imaging, and Photodynamic Therapy. ACS Appl. Bio Mater. 2020, 3, 1779–1786. [Google Scholar] [CrossRef]
- Chen, H.; Chan, J.Y.W.; Yang, X.; Wyman, I.W.; Bardelang, D.; Macartney, D.H.; Lee, S.M.Y.; Wang, R. Developmental and organ-specific toxicity of cucurbit[7]uril: In vivo study on zebrafish models. RSC Adv. 2015, 5, 30067–30074. [Google Scholar] [CrossRef]
- Oun, R.; Floriano, R.S.; Isaacs, L.; Rowan, E.G.; Wheate, N.J. The neurotoxic, myotoxic and cardiotoxic activity of cucurbituril-based macrocyclic drug delivery vehicles. Toxicol. Res. 2014, 3, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Hettiarachchi, G.; Nguyen, D.; Wu, J.; Lucas, D.; Ma, D.; Isaacs, L.; Briken, V. Toxicology and drug delivery by cucurbit[n]uril type molecular containers. PLoS ONE 2010, 5, e10514. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, X.; Li, S.; Wang, L.-H.; Zhang, J.; Wang, R. A systematic evaluation of the biocompatibility of cucurbit[7]uril in mice. Sci. Rep. 2018, 8, 8819. [Google Scholar] [CrossRef]
- Wu, X.; Gao, L.; Hu, X.-Y.; Wang, L. Supramolecular Drug Delivery Systems Based on Water-Soluble Pillar[n]arenes. Chem. Rec. 2016, 16, 1216–1227. [Google Scholar] [CrossRef]
- Hua, Y.; Chen, L.; Hou, C.; Liu, S.; Pei, Z.; Lu, Y. Supramolecular Vesicles Based on Amphiphilic Pillar[n]arenes for Smart Nano-Drug Delivery. Int. J. Nanomed. 2020, 15, 5873–5899. [Google Scholar] [CrossRef]
- Yu, G.; Zhou, X.; Zhang, Z.; Han, C.; Mao, Z.; Gao, C.; Huang, F. Pillar[6]arene/paraquat molecular recognition in water: High binding strength, pH-responsiveness, and application in controllable self-assembly, controlled release, and treatment of paraquat poisoning. J. Am. Chem. Soc. 2012, 134, 19489–19497. [Google Scholar] [CrossRef]
- Wheate, N.J.; Dickson, K.-A.; Kim, R.R.; Nematollahi, A.; Macquart, R.B.; Kayser, V.; Yu, G.; Church, W.B.; Marsh, D.J. Host-Guest Complexes of Carboxylated Pillar[n]arenes With Drugs. J. Pharm. Sci. 2016, 105, 3615–3625. [Google Scholar] [CrossRef]
- Parnham, M.J.; Wetzig, H. Toxicity screening of liposomes. Chem. Phys. Lipids 1993, 64, 263–274. [Google Scholar] [CrossRef]
- Liu, F.; Huang, H.; Gong, Y.; Li, J.; Zhang, X.; Cao, Y. Evaluation of in vitro toxicity of polymeric micelles to human endothelial cells under different conditions. Chem. Biol. Interact. 2017, 263, 46–54. [Google Scholar] [CrossRef]
- Wójcik, E.; Stańczyk, M.; Wojtasik, A.; Kowalska, J.D.; Nowakowska, M.; Łukasiak, M.; Bartnicka, M.; Kazimierczak, J.; Dastych, J. Comprehensive Evaluation of the Safety and Efficacy of BAFASAL® Bacteriophage Preparation for the Reduction of Salmonella in the Food Chain. Viruses 2020, 12, 742. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Pan, P.; Zheng, D.-W.; Bao, P.; Zeng, X.; Zhang, X.-Z. Bioinorganic hybrid bacteriophage for modulation of intestinal microbiota to remodel tumor-immune microenvironment against colorectal cancer. Sci. Adv. 2020, 6, eaba1590. [Google Scholar] [CrossRef] [PubMed]
- Vaks, L.; Benhar, I. In vivo characteristics of targeted drug-carrying filamentous bacteriophage nanomedicines. J. Nanobiotechnol. 2011, 9, 58. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Roose, B.W.; Zemerov, S.D.; Stringer, M.A.; Dmochowski, I.J. Detecting protein–protein interactions by Xe-129 NMR. Chem. Commun. 2020, 56, 11122–11125. [Google Scholar] [CrossRef]
- Leese, P.T.; Noveck, R.J.; Shorr, J.S.; Woods, C.M.; Flaim, K.E.; Keipert, P.E. Randomized safety studies of intravenous perflubron emulsion. I. Effects on coagulation function in healthy volunteers. Anesth. Analg. 2000, 91, 804–811. [Google Scholar] [CrossRef]
- Noveck, R.J.; Shannon, E.J.; Leese, P.T.; Shorr, J.S.; Flaim, K.E.; Keipert, P.E.; Woods, C.M. Randomized safety studies of intravenous perflubron emulsion. II. Effects on immune function in healthy volunteers. Anesth. Analg. 2000, 91, 812–822. [Google Scholar] [CrossRef]
- Nieuwoudt, M.; Engelbrecht, G.H.C.; Sentle, L.; Auer, R.; Kahn, D.; van der Merwe, S.W. Non-toxicity of IV injected perfluorocarbon oxygen carrier in an animal model of liver regeneration following surgical injury. Artif. Cells Blood Substit. Immobil. Biotechnol. 2009, 37, 117–124. [Google Scholar] [CrossRef]
- Shapiro, M.G.; Goodwill, P.W.; Neogy, A.; Yin, M.; Foster, F.S.; Schaffer, D.V.; Conolly, S.M. Biogenic gas nanostructures as ultrasonic molecular reporters. Nat. Nanotechnol. 2014, 9, 311–316. [Google Scholar] [CrossRef]
- Freeman, W.A.; Mock, W.L.; Shih, N.Y. Cucurbituril. J. Am. Chem. Soc. 1981, 103, 7367–7368. [Google Scholar] [CrossRef]
- Behrend, R.; Meyer, E.; Rusche, F.I. Ueber Condensationsproducte aus Glycoluril und Formaldehyd. Eur. J. Org. Chem. 1905, 339, 1–37. [Google Scholar] [CrossRef]
- Isaacs, L.; Park, S.-K.; Liu, S.; Ko, Y.H.; Selvapalam, N.; Kim, Y.; Kim, H.; Zavalij, P.Y.; Kim, G.-H.; Lee, H.-S.; et al. The Inverted Cucurbit[n]uril Family. J. Am. Chem. Soc. 2005, 127, 18000–18001. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-H.; Liu, S.; Zavalij, A.P.Y.; Isaacs, L. Nor-Seco-Cucurbit [10] uril Exhibits Homotropic Allosterism. J. Am. Chem. Soc. 2006, 128, 14744–14745. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-H.; Zavalij, P.Y.; Isaacs, L. Chiral Recognition inside a Chiral Cucurbituril. Angew. Chem. Int. Ed. 2007, 46, 7425–7427. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-H.; Zavalij, P.Y.; Isaacs, L. Cucurbit[n]uril Formation Proceeds by Step-Growth Cyclo-oligomerization. J. Am. Chem. Soc. 2008, 130, 8446–8454. [Google Scholar] [CrossRef] [PubMed]
- Petersen, H. Syntheses of Cyclic Ureas by α-Ureidoalkylation. Synthesis 1973, 1973, 243–292. [Google Scholar] [CrossRef]
- Grillon, E.; Gallo, R.; Pierrot, M.; Boileau, J.; Wimmer, E. Isolation and X-ray structure of the intermediate dihydroxyimidazolidine(DHI) in the synthesis of glycoluril from glyoxal and urea. Tetrahedron Lett. 1988, 29, 1015–1016. [Google Scholar] [CrossRef]
- Chakraborty, A.; Wu, A.; Witt, D.; Lagona, J.; Fettinger, J.C.; Isaacs, L. Diastereoselective Formation of Glycoluril Dimers: Isomerization Mechanism and Implications for Cucurbit[n]uril Synthesis. J. Am. Chem. Soc. 2002, 124, 8297–8306. [Google Scholar] [CrossRef]
- Wu, A.; Chakraborty, A.; Witt, D.; Lagona, J.; Damkaci, F.; Ofori, M.A.; Chiles, J.K.; Fettinger, J.C.; Isaacs, L. Methylene-Bridged Glycoluril Dimers: Synthetic Methods. J. Org. Chem. 2002, 67, 5817–5830. [Google Scholar] [CrossRef]
- Witt, D.; Lagona, J.; Damkaci, F.; Fettinger, J.C.; Isaacs, L. Diastereoselective Formation of Methylene-Bridged Glycoluril Dimers. Org. Lett. 2000, 2, 755–758. [Google Scholar] [CrossRef]
- Maslii, A.N.; Grishaeva, T.N.; Kuznetsov, A.M.; Bakovets, V.V. Quantum chemical investigation of structural and thermodynamic peculiarities of the formation of cucurbit[n]urils. J. Struct. Chem. 2007, 48, 552–557. [Google Scholar] [CrossRef]
- Bakovets, V.V. A thermodynamic analysis of the mechanism of formation of homologs of the cucurbit[n]uril family. Russ. J. Phys. Chem. A 2007, 81, 1586–1590. [Google Scholar] [CrossRef]
