
Cyclopentadienone Iron Tricarbonyl Complexes-Catalyzed Hydrogen Transfer in Water

 ,
, 
Abstract
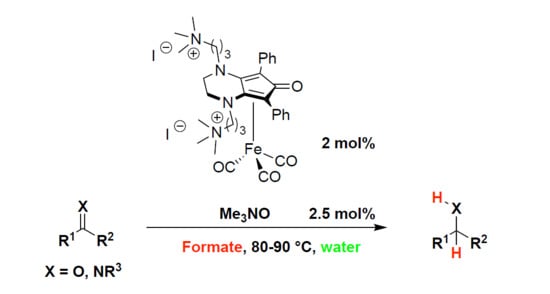
:
1. Introduction
2. Results and Discussion
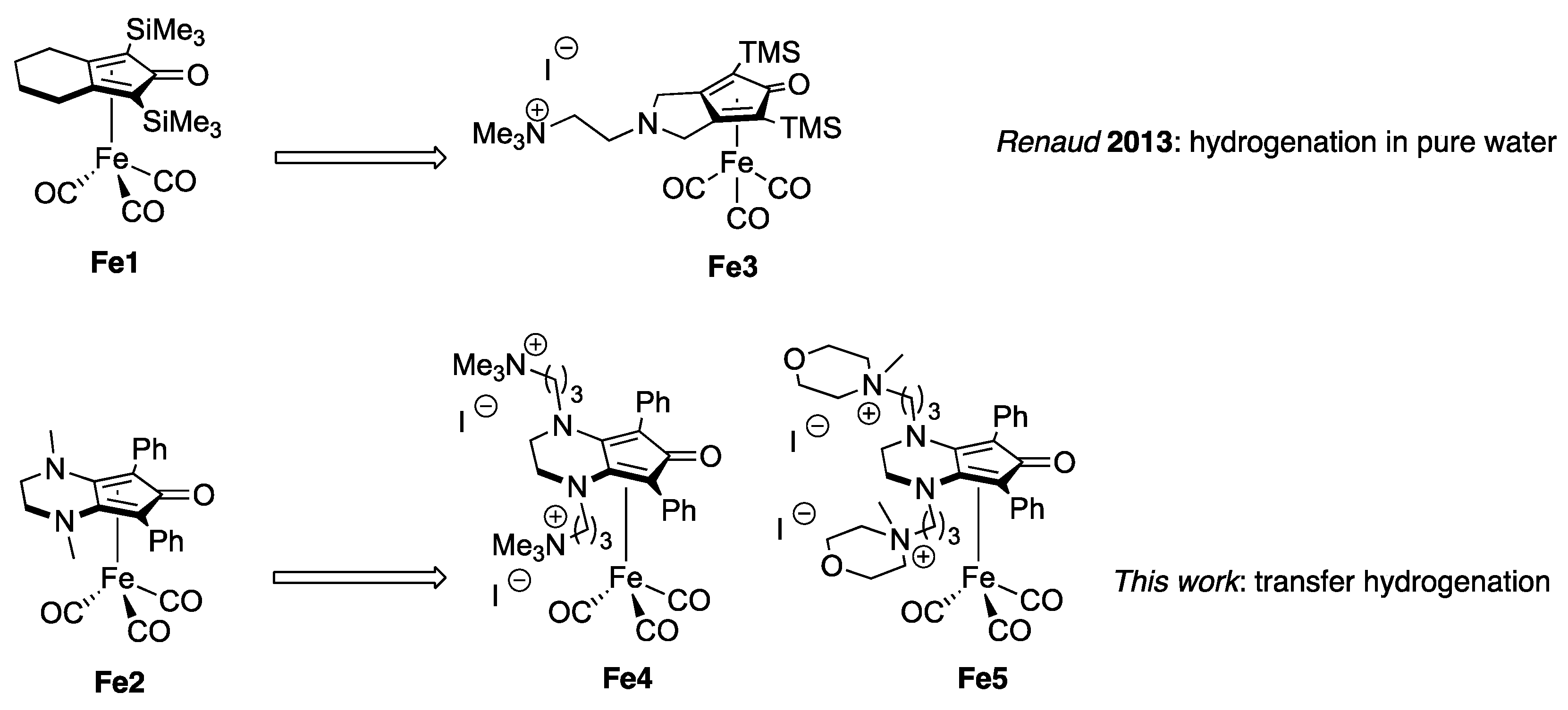
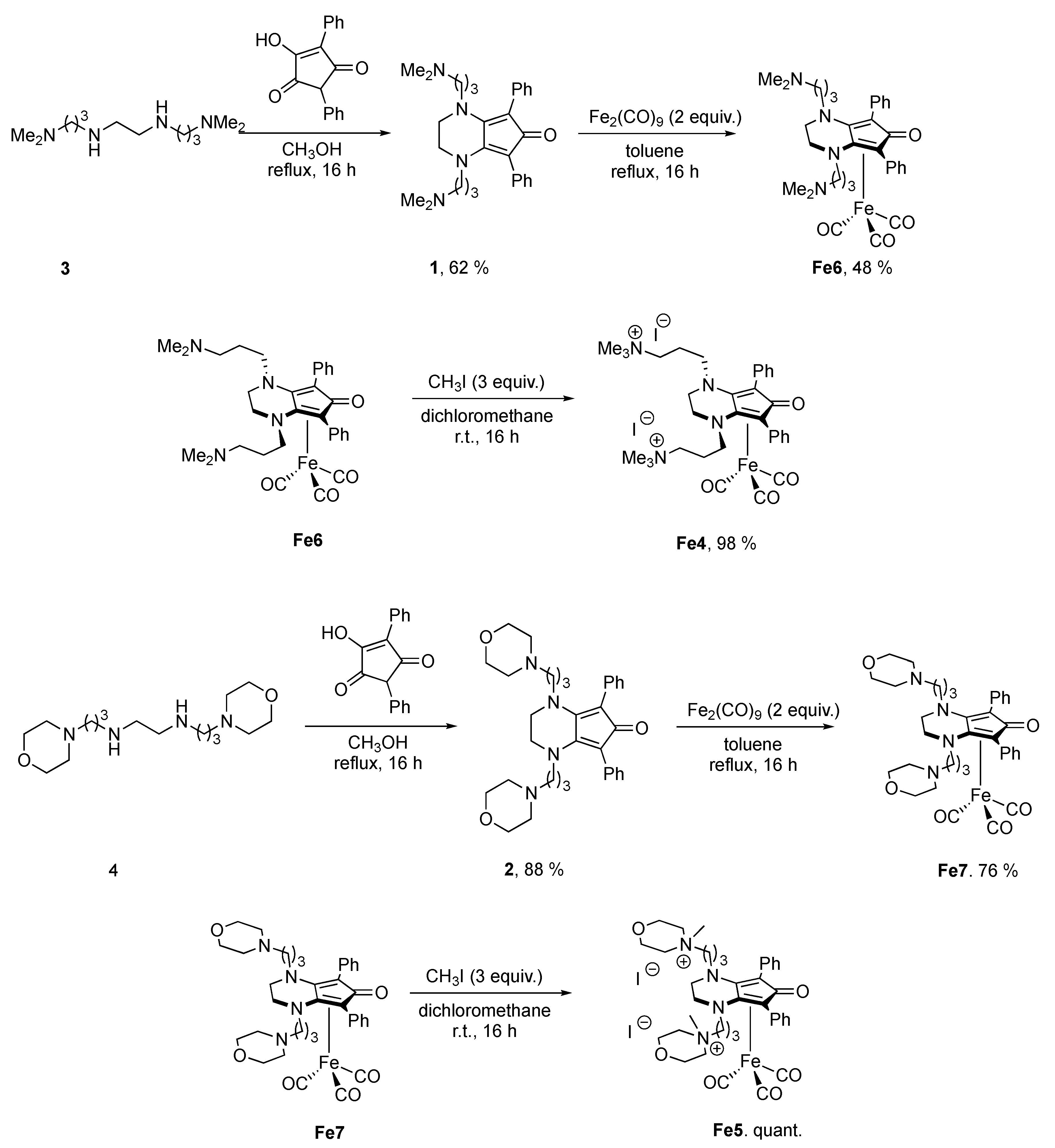
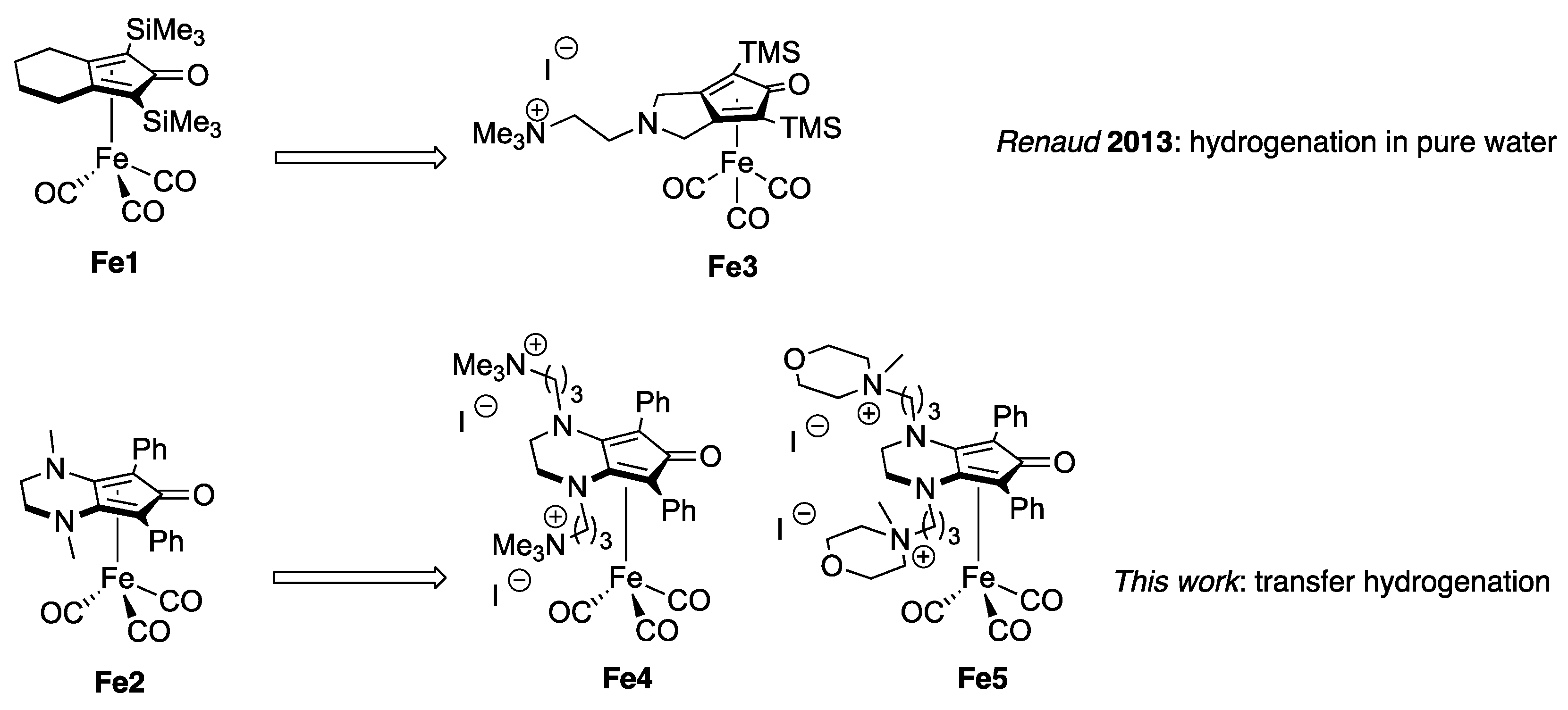
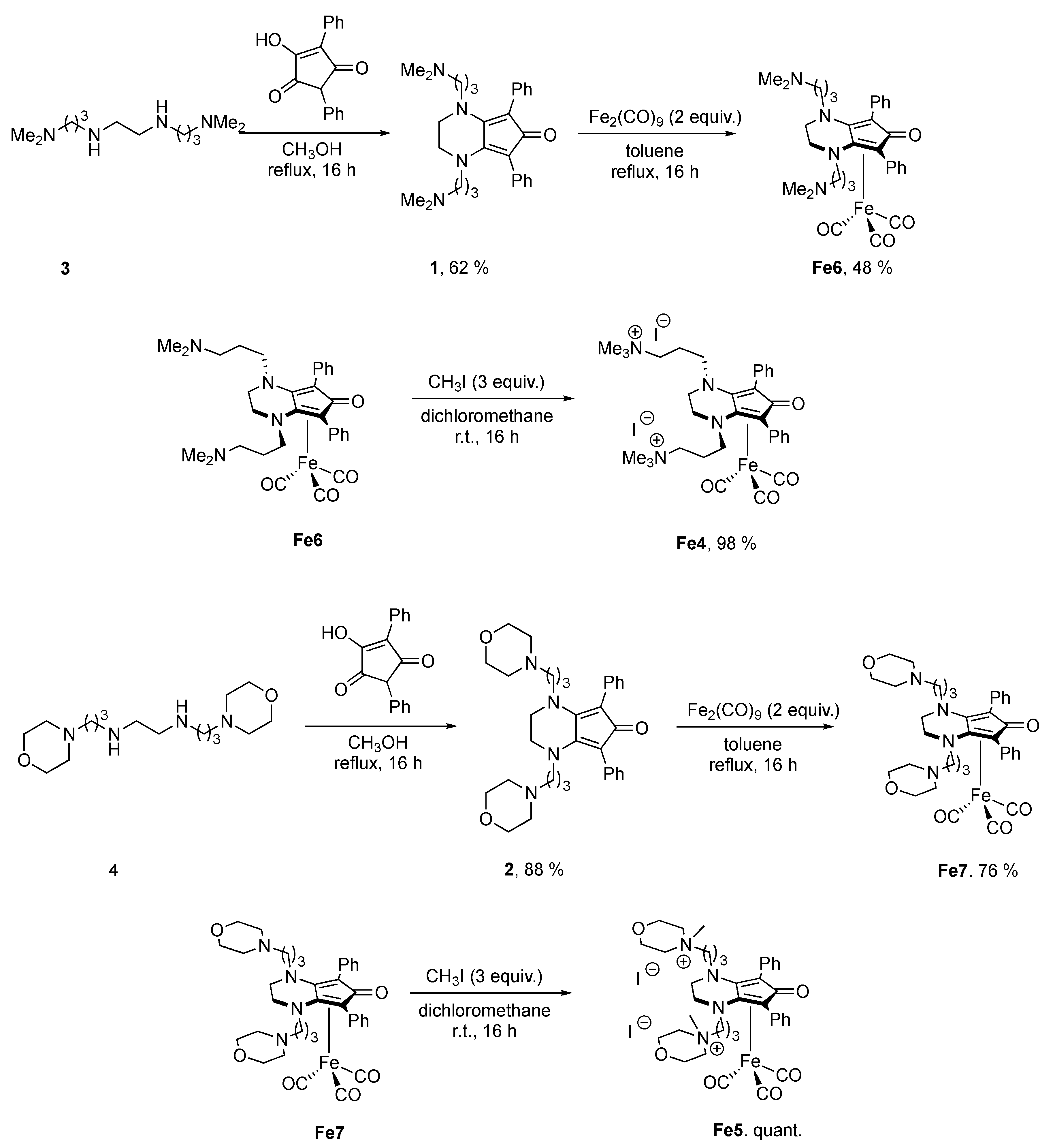
2.1. Synthesis of Complexes
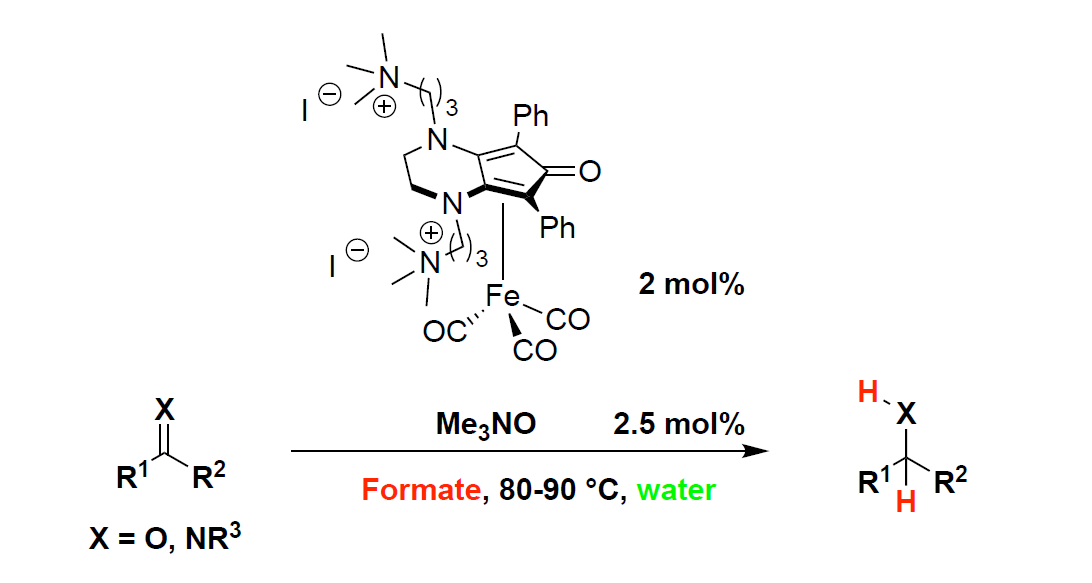
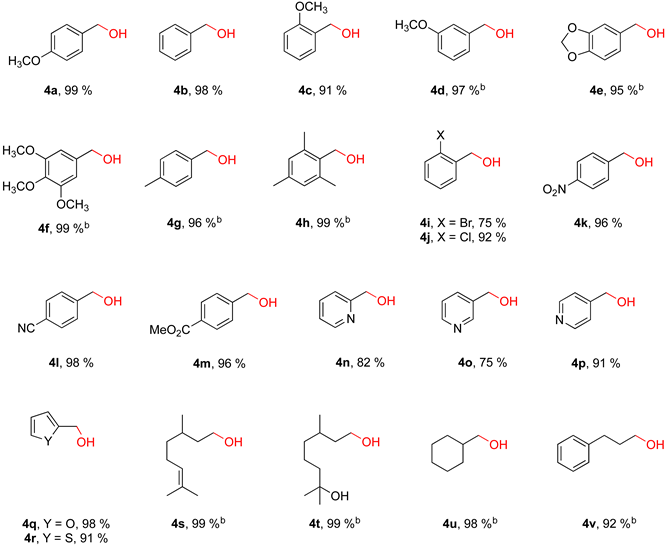
2.2. Iron-Catalyzed Reduction of Carbonyl Compounds
2.3. Iron-Catalyzed Reductive Amination
2.4. Recycling of the Water-Soluble Iron Complex
3. Materials and Methods
3.1. General Procedure for the Reduction of Aldehydes
3.2. General Procedure for the Reductive Amination of Aldehydes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Klingler, F.D. Asymmetric Hydrogenation of Prochiral Amino Ketones to Amino Alcohols for Pharmaceutical Use. Accounts Chem. Res. 2007, 40, 1367–1376. [Google Scholar] [CrossRef]
- Hems, W.P.; Groarke, M.; Zanotti-Gerosa, A.; Grasa, G.A. [(Bisphosphine) Ru(II) Diamine] Complexes in Asymmetric Hydrogenation: Expanding the Scope of the Diamine Ligand. Accounts Chem. Res. 2007, 40, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.B.; Lennon, I.C.; Moran, P.H.; Ramsden, J.A. Industrial-Scale Synthesis and Applications of Asymmetric Hydrogenation Catalysts. Accounts Chem. Res. 2007, 40, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Saudan, L.A. Hydrogenation Processes in the Synthesis of Perfumery Ingredients. Accounts Chem. Res. 2007, 40, 1309–1319. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Nagasaki, I.; Matsumura, K.; Sayo, N.; Saito, T. Developments in Asymmetric Hydrogenation from an Industrial Perspective. Accounts Chem. Res. 2007, 40, 1385–1393. [Google Scholar] [CrossRef] [PubMed]
- Blaser, H.-U.; Studer, M. Cinchona-Modified Platinum Catalysts: From Ligand Acceleration to Technical Processes. Accounts Chem. Res. 2007, 40, 1348–1356. [Google Scholar] [CrossRef]
- Blaser, H.-U.; Pugin, B.; Spindler, F.; Thommen, M. From a Chiral Switch to a Ligand Portfolio for Asymmetric Catalysis. Accounts Chem. Res. 2007, 40, 1240–1250. [Google Scholar] [CrossRef]
- Shultz, C.S.; Krska, S.W. Unlocking the Potential of Asymmetric Hydrogenation at Merck. Accounts Chem. Res. 2007, 40, 1320–1326. [Google Scholar] [CrossRef]
- Wang, N.; Astruc, D. The Golden Age of Transfer Hydrogenation. Chem. Rev. 2015, 115, 6621–6686. [Google Scholar] [CrossRef]
- Wang, C.; Wu, X.; Xiao, J. Broader, Greener, and More Efficient: Recent Advances in Asymmetric Transfer Hydrogenation. Chem. Asian J. 2008, 3, 1750–1770. [Google Scholar] [CrossRef]