- Oh, K.S.; Yoon, J.; Kim, K.S. Structural Stabilities and Self-Assembly of Cucurbit[n]uril (n = 4−7) and Decamethylcucurbit[n]uril (n = 4−6): A Theoretical Study. J. Phys. Chem. B 2001, 105, 9726–9731. [Google Scholar] [CrossRef]
- So, Y.-H. The effect of limited monomer solubility in heterogeneous step-growth polymerization. Acc. Chem. Res. 2001, 34, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Flory, P.J. Fundamental Principles of Condensation Polymerization. Chem. Rev. 1946, 39, 137–197. [Google Scholar] [CrossRef]
- Day, A.; Arnold, A.P.; Blanch, R.J.; Snushall, B. Controlling factors in the synthesis of cucurbituril and its homologues. J. Org. Chem. 2001, 66, 8094–8100. [Google Scholar] [CrossRef]
- Kim, J.; Jung, I.-S.; Kim, S.-Y.; Lee, E.; Kang, J.-K.; Sakamoto, S.; Yamaguchi, K.; Kim, K. New Cucurbituril Homologues: Syntheses, Isolation, Characterization, and X-ray Crystal Structures of Cucurbit[n]uril (n= 5, 7, and 8). J. Am. Chem. Soc. 2000, 122, 540–541. [Google Scholar] [CrossRef]
- Cheng, X.-J.; Liang, L.-L.; Chen, K.; Ji, N.-N.; Xiao, X.; Zhang, J.-X.; Zhang, Y.-Q.; Xue, S.-F.; Zhu, Q.-J.; Ni, X.-L.; et al. Twisted Cucurbit[14]uril. Angew. Chem. Int. Ed. 2013, 52, 7252–7255. [Google Scholar] [CrossRef]
- Liu, S.; Kim, K.; Isaacs, L. Mechanism of the Conversion of Inverted CB[6] to CB[6]. J. Org. Chem. 2007, 72, 6840–6847. [Google Scholar] [CrossRef]
- Lagona, J.; Mukhopadhyay, P.; Chakrabarti, S.; Isaacs, L. The Cucurbit[n]uril Family. Angew. Chem. 2005, 44, 4844–4870. [Google Scholar] [CrossRef]
- Zhao, J.; Kim, H.-J.; Oh, J.; Kim, S.-Y.; Lee, J.W.; Sakamoto, S.; Yamaguchi, K.; Kim, K. Cucurbit[n]uril Derivatives Soluble in Water and Organic Solvents. Angew. Chem. 2001, 40, 4233–4235. [Google Scholar] [CrossRef]
- Sasmal, S.; Sinha, M.K.; Keinan, E. Facile Purification of Rare Cucurbiturils by Affinity Chromatography. Org. Lett. 2004, 6, 1225–1228. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.-B.; Zhang, Y.-Q.; Zhu, Q.-J.; Xue, S.-F.; Tao, Z. Synthesis and X-ray Structure of the Inclusion Complex of Dodecamethylcucurbit [6] uril with 1,4-Dihydroxybenzene. Molecules 2007, 12, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.; Wu, L.-H.; Xiao, X.; Zhang, Y.; Xue, S.-F.; Tao, Z.; Day, A.I. Locating the Cyclopentano Cousins of the Cucurbit[n]uril Family. J. Org. Chem. 2011, 77, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Isobe, H.; Sato, S.; Nakamura, E. Synthesis of Disubstituted Cucurbit[6]uril and Its Rotaxane Derivative. Org. Lett. 2002, 4, 1287–1289. [Google Scholar] [CrossRef]
- Day, A.I.; Arnold, A.P.; Blanch, R.J. A Method for Synthesizing Partially Substituted Cucurbit[n]uril. Molecules 2003, 8, 74–84. [Google Scholar] [CrossRef]
- Zhao, Y. Synthesis of a symmetrical tetrasubstituted cucurbit[6]uril and its host-guest inclusion complex with 2,2′-bipyridine. Chin. Sci. Bull. 2004, 49, 1111. [Google Scholar] [CrossRef]
- Li-Mei, Z.; Qian-Jiang, Z.H.U.; Jian-Nan, Z.H.U.; Yun-Qian, Z.; Zhu, T.A.O.; Sai-Feng, X.U.E.; Zhan-Bing, W.E.I.; La-Sheng, L. Synthesis and Crystal Structure of a Novel Self-assembled (1,4-discyclohexyl cucurbituril) Sodium(Ⅰ) Complex. Chin. J. Inorg. Chem. 2005, 21, 1583–1588. [Google Scholar]
- Li, Z.-F.; Liang, L.-L.; Wu, F.; Zhou, F.-G.; Ni, X.-L.; Feng, X.; Xiao, X.; Zhang, Y.; Xue, S.-F.; Zhu, Q.-J.; et al. An approach to networks based on coordination of alkyl-substituted cucurbit[5]urils and potassium ions. CrystEngComm 2013, 15, 1994. [Google Scholar] [CrossRef]
- Masson, E.; Ling, X.; Joseph, R.; Kyeremeh-Mensah, L.; Lu, X. Cucurbituril chemistry: A tale of supramolecular success. RSC Adv. 2012, 2, 1213–1247. [Google Scholar] [CrossRef]
- Ni, X.-L.; Xiao, X.; Cong, H.; Liang, L.-L.; Cheng, K.; Cheng, X.-J.; Ji, N.-N.; Zhu, Q.-J.; Xue, S.-F.; Tao, Z. Cucurbit[n]uril-based coordination chemistry: From simple coordination complexes to novel poly-dimensional coordination polymers. Chem. Soc. Rev. 2013, 42, 9480–9508. [Google Scholar] [CrossRef]
- Jeon, Y.-M.; Kim, J.; Whang, D.; Kim, K. Molecular Container Assembly Capable of Controlling Binding and Release of Its Guest Molecules: Reversible Encapsulation of Organic Molecules in Sodium Ion Complexed Cucurbituril. J. Am. Chem. Soc. 1996, 118, 9790–9791. [Google Scholar] [CrossRef]
- Buschmann, H.-J.; Cleve, E.; Schollmeyer, E. Cucurbituril as a ligand for the complexation of cations in aqueous solutions. Inorg. Chim. Acta 1992, 193, 93–97. [Google Scholar] [CrossRef]
- Hoffmann, R.; Knoche, W.; Fenn, C.; Buschmann, H.-J. Host–guest complexes of cucurbituril with the 4-methylbenzylammonium lon, alkali-metal cations and NH4+. J. Chem. Soc. Faraday Trans. 1994, 90, 1507–1511. [Google Scholar] [CrossRef]
- Whang, D.; Heo, J.; Park, J.H.; Kim, K. A Molecular Bowl with Metal Ion as Bottom: Reversible Inclusion of Organic Molecules in Cesium Ion Complexed Cucurbituril. Angew. Chem. Int. Ed. 1998, 37, 78–80. [Google Scholar] [CrossRef]
- Kim, B.S.; Ko, Y.H.; Kim, Y.; Lee, H.J.; Selvapalam, N.; Lee, H.C.; Kim, K. Water soluble cucurbit [6] uril derivative as a potential Xe carrier for 129Xe NMR-based biosensors. Chem. Commun. 2008, 2756. [Google Scholar] [CrossRef]
- Jiang, W.; Winkler, H.D.F.; Schalley, C.A. Integrative Self-Sorting: Construction of a Cascade-Stoppered Hetero[3]rotaxane. J. Am. Chem. Soc. 2008, 130, 13852–13853. [Google Scholar] [CrossRef]
- Yang, W.; Li, Y.; Liu, H.; Chi, L.; Li, Y. Design and Assembly of Rotaxane-Based Molecular Switches and Machines. Small 2012, 8, 504–516. [Google Scholar] [CrossRef]
- Finbloom, J.A.; Slack, C.C.; Bruns, C.J.; Jeong, K.; Wemmer, D.E.; Pines, A.; Francis, M.B. Rotaxane-mediated suppression and activation of cucurbit[6]uril for molecular detection by 129Xe hyperCEST NMR. Chem. Commun. 2016, 52, 3119–3122. [Google Scholar] [CrossRef]
- Ke, C.; Smaldone, R.A.; Kikuchi, T.; Li, H.; Davis, A.P.; Stoddart, J.F. Quantitative emergence of hetero[4]rotaxanes by template-directed click chemistry. Angew. Chem. Int. Ed. Engl. 2013, 52, 381–387. [Google Scholar] [CrossRef]
- Hou, X.; Ke, C.; Bruns, C.J.; McGonigal, P.R.; Pettman, R.B.; Stoddart, J.F. Tunable solid-state fluorescent materials for supramolecular encryption. Nat. Commun. 2015, 6, 6884. [Google Scholar] [CrossRef]
- Slack, C.C.; Finbloom, J.A.; Jeong, K.; Bruns, C.J.; Wemmer, D.E.; Pines, A.; Francis, M.B. Rotaxane probes for protease detection by 129 Xe hyperCEST NMR. Chem. Commun. 2017, 53, 1076–1079. [Google Scholar] [CrossRef] [PubMed]
- Niethammer, P.; Grabher, C.; Look, A.T.; Mitchison, T.J. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 2009, 459, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Weinstain, R.; Savariar, E.N.; Felsen, C.N.; Tsien, R.Y. In Vivo Targeting of Hydrogen Peroxide by Activatable Cell-Penetrating Peptides. J. Am. Chem. Soc. 2014, 136, 874–877. [Google Scholar] [CrossRef] [PubMed]
- Finbloom, J.A.; Aanei, I.L.; Bernard, J.M.; Klass, S.H.; Elledge, S.K.; Han, K.; Ozawa, T.; Nicolaides, T.P.; Berger, M.S.; Francis, M.B. Evaluation of Three Morphologically Distinct Virus-Like Particles as Nanocarriers for Convection-Enhanced Drug Delivery to Glioblastoma. Nanomaterials 2018, 8, 1007. [Google Scholar] [CrossRef]