- Wei, D.; Darcel, C. Iron Catalysis in Reduction and Hydrometalation Reactions. Chem. Rev. 2018, 119, 2550–2610. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Sahoo, B.; Junge, K.; Beller, M. Cobalt Complexes as an Emerging Class of Catalysts for Homogeneous Hydrogenations. Accounts Chem. Res. 2018, 51, 1858–1869. [Google Scholar] [CrossRef] [PubMed]
- Ai, W.; Zhong, R.; Liu, X.; Liu, Q. Hydride Transfer Reactions Catalyzed by Cobalt Complexes. Chem. Rev. 2018, 119, 2876–2953. [Google Scholar] [CrossRef] [PubMed]
- Filonenko, G.A.; Van Putten, R.; Hensen, E.J.M.; Pidko, E.A. Catalytic (de)hydrogenation promoted by non-precious metals—Co, Fe and Mn: Recent advances in an emerging field. Chem. Soc. Rev. 2018, 47, 1459–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindström, U.M. Stereoselective Organic Reactions in Water. Chem. Rev. 2002, 102, 2751–2772. [Google Scholar] [CrossRef]
- Cornils, B.; Hermann, W.A. Aqueous-Phase Organometallic Catalysis; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Robertson, A.; Matsumoto, T.; Ogo, S. The development of aqueous transfer hydrogenation catalysts. Dalton Trans. 2011, 40, 10304–10310. [Google Scholar] [CrossRef]
- Wu, X.; Xiao, J. Aqueous Phase Asymmetric transfer hydrogenation of ketones-a greener approach to chiral alcohols. Chem. Commun. 2007, 24, 2449–2466. [Google Scholar] [CrossRef]
- Mérel, D.S.; Elie, M.; Lohier, J.-F.; Gaillard, S.; Renaud, J.-L. Bifunctional Iron Complexes: Efficient Catalysts for C=O and C=N Reduction in Water. ChemCatChem 2013, 5, 2939–2945. [Google Scholar] [CrossRef]
- Bohr, M.D.; Bhanushali, M.J.; Nandurkar, N.S.; Bhanage, B.M. Direct Reductive Amination of Carbonyl Compounds with Primary/Secondary Amines Using Recyclable Water-Soluble FeII/EDTA Complex as Catalyst. Tetrahedron Lett. 2008, 49, 965–969. [Google Scholar] [CrossRef]
- Thai, T.-T.; Mérel, D.S.; Poater, A.; Gaillard, S.; Renaud, J.-L. Highly active phosphine-free bifunctional iron complex for hydrogenation of bicarbonate and reductive amination. Chem. Eur. J. 2015, 21, 7066–7070. [Google Scholar] [CrossRef]
- Lator, A.; Gaillard, S.; Poater, A.; Renaud, J.-L. Iron-Catalyzed Chemoselective Reduction of α,β-Unsaturated Ketones. Chem. Eur. J. 2018, 24, 5770–5774. [Google Scholar] [CrossRef] [PubMed]
- Coufourier, S.; Gaillard, S.; Clet, G.; Serre, C.; Daturi, M.; Renaud, J.-L. A MOF-assisted phosphine free bifunctional iron complex for the hydrogenation of carbon dioxide, sodium bicarbonate and carbonate to formate. Chem. Commun. 2019, 55, 4977–4980. [Google Scholar] [CrossRef] [PubMed]
- Seck, C.; Mbaye, M.D.; Coufourier, S.; Lator, A.; Lohier, J.-F.; Poater, A.; Ward, T.R.; Gaillard, S.; Renaud, J. Alkylation of Ketones Catalyzed by Bifunctional Iron Complexes: From Mechanistic Understanding to Application. ChemCatChem 2017, 9, 4410–4416. [Google Scholar] [CrossRef]