- Adriani, G.; De Tullio, M.D.; Ferrari, M.; Hussain, F.; Pascazio, G.; Liu, X.; Decuzzi, P. The preferential targeting of the diseased microvasculature by disk-like particles. Biomaterials 2012, 33, 5504–5513. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Methods 2013, 10, 9–17. [Google Scholar] [CrossRef]
- Martinez-Outschoorn, U.; Lin, Z.; Trimmer, C.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect. Cell Cycle 2011, 10, 2504–2520. [Google Scholar] [CrossRef]
- Klass, S.H.; Truxal, A.E.; Fiala, T.A.; Kelly, J.; Nguyen, D.; Finbloom, J.A.; Wemmer, D.E.; Pines, A.; Francis, M.B. Rotaxane Probes for the Detection of Hydrogen Peroxide by 129Xe HyperCEST NMR Spectroscopy. Angew. Chem. 2019, 58, 9948–9953. [Google Scholar] [CrossRef]
- Finbloom, J.A.; Han, K.; Slack, C.C.; Furst, A.L.; Francis, M.B. Cucurbit[6]uril-Promoted Click Chemistry for Protein Modification. J. Am. Chem. Soc. 2017, 139, 9691–9697. [Google Scholar] [CrossRef]
- Cao, L.; Hettiarachchi, G.; Briken, V.; Isaacs, L. Cucurbit[7]uril Containers for Targeted Delivery of Oxaliplatin to Cancer Cells. Angew. Chem. Int. Ed. 2013, 52, 12033–12037. [Google Scholar] [CrossRef]
- Truxal, A.E.; Cao, L.; Isaacs, L.; Wemmer, D.E.; Pines, A. Directly Functionalized Cucurbit[7]uril as a Biosensor for the Selective Detection of Protein Interactions by Xe hyperCEST NMR. Chemistry 2019, 25, 6108–6112. [Google Scholar] [CrossRef] [PubMed]
- Ogoshi, T.; Kanai, S.; Fujinami, S.; Yamagishi, T.-A.; Nakamoto, Y. para-Bridged Symmetrical Pillar[5]arenes: Their Lewis Acid Catalyzed Synthesis and Host–Guest Property. J. Am. Chem. Soc. 2008, 130, 5022–5023. [Google Scholar] [CrossRef] [PubMed]
- Ogoshi, T.; Yamagishi, T.-A.; Nakamoto, Y. Pillar-Shaped Macrocyclic Hosts Pillar[n]arenes: New Key Players for Supramolecular Chemistry. Chem. Rev. 2016, 116, 7937–8002. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Yang, Y.; Chi, X.; Zhang, Z.; Huang, F. Pillararenes, A New Class of Macrocycles for Supramolecular Chemistry. Acc. Chem. Res. 2012, 45, 1294–1308. [Google Scholar] [CrossRef] [PubMed]
- Strutt, N.L.; Zhang, H.; Schneebeli, S.T.; Stoddart, J.F. Functionalizing Pillar[n]arenes. Acc. Chem. Res. 2014, 47, 2631–2642. [Google Scholar] [CrossRef] [PubMed]
- Ogoshi, T.; Hashizume, M.; Yamagishi, T.-A.; Nakamoto, Y. Synthesis, conformational and host–guest properties of water-soluble pillar[5]arene. Chem. Commun. 2010, 46, 3708. [Google Scholar] [CrossRef]
- Murray, J.; Kim, K.; Ogoshi, T.; Yao, W.; Gibb, B.C. The aqueous supramolecular chemistry of cucurbit[n]urils, pillar[n]arenes and deep-cavity cavitands. Chem. Soc. Rev. 2017, 46, 2479–2496. [Google Scholar] [CrossRef]
- Adiri, T.; Marciano, D.; Cohen, Y. Potential 129Xe-NMR biosensors based on secondary and tertiary complexes of a water-soluble pillar[5]arene derivative. Chem. Commun. 2013, 49, 7082. [Google Scholar] [CrossRef]
- Talapaneni, S.N.; Kim, D.; Barin, G.; Buyukcakir, O.; Je, S.H.; Coskun, A. Pillar [5] arene Based Conjugated Microporous Polymers for Propane/Methane Separation through Host–Guest Complexation. Chem. Mater. 2016, 28, 4460–4466. [Google Scholar] [CrossRef]
- Mal, P.; Schultz, D.; Beyeh, N.K.; Rissanen, K.; Nitschke, J.R. An Unlockable-Relockable Iron Cage by Subcomponent Self-Assembly. Angew. Chem. Int. Ed. 2008, 47, 8297–8301. [Google Scholar] [CrossRef]
- Mal, P.; Breiner, B.; Rissanen, K.; Nitschke, J.R. White Phosphorus Is Air-Stable Within a Self-Assembled Tetrahedral Capsule. Science 2009, 324, 1697–1699. [Google Scholar] [CrossRef] [PubMed]
- Riddell, I.A.; Smulders, M.M.J.; Clegg, J.K.; Nitschke, J.R. Encapsulation, storage and controlled release of sulfur hexafluoride from a metal–organic capsule. Chem. Commun. 2011, 47, 457–459. [Google Scholar] [CrossRef] [PubMed]
- Ronson, T.K.; Giri, C.; Beyeh, N.K.; Minkkinen, A.; Topić, F.; Holstein, J.J.; Rissanen, K.; Nitschke, J.R. Size-Selective Encapsulation of Hydrophobic Guests by Self-Assembled M4L6Cobalt and Nickel Cages. Chemistry 2013, 19, 3374–3382. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.V.; Raymond, K.N. The Big Squeeze: Guest Exchange in an M4L6Supramolecular Host. J. Am. Chem. Soc. 2005, 127, 7912–7919. [Google Scholar] [CrossRef]
- Roukala, J.; Zhu, J.; Giri, C.; Rissanen, K.; Lantto, P.; Telkki, V.-V.A. Encapsulation of Xenon by a Self-Assembled Fe4L6 Metallosupramolecular Cage. J. Am. Chem. Soc. 2015, 137, 2464–2467. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, J.-P.; Fujita, M.; Kawano, M.; Sakamoto, A.S.; Yamaguchi, K. A Cationic Guest in a 24+Cationic Host. J. Am. Chem. Soc. 2003, 125, 9260–9261. [Google Scholar] [CrossRef]
- Bruns, C.J.; Fujita, D.; Hoshino, M.; Sato, S.; Stoddart, J.F.; Fujita, M. Emergent Ion-Gated Binding of Cationic Host–Guest Complexes within Cationic M12L24 Molecular Flasks. J. Am. Chem. Soc. 2014, 136, 12027–12034. [Google Scholar] [CrossRef]
- Ruiz, E.J.; Sears, D.N.; Pines, A.; Jameson, C.J. Diastereomeric Xe Chemical Shifts in Tethered Cryptophane Cages. J. Am. Chem. Soc. 2006, 128, 16980–16988. [Google Scholar] [CrossRef]
- Hardie, M.J. Recent advances in the chemistry of cyclotriveratrylene. Chem. Soc. Rev. 2010, 39, 516–527. [Google Scholar] [CrossRef]
- Wang, Y.; Fang, H.; Tranca, I.; Qu, H.; Wang, X.; Markvoort, A.J.; Tian, Z.; Cao, X.-Y. Elucidation of the origin of chiral amplification in discrete molecular polyhedra. Nat. Commun. 2018, 9, 488. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Yang, H.; Fang, H.; Chen, R.; Sun, Y.; Zheng, N.; Tan, K.; Lu, X.; Tian, Z.; et al. Assembled molecular face-rotating polyhedra to transfer chirality from two to three dimensions. Nat. Commun. 2016, 7, 12469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Ronson, T.K.; Greenfield, J.L.; Brotin, T.; Berthault, P.; Leonce, E.; Zhu, J.-L.; Xu, L.; Nitschke, J.R. Enantiopure [Cs+/Xe⊂Cryptophane]⊂FeII4L4 Hierarchical Superstructures. J. Am. Chem. Soc. 2019, 141, 8339–8345. [Google Scholar] [CrossRef] [PubMed]
- Pattni, B.S.; Chupin, V.V.; Torchilin, V.P. New Developments in Liposomal Drug Delivery. Chem. Rev. 2015, 115, 10938–10966. [Google Scholar] [CrossRef]
- Yue, X.; Dai, Z. Liposomal Nanotechnology for Cancer Theranostics. Curr. Med. Chem. 2018, 25, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Wang, X.; Cheng, H.; Fang, M.; Ning, P.; Zhou, Y.; Chen, W.; Song, H. A polycation coated liposome as efficient siRNA carrier to overcome multidrug resistance. Colloids Surf. B Biointerfaces 2017, 159, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Tian, J.; Chen, X. Effect of surface properties on liposomal siRNA delivery. Biomaterials 2016, 79, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Fang, M.; Dong, J.; Xu, C.; Liao, Z.; Ning, P.; Zeng, Q. pH sensitive liposomes delivering tariquidar and doxorubicin to overcome multidrug resistance of resistant ovarian cancer cells. Colloids Surf. B Biointerfaces 2018, 170, 514–520. [Google Scholar] [CrossRef]
- Silindir, M.; Erdoğan, S.; Ozer, A.Y.; Maia, S. Liposomes and their applications in molecular imaging. J. Drug Target. 2012, 20, 401–415. [Google Scholar] [CrossRef]