- Polidano, K.; Allen, B.D.W.; Williams, J.M.J.; Morrill, L.C. Iron-Catalyzed Methylation Using the Borrowing Hydrogen Approach. ACS Catal. 2018, 8, 6440–6445. [Google Scholar] [CrossRef]
- Bettoni, L.; Seck, C.; Mbaye, M.D.; Gaillard, S.; Renaud, J.-L. Iron-Catalyzed Tandem Three-Component Alkylation: Access to α-Methylated Substituted Ketones. Org. Lett. 2019, 21, 3057–3061. [Google Scholar] [CrossRef]
- Latham, D.E.; Polidano, K.; Williams, J.M.J.; Morrill, L.C. One-Pot Conversion of Allylic Alcohols to α-Methyl Ketones via Iron-Catalyzed Isomerization-Methylation. Org. Lett. 2019, 21, 7914–7918. [Google Scholar] [CrossRef]
- Lator, A.; Gaillard, S.; Poater, A.; Renaud, J.-L. Well-Defined Phosphine-Free Iron-Catalyzed N-Ethylation and N-Methylation of Amines with Ethanol and Methanol. Org. Lett. 2018, 20, 5985–5990. [Google Scholar] [CrossRef]
- Dambatta, M.B.; Polidano, K.; Northey, A.D.; Williams, J.M.J.; Morrill, L.C. Iron-Catalyzed Borrowing Hydrogen C-Alkylation of Oxindoles with Alcohols. ChemSusChem 2019, 12, 2345–2349. [Google Scholar] [CrossRef] [Green Version]
- Seck, C.; Mbaye, M.D.; Gaillard, S.; Renaud, J.-L.; Seck-Diouf, C. Bifunctional Iron Complexes Catalyzed Alkylation of Indoles. Adv. Synth. Catal. 2018, 360, 4640–4645. [Google Scholar] [CrossRef]
- Bettoni, L.; Gaillard, S.; Renaud, J.-L. Iron-Catalyzed β-Alkylation of Alcohols. Org. Lett. 2019, 21, 8404–8408. [Google Scholar] [CrossRef]
- Polidano, K.; Williams, J.M.J.; Morrill, L.C. Iron-Catalyzed Borrowing Hydrogen β-C(sp3)-Methylation of Alcohols. ACS Catal. 2019, 9, 8575–8580. [Google Scholar] [CrossRef] [Green Version]
- Coufourier, S.; Gaillard, Q.G.; Lohier, J.-F.; Poater, A.; Gaillard, S.; Renaud, J.-L. Hydrogenation of CO2, Hydrogenocarbonate and Carbonate to Formate in Water using Phosphine Free Bifunctional Iron Complexes. ACS Catal. 2020. [Google Scholar] [CrossRef]
- Pagnoux-Ozherelyeva, A.; Pannetier, N.; Mbaye, D.M.; Gaillard, S.; Renaud, J.-L. Knölker’s Iron Complex: An Efficient In Situ Generated Catalyst for Reductive Amination of Alkyl Aldehydes and Amines. Angew. Chem. Int. Ed. 2012, 51, 4976–4980. [Google Scholar] [CrossRef] [PubMed]
- Moulin, S.; Dentel, H.; Gaillard, S.; Poater, A.; Cavallo, L.; Lohier, J.-F.; Renaud, J.-L.; Pagnoux-Ozherelyeva, A. Bifunctional (Cyclopentadienone)Iron-Tricarbonyl Complexes: Synthesis, Computational Studies and Application in Reductive Amination. Chem. A Eur. J. 2013, 19, 17881–17890. [Google Scholar] [CrossRef] [PubMed]
- Luh, T.-Y. Trimethylamine N-oxide—A versatile reagent for organometallic chemistry. Coord. Chem. Rev. 1984, 60, 255–276. [Google Scholar] [CrossRef]