- Lamichhane, N.; Udayakumar, T.S.; D’Souza, W.D.; Simone, C.B.; Raghavan, S.R.; Polf, J.; Mahmood, J.; Ii, C.S. Liposomes: Clinical Applications and Potential for Image-Guided Drug Delivery. Molecules 2018, 23, 288. [Google Scholar] [CrossRef]
- Xia, Y.; Xu, C.; Zhang, X.; Ning, P.; Wang, Z.; Tian, J.; Chen, X. Liposome-based probes for molecular imaging: From basic research to the bedside. Nanoscale 2019, 11, 5822–5838. [Google Scholar] [CrossRef]
- Navon, G.; Panigel, R.; Valensin, G. Liposomes containing paramagnetic macromolecules as MRI contrast agents. Magn. Reson. Med. 1986, 3, 876–880. [Google Scholar] [CrossRef] [PubMed]
- Ding, N.; Lu, Y.; Lee, R.J.; Yang, C.; Huang, L.; Liu, J.; Xiang, G. Folate receptor-targeted fluorescent paramagnetic bimodal liposomes for tumor imaging. Int. J. Nanomed. 2011, 2011, 2513–2520. [Google Scholar] [CrossRef] [PubMed]
- Kneepkens, E.; Fernandes, A.; Nicolay, K.; Grüll, H. Iron (III)-Based Magnetic Resonance–Imageable Liposomal T1 Contrast Agent for Monitoring Temperature-Induced Image-Guided Drug Delivery. Investig. Radiol. 2016, 51, 735–745. [Google Scholar] [CrossRef]
- Mikhaylov, G.; Mikac, U.; Magaeva, A.A.; Itin, V.I.; Naiden, E.P.; Psakhye, I.; Babes, L.; Reinheckel, T.; Peters, C.; Zeiser, R.; et al. Ferri-liposomes as an MRI-visible drug-delivery system for targeting tumours and their microenvironment. Nat. Nanotechnol. 2011, 6, 594–602. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Yan, X.; Jacobson, O.; Sun, W.; Wang, Z.; Tong, X.; Xia, Y.; Ling, D.; Chen, X. Improved Tumor Uptake by Optimizing Liposome Based RES Blockade Strategy. Theranostics 2017, 7, 319–328. [Google Scholar] [CrossRef]
- Castelli, D.D.; Terreno, E.; Longo, D.L.; Aime, S.; Mulder, W.J.M.; McMahon, M.T.; Nicolay, K. Nanoparticle-based chemical exchange saturation transfer (CEST) agents. NMR Biomed. 2013, 26, 839–849. [Google Scholar] [CrossRef]
- Terreno, E.; Castelli, D.D.; Violante, E.; Sanders, H.M.H.F.; Sommerdijk, N.A.J.M.; Aime, S. Osmotically Shrunken LIPOCEST Agents: An Innovative Class of Magnetic Resonance Imaging Contrast Media Based on Chemical Exchange Saturation Transfer. Chemistry 2009, 15, 1440–1448. [Google Scholar] [CrossRef]
- Ferrauto, G.; Castelli, D.D.; Di Gregorio, E.; Terreno, E.; Aime, S. LipoCEST and cellCEST imaging agents: Opportunities and challenges. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2016, 8, 602–618. [Google Scholar] [CrossRef]
- Meldrum, T.; Schröder, L.; Denger, P.; Wemmer, D.E.; Pines, A. Xenon-based molecular sensors in lipid suspensions. J. Magn. Reson. 2010, 205, 242–246. [Google Scholar] [CrossRef]
- Leupold, E.; Nikolenko, H.; Dathe, M. Apolipoprotein E peptide-modified colloidal carriers: The design determines the mechanism of uptake in vascular endothelial cells. Biochim. Biophys. Acta Biomembr. 2009, 1788, 442–449. [Google Scholar] [CrossRef]
- Leupold, E.; Nikolenko, H.; Beyermann, M.; Dathe, M. Insight into the role of HSPG in the cellular uptake of apolipoprotein E-derived peptide micelles and liposomes. Biochim. Biophys. Acta Biomembr. 2008, 1778, 2781–2789. [Google Scholar] [CrossRef] [PubMed]
- Sydow, K.; Torchilin, V.P.; Dathe, M. Lipopeptide-modified PEG-PE-based pharmaceutical nanocarriers for enhanced uptake in blood-brain barrier cells and improved cytotoxicity against glioma cells. Eur. J. Lipid Sci. Technol. 2014, 116, 1174–1183. [Google Scholar] [CrossRef]
- Kreuter, J.; Shamenkov, D.; Petrov, V.; Ramge, P.; Cychutek, K.; Koch-Brandt, C.; Alyautdin, R.N. Apolipoprotein-mediated Transport of Nanoparticle-bound Drugs Across the Blood-Brain Barrier. J. Drug Target. 2002, 10, 317–325. [Google Scholar] [CrossRef]
- Schnurr, M.; Volk, I.; Nikolenko, H.; Winkler, L.; Dathe, M.; Schröder, L. Functionalized Lipopeptide Micelles as Highly Efficient NMR Depolarization Seed Points for Targeted Cell Labelling in Xenon MRI. Adv. Biosyst. 2020, 4, e1900251. [Google Scholar] [CrossRef] [PubMed]
- Booker, R.D.; Sum, A.K. Biophysical changes induced by xenon on phospholipid bilayers. Biochim. Biophys. Acta Biomembr. 2013, 1828, 1347–1356. [Google Scholar] [CrossRef]
- Zaiss, M.; Schnurr, M.; Bachert, P. Analytical solution for the depolarization of hyperpolarized nuclei by chemical exchange saturation transfer between free and encapsulated xenon (HyperCEST). J. Chem. Phys. 2012, 136, 144106. [Google Scholar] [CrossRef]
- Schnurr, M.; Witte, C.; Schröder, L. Functionalized 129Xe as a potential biosensor for membrane fluidity. Phys. Chem. Chem. Phys. 2013, 15, 14178–14181. [Google Scholar] [CrossRef]
- Lafleur, M.; Cullis, P.R.; Bloom, M. Modulation of the orientational order profile of the lipid acyl chain in the Lalpha phase. Eur. Biophys. J. 1990, 19, 55–62. [Google Scholar] [CrossRef]
- Marsh, D. Liquid-ordered phases induced by cholesterol: A compendium of binary phase diagrams. Biochim. Biophys. Acta 2010, 1798, 688–699. [Google Scholar] [CrossRef]
- Schnurr, M.; Witte, C.; Schröder, L. Depolarization Laplace Transform Analysis of Exchangeable Hyperpolarized 129Xe for Detecting Ordering Phases and Cholesterol Content of Biomembrane Models. Biophys. J. 2014, 106, 1301–1308. [Google Scholar] [CrossRef][Green Version]
- Svetlovics, J.A.; Wheaten, S.A.; Almeida, P.F. Phase Separation and Fluctuations in Mixtures of a Saturated and an Unsaturated Phospholipid. Biophys. J. 2012, 102, 2526–2535. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Chen, Y.; Li, S.; Nguyen, H.G.; Niu, Z.; You, S.; Mello, C.M.; Lu, X.; Wang, Q. Chemical Modification of M13 Bacteriophage and Its Application in Cancer Cell Imaging. Bioconjug. Chem. 2010, 21, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Jayanna, P.K.; Bedi, D.; DeInnocentes, P.; Bird, R.C.; Petrenko, V.A. Landscape phage ligands for PC3 prostate carcinoma cells. Protein Eng. Des. Sel. 2010, 23, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Carrico, Z.M.; Farkas, M.E.; Zhou, Y.; Hsiao, S.C.; Marks, J.D.; Chokhawala, H.; Clark, U.S.; Francis, M.B. N-Terminal Labeling of Filamentous Phage to Create Cancer Marker Imaging Agents. ACS Nano 2012, 6, 6675–6680. [Google Scholar] [CrossRef]
- Dwyer, M.A.; Lu, W.; Dwyer, J.J.; Kossiakoff, A.A. Biosynthetic phage display: A novel protein engineering tool combining chemical and genetic diversity. Chem. Boil. 2000, 7, 263–274. [Google Scholar] [CrossRef][Green Version]
- Tian, F.; Tsao, M.-L.; Schultz, P.G. A Phage Display System with Unnatural Amino Acids. J. Am. Chem. Soc. 2004, 126, 15962–15963. [Google Scholar] [CrossRef]
- Liu, C.C.; Mack, A.V.; Tsao, M.-L.; Mills, J.H.; Lee, H.S.; Choe, H.; Farzan, M.; Schultz, P.G.; Smider, V.V. Protein evolution with an expanded genetic code. Proc. Natl. Acad. Sci. USA 2008, 105, 17688–17693. [Google Scholar] [CrossRef]
- Welsh, L.C.; Symmons, M.F.; Sturtevant, J.M.; A Marvin, D.; Perham, R.N. Structure of the capsid of pf3 filamentous phage determined from X-ray fibre diffraction data at 3.1 Å resolution. J. Mol. Boil. 1998, 283, 155–177. [Google Scholar] [CrossRef]
- Bacteriophage MS2 Capsid Protein—Stock Image—C013/9330. Available online: https://www.sciencephoto.com/media/471076/view/bacteriophage-ms2-capsid-protein (accessed on 4 October 2019).