- Moyer, S.A.; Funk, T.W. Air-Stable iron catalyse for the oppenauer type oxidation of alcohols. Tetrahedron Lett. 2010, 51, 5430–5433. [Google Scholar] [CrossRef]
- Johnson, T.C.; Clarkson, G.J.; Wills, M. (Cyclopentadienone)iron Shvo Complexes: Synthesis and applications to hydrogen transfer reactions. Organometallics 2011, 30, 1859–1868. [Google Scholar] [CrossRef]
- Plank, T.N.; Drake, J.L.; Kim, D.K.; Funk, T.W. Air-Stable, Nitrile-Ligated (Cyclopentadienone)iron Dicarbonyl Compounds as Transfer Reduction and Oxidation Catalysts. Adv. Synth. Catal. 2012, 354, 597–601. [Google Scholar] [CrossRef]
- Knölker, H.-J.; Heber, J. Transition Metal-Diene Complexes in Organic Synthesis, Part 18.1 Iron-Mediated [2 + 2 + 1] Cycloadditions of Diynes and Carbon Monoxide: Selective Demetalation Reactions. Synlett 1993, 12, 924–926. [Google Scholar] [CrossRef]
- Knölker, H.-J.; Baum, E.; Heber, J. Transition Metal-Diene Complexes in Organic Synthesis, Part 25.1 Cycloadditions of Annulated 2,5-Bis(trimethylsilyl)cyclopentadienones. Tetrahedron Lett. 1995, 36, 7647–7650. [Google Scholar] [CrossRef]
- Knölker, H. Trimethylamine N-Oxide—A useful oxidizing reagent. J. Für Prakt. Chem. 1996, 338, 190–192. [Google Scholar] [CrossRef]