- Tong, G.J.; Hsiao, S.C.; Carrico, Z.M.; Francis, M.B. Viral Capsid DNA Aptamer Conjugates as Multivalent Cell-Targeting Vehicles. J. Am. Chem. Soc. 2009, 131, 11174–11178. [Google Scholar] [CrossRef]
- Carrico, Z.M.; Romanini, D.W.; Mehl, R.A.; Francis, M.B. Oxidative coupling of peptides to a virus capsid containing unnatural amino acids. Chem. Commun. 2008, 1205. [Google Scholar] [CrossRef]
- Stephanopoulos, N.; Tong, G.J.; Hsiao, S.C.; Francis, M.B. Dual-Surface Modified Virus Capsids for Targeted Delivery of Photodynamic Agents to Cancer Cells. ACS Nano 2010, 4, 6014–6020. [Google Scholar] [CrossRef] [PubMed]
- Caravan, P. Strategies for increasing the sensitivity of gadolinium based MRI contrast agents. Chem. Soc. Rev. 2006, 35, 512. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J.; Yang, J.; Wei, L.; Zurkiya, O.; Yang, W.; Li, S.; Zou, J.; Zhou, Y.; Maniccia, A.L.W.; Mao, H.; et al. Rational Design of Protein-Based MRI Contrast Agents. J. Am. Chem. Soc. 2008, 130, 9260–9267. [Google Scholar] [CrossRef] [PubMed]
- Pierre, V.C.; Botta, M.; Raymond, K.N. Dendrimeric Gadolinium Chelate with Fast Water Exchange and High Relaxivity at High Magnetic Field Strength. J. Am. Chem. Soc. 2005, 127, 504–505. [Google Scholar] [CrossRef] [PubMed]
- Langereis, S.; De Lussanet, Q.G.; Van Genderen, M.H.P.; Meijer, E.W.; Beets-Tan, R.G.H.; Griffioen, A.W.; Van Engelshoven, J.M.A.; Backes, W. Evaluation of Gd(III)DTPA-terminated poly(propylene imine) dendrimers as contrast agents for MR imaging. NMR Biomed. 2006, 19, 133–141. [Google Scholar] [CrossRef]
- Strijkers, G.; Mulder, W.J.M.; Van Heeswijk, R.B.; Frederik, P.; Bomans, P.; Magusin, P.; Nicolay, K. Relaxivity of liposomal paramagnetic MRI contrast agents. Magn. Reson. Mater. Phys. Boil. Med. 2005, 18, 186–192. [Google Scholar] [CrossRef]
- Allen, M.; Bulte, J.W.M.; Liepold, L.; Basu, G.; Zywicke, H.A.; Frank, J.A.; Young, M.; Douglas, T. Paramagnetic viral nanoparticles as potential high-relaxivity magnetic resonance contrast agents. Magn. Reson. Med. 2005, 54, 807–812. [Google Scholar] [CrossRef]
- Anderson, E.A.; Isaacman, S.; Peabody, D.S.; Wang, E.Y.; Canary, J.W.; Kirshenbaum, K. Viral Nanoparticles Donning a Paramagnetic Coat: Conjugation of MRI Contrast Agents to the MS2 Capsid. Nano Lett. 2006, 6, 1160–1164. [Google Scholar] [CrossRef]
- Hooker, J.M.; Datta, A.; Botta, M.; Raymond, K.N.; Francis, M.B. Magnetic Resonance Contrast Agents from Viral Capsid Shells: A Comparison of Exterior and Interior Cargo Strategies. Nano Lett. 2007, 7, 2207–2210. [Google Scholar] [CrossRef]
- Datta, A.; Hooker, J.M.; Botta, M.; Francis, M.B.; Aime, S.; Raymond, K.N. High Relaxivity Gadolinium Hydroxypyridonate−Viral Capsid Conjugates: Nanosized MRI Contrast Agents1. J. Am. Chem. Soc. 2008, 130, 2546–2552. [Google Scholar] [CrossRef]
- Garimella, P.D.; Datta, A.; Romanini, D.W.; Raymond, K.N.; Francis, M.B. Multivalent, High-Relaxivity MRI Contrast Agents Using Rigid Cysteine-Reactive Gadolinium Complexes. J. Am. Chem. Soc. 2011, 133, 14704–14709. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Schultz, P.G. Expanding the genetic code. Angew. Chem. 2004, 44, 34–66. [Google Scholar] [CrossRef] [PubMed]
- Mehl, R.A.; Anderson, J.C.; Santoro, S.W.; Wang, L.; Martin, A.B.; King, D.S.; Horn, D.M.; Schultz, P.G. Generation of a Bacterium with a 21 Amino Acid Genetic Code. J. Am. Chem. Soc. 2003, 125, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Netirojjanakul, C.; Witus, L.S.; Behrens, C.R.; Weng, C.-H.; Iavarone, A.T.; Francis, M.B. Synthetically modified Fc domains as building blocks for immunotherapy applications. Chem. Sci. 2013, 4, 266–272. [Google Scholar] [CrossRef]
- Mallikaratchy, P.R.; Tang, Z.; Kwame, S.; Meng, L.; Shangguan, D.; Tan, W. Aptamer Directly Evolved from Live Cells Recognizes Membrane Bound Immunoglobin Heavy Mu Chain in Burkitt’s Lymphoma Cells. Mol. Cell. Proteom. 2007, 6, 2230–2238. [Google Scholar] [CrossRef] [PubMed]
- Jeong, K.; Netirojjanakul, C.; Munch, H.K.; Sun, J.; Finbloom, J.A.; Wemmer, D.E.; Pines, A.; Francis, M.B. Targeted Molecular Imaging of Cancer Cells Using MS2-Based 129Xe NMR. Bioconjug. Chem. 2016, 27, 1796–1801. [Google Scholar] [CrossRef] [PubMed]
- Tzagoloff, H.; Pratt, D. The initial steps in infection with coliphage M13. Virology 1964, 24, 372–380. [Google Scholar] [CrossRef]
- Chung, W.-J.; Lee, D.-Y.; Yoo, S.Y. Chemical modulation of M13 bacteriophage and its functional opportunities for nanomedicine. Int. J. Nanomed. 2014, 9, 5825–5836. [Google Scholar]
- Hess, G.T.; Cragnolini, J.J.; Popp, M.W.; Allen, M.A.; Dougan, S.K.; Spooner, E.; Ploegh, H.L.; Belcher, A.M.; Guimaraes, C.P. M13 Bacteriophage Display Framework That Allows Sortase-Mediated Modification of Surface-Accessible Phage Proteins. Bioconjug. Chem. 2012, 23, 1478–1487. [Google Scholar] [CrossRef]
- Popp, M.W.; Antos, J.M.; Grotenbreg, G.M.; Spooner, E.; Ploegh, H.L. Sortagging: A versatile method for protein labeling. Nat. Methods 2007, 3, 707–708. [Google Scholar] [CrossRef]
- Lubkowski, J.; Hennecke, F.; Plückthun, A.; Wlodawer, A. The structural basis of phage display elucidated by the crystal structure of the N-terminal domains of g3p. Nat. Genet. 1998, 5, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, S.S.; Weiss, G.A.; Wells, J.A. High copy display of large proteins on phage for functional selections 1 1Edited by P. E. Wright. J. Mol. Boil. 2000, 296, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Hemminga, M.; Vos, W.L.; Nazarov, P.V.; Koehorst, R.B.M.; Wolfs, C.J.A.M.; Spruijt, R.B.; Stopar, D. Viruses: Incredible nanomachines. New advances with filamentous phages. Eur. Biophys. J. 2009, 39, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wu, Y.; Lin, Y.; Wang, Q. Virus-templated FRET platform for the rational design of ratiometric fluorescent nanosensors. Chem. Commun. 2015, 51, 10190–10193. [Google Scholar] [CrossRef]
- Nam, K.T.; Kim, N.-W.; Yoo, P.J.; Chiang, C.-Y.; Meethong, N.; Hammond, P.T.; Belcher, A.M. Virus-Enabled Synthesis and Assembly of Nanowires for Lithium Ion Battery Electrodes. Science 2006, 312, 885–888. [Google Scholar] [CrossRef]
- Nam, K.T.; Wartena, R.; Yoo, P.J.; Liau, F.W.; Lee, Y.J.; Chiang, Y.-M.; Hammond, P.T.; Belcher, A.M. Stamped microbattery electrodes based on self-assembled M13 viruses. Proc. Natl. Acad. Sci. USA 2008, 105, 17227–17231. [Google Scholar] [CrossRef]
- Lee, Y.J.; Yi, H.; Kim, W.-J.; Kang, K.; Yun, D.S.; Strano, M.S.; Ceder, G.; Belcher, A.M. Fabricating Genetically Engineered High-Power Lithium Ion Batteries Using Multiple Virus Genes. Science 2009, 324. [Google Scholar] [CrossRef]
- Merzlyak, A.; Indrakanti, S.; Lee, S.-W. Genetically Engineered Nanofiber-Like Viruses for Tissue Regenerating Materials. Nano Lett. 2009, 9, 846–852. [Google Scholar] [CrossRef]
- Avery, K.N.; Schaak, J.E.; Schaak, R.E. M13 Bacteriophage as a Biological Scaffold for Magnetically-Recoverable Metal Nanowire Catalysts: Combining Specific and Nonspecific Interactions To Design Multifunctional Nanocomposites. Chem. Mater. 2009, 21, 2176–2178. [Google Scholar] [CrossRef]
- Yang, L.-M.C.; Diaz, J.E.; McIntire, T.M.; Weiss, G.A.; Penner, R.M. Covalent Virus Layer for Mass-Based Biosensing. Anal. Chem. 2008, 80, 933–943. [Google Scholar] [CrossRef]
- Yang, L.-M.C.; Diaz, J.E.; McIntire, T.M.; Weiss, G.A.; Penner, R.M. Direct Electrical Transduction of Antibody Binding to a Covalent Virus Layer Using Electrochemical Impedance. Anal. Chem. 2008, 80, 5695–5705. [Google Scholar] [CrossRef] [PubMed]
- Kassner, P.D.; Burg, M.A.; Baird, A.; LaRocca, D. Genetic Selection of Phage Engineered for Receptor-Mediated Gene Transfer to Mammalian Cells. Biochem. Biophys. Res. Commun. 1999, 264, 921–928. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, D.; Baird, A. Receptor-mediated gene transfer by phage-display vectors: Applications in functional genomics and gene therapy. Drug Discov. Today 2001, 6, 793–801. [Google Scholar] [CrossRef]
- Nielsen, U.B.; Marks, J.D. Internalizing antibodies and targeted cancer therapy: Direct selection from phage display libraries. Pharm. Sci. Technol. Today 2000, 3, 282–291. [Google Scholar] [CrossRef]
- Yacoby, I.; Benhar, I. Targeted filamentous bacteriophages as therapeutic agents. Expert Opin. Drug Deliv. 2008, 5, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Hilderbrand, S.A.; Kelly, K.A.; Niedre, M.; Weissleder, R. Near Infrared Fluorescence-Based Bacteriophage Particles for Ratiometric pH Imaging. Bioconjug. Chem. 2008, 19, 1635–1639. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.; Jafari, M.R.; Matochko, W.L.; Derda, R. Quantitative Synthesis of Genetically Encoded Glycopeptide Libraries Displayed on M13 Phage. ACS Chem. Boil. 2012, 7, 1482–1487. [Google Scholar] [CrossRef]
- Chen, I.; Choi, A.Y.-A.; Ting, A.Y. Phage Display Evolution of a Peptide Substrate for Yeast Biotin Ligase and Application to Two-Color Quantum Dot Labeling of Cell Surface Proteins. J. Am. Chem. Soc. 2007, 129, 6619–6625. [Google Scholar] [CrossRef]
- Gilmore, J.M.; Scheck, R.A.; Esser-Kahn, A.P.; Joshi, N.S.; Francis, M.B. N-Terminal Protein Modification through a Biomimetic Transamination Reaction. Angew. Chem. Int. Ed. 2008, 47, 7788. [Google Scholar] [CrossRef]
- Scheck, R.A.; DeDeo, M.T.; Iavarone, A.T.; Francis, M.B. Optimization of a Biomimetic Transamination Reaction. J. Am. Chem. Soc. 2008, 130, 11762–11770. [Google Scholar] [CrossRef]
- Dixon, H.B.F. N-terminal modification of proteins? A review. Protein J. 1984, 3, 99–108. [Google Scholar] [CrossRef]
- Snell, E.E. The Vitamin B6Group. V. The Reversible Interconversion of Pyridoxal and Pyridoxamine by Transamination Reactions. J. Am. Chem. Soc. 1945, 67, 194–197. [Google Scholar] [CrossRef]
- Witus, L.S.; Moore, T.; Thuronyi, B.W.; Esser-Kahn, A.P.; Scheck, R.A.; Iavarone, A.T.; Francis, M.B. Identification of Highly Reactive Sequences For PLP-Mediated Bioconjugation Using a Combinatorial Peptide Library. J. Am. Chem. Soc. 2010, 132, 16812–16817. [Google Scholar] [CrossRef] [PubMed]
- Stevens, T.K.; Palaniappan, K.K.; Ramirez, R.M.; Francis, M.B.; Wemmer, D.E.; Pines, A. HyperCEST detection of a129Xe-based contrast agent composed of cryptophane-A molecular cages on a bacteriophage scaffold. Magn. Reson. Med. 2012, 69, 1245–1252. [Google Scholar] [CrossRef]
- Dirksen, A.; Hackeng, T.M.; Dawson, P.E. Nucleophilic Catalysis of Oxime Ligation. Angew. Chem. Int. Ed. 2006, 45, 7581–7584. [Google Scholar] [CrossRef]
- Laskin, J.; Sandler, A.B. Epidermal growth factor receptor: A promising target in solid tumours. Cancer Treat. Rev. 2004, 30, 1–17. [Google Scholar] [CrossRef]
- Nicholson, R.I.; Gee, J.M.; Harper, M.E. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37, 9–15. [Google Scholar] [CrossRef]
- Palaniappan, K.K.; Ramirez, R.M.; Bajaj, V.S.; Wemmer, D.E.; Pines, A.; Francis, M.B. Molecular Imaging of Cancer Cells Using a Bacteriophage-Based129Xe NMR Biosensor. Angew. Chem. 2013, 125, 4949–4953. [Google Scholar] [CrossRef]
- Wang, Y.; Roose, B.W.; Palovcak, E.J.; Carnevale, V.; Dmochowski, I.J. A Genetically Encoded β-Lactamase Reporter for Ultrasensitive (129) Xe NMR in Mammalian Cells. Angew. Chem. Int. Ed. 2016, 55, 8984–8987. [Google Scholar] [CrossRef]
- Wang, Y.; Roose, B.W.; Palovcak, E.J.; Carnevale, V.; Dmochowski, I.J. A Genetically Encoded β-Lactamase Reporter for Ultrasensitive129Xe NMR in Mammalian Cells. Angew. Chem. 2016, 128, 9130–9133. [Google Scholar] [CrossRef]
- Roose, B.W.; Zemerov, S.D.; Wang, Y.; Kasimova, M.A.; Carnevale, V.; Dmochowski, I.J. A Structural Basis for 129 Xe Hyper-CEST Signal in TEM-1 β-Lactamase. ChemPhysChem 2018, 20, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Bonomo, R. Tazobactam is a potent inactivator of selected inhibitor-resistant class A β-lactamases. FEMS Microbiol. Lett. 1997, 148, 59–62. [Google Scholar] [CrossRef]
- Roose, B.W.; Dmochowski, I.J. Crystal structure of TEM1 beta-lactamase mutant I263L in the presence of 1.2 MPa xenon 2017. ChemPhysChem 2019, 20, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Evenäs, J.; Tugarinov, V.; Skrynnikov, N.R.; Goto, N.K.; Muhandiram, R.; Kay, L.E. Ligand-induced structural changes to maltodextrin-binding protein as studied by solution NMR spectroscopy. J. Mol. Boil. 2001, 309, 961–974. [Google Scholar] [CrossRef] [PubMed]
- Roose, B.W.; Zemerov, S.D.; Dmochowski, I.J. Nanomolar small-molecule detection using a genetically encoded129Xe NMR contrast agent. Chem. Sci. 2017, 8, 7631–7636. [Google Scholar] [CrossRef] [PubMed]
- Riess, J.G. Perfluorocarbon-based Oxygen Delivery. Artif. Cells Blood Substit. Biotechnol. 2006, 34, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Wolber, J.; Rowland, I.J.; Leach, M.O.; Bifone, A. Perfluorocarbon emulsions as intravenous delivery media for hyperpolarized xenon. Magn. Reson. Med. 1999, 41, 442–449. [Google Scholar] [CrossRef]
- Stevens, T.K.; Ramirez, R.M.; Pines, A. Nanoemulsion Contrast Agents with Sub-picomolar Sensitivity for Xenon NMR. J. Am. Chem. Soc. 2013, 135, 9576–9579. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, S.; Yuan, Y.; Li, Y.; Jiang, Z.-X.; Zhou, X. 129Xe Hyper-CEST/19F MRI Multimodal Imaging System for Sensitive and Selective Tumor Cells Detection. ACS Appl. Bio Mater. 2018, 2, 27–32. [Google Scholar] [CrossRef]
- Nienhaus, F.; Colley, D.; Jahn, A.; Pfeiler, S.; Flocke, V.; Temme, S.; Kelm, M.; Gerdes, N.; Flögel, U.; Bönner, F. Phagocytosis of a PFOB-Nanoemulsion for 19F Magnetic Resonance Imaging: First Results in Monocytes of Patients with Stable Coronary Artery Disease and ST-Elevation Myocardial Infarction. Molecules 2019, 24, 2058. [Google Scholar] [CrossRef]
- Jacoby, C.; Temme, S.; Mayenfels, F.; Benoit, N.; Krafft, M.P.; Schubert, R.; Schrader, J.; Flögel, U. Probing different perfluorocarbons forin vivoinflammation imaging by19F MRI: Image reconstruction, biological half-lives and sensitivity. NMR Biomed. 2013, 27, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Walsby, A.E. Gas vesicles. Microbiol. Rev. 1994, 58, 94–144. [Google Scholar] [CrossRef] [PubMed]
- Walsby, A.E. The permeability of blue-green algal gas-vacuole membranes to gas. R. Soc. Lond. Ser. B Biol. Sci. 1969, 173, 235–255. [Google Scholar]
- Oliver, R.L.; Waisby, A.E. Direct evidence for the role of light-mediated gas vesicle collapse in the buoyancy regulation of Anabaena flos-aquae (cyanobacteria)1. Limnol. Oceanogr. 1984, 29, 879–886. [Google Scholar] [CrossRef]
- Thomas, R.; Walsby, A. Buoyancy Regulation in a Strain of Microcystis. Microbiology 1985, 131, 799–809. [Google Scholar] [CrossRef]
- Daviso, E.; Belenky, M.; Griffin, R.G.; Herzfeld, J. Gas Vesicles across Kingdoms: A Comparative Solid-State Nuclear Magnetic Resonance Study. J. Mol. Microbiol. Biotechnol. 2013, 23, 281–289. [Google Scholar] [CrossRef]
- Pfeifer, F. Distribution, formation and regulation of gas vesicles. Nat. Rev. Genet. 2012, 10, 705–715. [Google Scholar] [CrossRef]
- Belenky, M.; Meyers, R.; Herzfeld, J. Subunit Structure of Gas Vesicles: A MALDI-TOF Mass Spectrometry Study. Biophys. J. 2004, 86, 499–505. [Google Scholar] [CrossRef]
- Offner, S.; Ziese, U.; Wanner, G.; Typke, D.; Pfeifer, F. Structural characteristics of halobacterial gas vesicles. Microbiology 1998, 144, 1331–1342. [Google Scholar] [CrossRef]
- Hayes, P.K.; Walsby, A.E.; Walker, J.E. Complete amino acid sequence of cyanobacterial gas-vesicle protein indicates a 70-residue molecule that corresponds in size to the crystallographic unit cell. Biochem. J. 1986, 236, 31–36. [Google Scholar] [CrossRef]
- Hayes, P.K.; Lazarus, C.M.; Bees, A.; Walker, J.E.; Walsby, A.E. The protein encoded by gvpC is a minor component of gas vesicles isolated from the cyanobacteria Anabaena flos-aquae and Microcyctis sp. Mol. Microbiol. 1988, 2, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Hayes, P.K.; Buchholz, B.; Walsby, A.E. Gas vesicles are strengthened by the outer-surface protein, GvpC. Arch. Microbiol. 1992, 157, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Shukla, H.D.; DasSarma, S. Complexity of Gas Vesicle Biogenesis in Halobacterium sp. Strain NRC-1: Identification of Five New Proteins. J. Bacteriol. 2004, 186, 3182–3186. [Google Scholar] [CrossRef] [PubMed]
- Lakshmanan, A.; Farhadi, A.; Nety, S.P.; Lee-Gosselin, A.; Bourdeau, R.W.; Maresca, D.; Shapiro, M.G. Molecular Engineering of Acoustic Protein Nanostructures. ACS Nano 2016, 10, 7314–7322. [Google Scholar] [CrossRef] [PubMed]
- Maresca, D.; Lakshmanan, A.; Lee-Gosselin, A.; Melis, J.M.; Ni, Y.L.; Bourdeau, R.W.; Kochmann, D.M.; Shapiro, M.G. Nonlinear ultrasound imaging of nanoscale acoustic biomolecules. Appl. Phys. Lett. 2017, 110, 073704. [Google Scholar] [CrossRef] [PubMed]
- Maresca, D.; Sawyer, D.P.; Renaud, G.; Lee-Gosselin, A.; Shapiro, M.G. Nonlinear X-Wave Ultrasound Imaging of Acoustic Biomolecules. Phys. Rev. X 2018, 8, 041002. [Google Scholar] [CrossRef]
- Lu, G.J.; Farhadi, A.; Mukherjee, A.; Shapiro, M.G. Proteins, air and water: Reporter genes for ultrasound and magnetic resonance imaging. Curr. Opin. Chem. Boil. 2018, 45, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Bourdeau, R.W.; Lee-Gosselin, A.; Lakshmanan, A.; Farhadi, A.; Kumar, S.R.; Nety, S.P.; Shapiro, M.G. Acoustic reporter genes for noninvasive imaging of microorganisms in mammalian hosts. Nature 2018, 553, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Cannon, M.C. Gas Vesicle Genes Identified in Bacillus megaterium and Functional Expression in Escherichia coli. J. Bacteriol. 1998, 180, 2450–2458. [Google Scholar] [CrossRef] [PubMed]
- Anet, F.A.; Basus, V. Limiting equations for exchange broadening in two-site NMR systems with very unequal populations. J. Magn. Reson. 1978, 32, 339–343. [Google Scholar] [CrossRef]
- Soesbe, T.C.; Merritt, M.E.; Green, K.N.; Rojas-Quijano, F.A.; Sherry, A.D. T 2 exchange agents: A new class of paramagnetic MRI contrast agent that shortens water T 2 by chemical exchange rather than relaxation. Magn. Reson. Med. 2011, 66, 1697–1703. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, M.G.; Ramirez, R.M.; Sperling, L.J.; Sun, G.; Sun, J.; Pines, A.; Schaffer, D.V.; Bajaj, V.S. Genetically encoded reporters for hyperpolarized xenon magnetic resonance imaging. Nat. Chem. 2014, 6, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Fernando, A.; Gariépy, J. Light-Activated Nanoscale Gas Vesicles Selectively Kill Tumor Cells. BioRxiv 2019. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.L.; Harada, T.; Christian, D.A.; Pantano, D.A.; Tsai, R.K.; Discher, D.E. Minimal “Self” Peptides That Inhibit Phagocytic Clearance and Enhance Delivery of Nanoparticles. Science 2013, 339, 971–975. [Google Scholar] [CrossRef] [PubMed]
- Brock, R. The Uptake of Arginine-Rich Cell-Penetrating Peptides: Putting the Puzzle Together. Bioconjug. Chem. 2014, 25, 863–868. [Google Scholar] [CrossRef]
- Zakeri, B.; Fierer, J.O.; Celik, E.; Chittock, E.C.; Schwarz-Linek, U.; Moy, V.T.; Howarth, M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. USA 2012, 109, E690–E697. [Google Scholar] [CrossRef]
- Lu, G.J.; Farhadi, A.; Szablowski, J.O.; Lee-Gosselin, A.; Barnes, S.R.; Lakshmanan, A.; Bourdeau, R.W.; Shapiro, M.G. Acoustically modulated magnetic resonance imaging of gas-filled protein nanostructures. Nat. Mater. 2018, 17, 456–463. [Google Scholar] [CrossRef]
- McMahon, M.T.; Gilad, A.A.; Zhou, J.; Sun, P.Z.; Bulte, J.W.M.; Van Zijl, P.C.M. Quantifying exchange rates in chemical exchange saturation transfer agents using the saturation time and saturation power dependencies of the magnetization transfer effect on the magnetic resonance imaging signal (QUEST and QUESP): Ph calibration for poly-L-lysine and a starburst dendrimer. Magn. Reson. Med. 2006, 55, 836–847. [Google Scholar] [CrossRef]
- Sun, P.Z. Simplified quantification of labile proton concentration-weighted chemical exchange rate (k(ws) ) with RF saturation time dependent ratiometric analysis (QUESTRA): Normalization of relaxation and RF irradiation spillover effects for improved quantitative chemical exchange saturation transfer (CEST) MRI. Magn. Reson. Med. 2012, 67, 936–942. [Google Scholar] [PubMed]
- Randtke, E.A.; Chen, L.Q.; Corrales, L.R.; Pagel, M.D.; Corrales, R.L. The Hanes-Woolf linear QUESP method improves the measurements of fast chemical exchange rates with CEST MRI. Magn. Reson. Med. 2013, 71, 1603–1612. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, M.; Angelovski, G.; Demetriou, E.; McMahon, M.T.; Golay, X.; Scheffler, K. QUESP and QUEST revisited—Fast and accurate quantitative CEST experiments. Magn. Reson. Med. 2017, 79, 1708–1721. [Google Scholar] [CrossRef] [PubMed]
- Korchak, S.; Kilian, W.; Mitschang, L. Degeneracy in cryptophane–xenon complex formation in aqueous solution. Chem. Commun. 2015, 51, 1721–1724. [Google Scholar] [CrossRef] [PubMed]
- Korchak, S.; Kilian, W.; Schröder, L.; Mitschang, L. Design and comparison of exchange spectroscopy approaches to cryptophane-xenon host-guest kinetics. J. Magn. Reson. 2016, 265, 139–145. [Google Scholar] [CrossRef]
- Xu, X.; Lee, J.-S.; Jerschow, A. Ultrafast scanning of exchangeable sites by NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 2013, 52, 8281–8284. [Google Scholar] [CrossRef]
- Döpfert, J.; Zaiss, M.; Witte, C.; Schröder, L. Ultrafast CEST imaging. J. Magn. Reson. 2014, 243, 47–53. [Google Scholar] [CrossRef]
- Döpfert, J.; Witte, C.; Schröder, L. Fast gradient-encoded CEST spectroscopy of hyperpolarized xenon. ChemPhysChem 2014, 15, 261–264. [Google Scholar] [CrossRef]
- Boutin, C.; Léonce, E.; Brotin, T.; Jerschow, A.; Berthault, P. Ultrafast Z-Spectroscopy for Xe NMR-Based Sensors. J. Phys. Chem. Lett. 2013, 4, 4172–4176. [Google Scholar] [CrossRef][Green Version]
- Döpfert, J.; Schnurr, M.; Kunth, M.; Rose, H.M.; Hennig, A.; Schröder, L. Time-resolved monitoring of enzyme activity with ultrafast Hyper-CEST spectroscopy. Magn. Reson. Chem. 2018, 56, 679–688. [Google Scholar] [CrossRef]
- Hennig, A.; Bakirci, H.; Nau, W.M. Label-free continuous enzyme assays with macrocycle-fluorescent dye complexes. Nat. Methods 2007, 4, 629–632. [Google Scholar] [CrossRef]
- Goldenberg, J.M.; Pagel, M.D. Assessments of tumor metabolism with CEST MRI. NMR Biomed. 2018, 32, 3943. [Google Scholar] [CrossRef] [PubMed]
- Ardenkjær-Larsen, J.H.; Fridlund, B.; Gram, A.; Hansson, G.; Lerche, M.H.; Servin, R.; Thaning, M.; Golman, K.; Hansson, L. Increase in signal-to-noise ratio of > 10,000 times in liquid-state NMR. Proc. Natl. Acad. Sci. USA 2003, 100, 10158–10163. [Google Scholar] [CrossRef] [PubMed]
- Keshari, K.R.; Wilson, D.M. ChemInform Abstract: Chemistry and Biochemistry of 13 C Hyperpolarized Magnetic Resonance Using Dynamic Nuclear Polarization. Chemin 2014, 45, 1627–1659. [Google Scholar] [CrossRef]
- Wang, Z.J.; Ohliger, M.A.; Larson, P.E.Z.; Gordon, J.W.; Bok, R.A.; Slater, J.; Villanueva-Meyer, J.E.; Hess, C.P.; Kurhanewicz, J.; Vigneron, D.B. Hyperpolarized C MRI: State of the Art and Future Directions. Radiology 2019, 291, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Le Page, L.M.; Guglielmetti, C.; Taglang, C.; Chaumeil, M.M. Imaging Brain Metabolism Using Hyperpolarized 13C Magnetic Resonance Spectroscopy. Trends Neurosci. 2020, 43, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Doganay, O.; Chen, M.; Matin, T.; Rigolli, M.; Phillips, J.-A.; McIntyre, A.; Gleeson, F.V. Magnetic resonance imaging of the time course of hyperpolarized Xe gas exchange in the human lungs and heart. Eur. Radiol. 2019, 29, 2283–2292. [Google Scholar] [CrossRef]
- Ruppert, K.; Qing, K.; Patrie, J.T.; Altes, T.A.; Mugler, J.P., 3rd. Using Hyperpolarized Xenon-129 MRI to Quantify Early-Stage Lung Disease in Smokers. Acad. Radiol. 2019, 26, 355–366. [Google Scholar] [CrossRef]
- Rao, M.R.; Norquay, G.; Stewart, N.J.; Hoggard, N.; Griffiths, P.D.; Wild, J.M. Assessment of brain perfusion using hyperpolarized Xe MRI in a subject with established stroke. J. Magn. Reson. Imaging 2019. [Google Scholar] [CrossRef]
- Chahal, S.; Prete, B.R.J.; Wade, A.; Hane, F.T.; Albert, M.S. Brain Imaging Using Hyperpolarized Xe Magnetic Resonance Imaging. Methods Enzymol. 2018, 603, 305–320. [Google Scholar]
- Nikolaou, P.; Coffey, A.M.; Walkup, L.L.; Gust, B.M.; Whiting, N.; Newton, H.; Muradyan, I.; Dabaghyan, M.; Ranta, K.; Moroz, G.D.; et al. XeNA: An automated “open-source” 129Xe hyperpolarizer for clinical use. Magn. Reson. Imaging 2014, 32, 541–550. [Google Scholar] [CrossRef]
- Rao, M.; Stewart, N.J.; Norquay, G.; Griffiths, P.D.; Wild, J.M. High resolution spectroscopy and chemical shift imaging of hyperpolarized 129Xe dissolved in the human brain in vivo at 1.5 tesla. Magn. Reson. Med. 2016, 75, 2227–2234. [Google Scholar] [CrossRef] [PubMed]
- Asfour, A. A three-coil RF probe-head at 2.35 T: Potential applications to the 23Na and to the hyperpolarized 129Xe MRI in small animals. In Proceedings of the 2010 Annual International Conference of the IEEE Engineering in Medicine and Biology, Buenos Aires, Argentina, 31 August–4 September 2010; pp. 5693–5699. [Google Scholar]
- Virgincar, R.S.; Nouls, J.C.; Wang, Z.; Degan, S.; Qi, Y.; Xiong, X.; Rajagopal, S.; Driehuys, B. Quantitative Xe MRI detects early impairment of gas-exchange in a rat model of pulmonary hypertension. Sci. Rep. 2020, 10, 7385. [Google Scholar] [CrossRef] [PubMed]
- Hane, F.T.; Li, T.; Smylie, P.; Pellizzari, R.M.; Plata, J.A.; DeBoef, B.; Albert, M.S. In vivo detection of cucurbit[6]uril, a hyperpolarized xenon contrast agent for a xenon magnetic resonance imaging biosensor. Sci. Rep. 2017, 7, 41027. [Google Scholar] [CrossRef] [PubMed]
- XeMat 2018 Speaker Abstracts. Available online: https://cpb-us-w2.wpmucdn.com/web.sas.upenn.edu/dist/a/374/files/2018/05/SPEAKERS-XEMAT-2018-274k1mu.pdf (accessed on 9 February 2020).















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jayapaul, J.; Schröder, L. Molecular Sensing with Host Systems for Hyperpolarized 129Xe. Molecules 2020, 25, 4627. https://doi.org/10.3390/molecules25204627
Jayapaul J, Schröder L. Molecular Sensing with Host Systems for Hyperpolarized 129Xe. Molecules. 2020; 25(20):4627. https://doi.org/10.3390/molecules25204627
Chicago/Turabian StyleJayapaul, Jabadurai, and Leif Schröder. 2020. "Molecular Sensing with Host Systems for Hyperpolarized 129Xe" Molecules 25, no. 20: 4627. https://doi.org/10.3390/molecules25204627
APA StyleJayapaul, J., & Schröder, L. (2020). Molecular Sensing with Host Systems for Hyperpolarized 129Xe. Molecules, 25(20), 4627. https://doi.org/10.3390/molecules25204627