- Knölker, H.-J.; Goesmann, H.; Klauss, R. A Novel Method for the Demetalation of Tricarbonyliron-Diene Complexes by a Photolytically Induced Ligand Exchange Reaction with Acetonitrile. Angew. Chem. Int. Ed. 1999, 38, 702–705. [Google Scholar] [CrossRef]
- Knölker, H.-J.; Baum, E.; Goesmann, H.; Klauss, R. Demetalation of Tricarbonyl(cyclopentadienone)iron Complexes Initiated by a Ligand Exchange Reaction with NaOH—X-Ray Analysis of a Complex with Nearly Square-Planar Coordinated Sodium. Angew. Chem. Int. Ed. 1999, 38, 2064–2066. [Google Scholar] [CrossRef]
- Abdel-Magid, A.F.; Mehrman, S.J. A Review on the Use of Sodium Triacetoxyborohydride in the Reductive Amination of Ketones and Aldehydes. Org. Process. Res. Dev. 2006, 10, 971–1031. [Google Scholar] [CrossRef]
- Nugent, T.C.; El-Shazly, M. Chiral amine synthesis—Recent developments and trends for enamide reduction, reductive amination, and imine reduction. Adv. Synth. Catal. 2010, 352, 753–819. [Google Scholar] [CrossRef]
- Alinezhad, H.; Yavari, H.; Salehian, F. Recent Advances in Reductive Amination Catalysis and Its Applications. Curr. Org. Chem. 2015, 19, 1021–1049. [Google Scholar] [CrossRef]
- Gusak, K.N.; Ignatovich, Z.V.; Koroleva, E.V. New potential of the reductive alkylation of amines. Russ. Chem. Rev. 2015, 84, 288–309. [Google Scholar] [CrossRef]
- Fleischer, S.; Zhou, S.; Junge, K.; Beller, M. An Easy and General Iron-catalyzed Reductive Amination of Aldehydes and Ketones with Anilines. Chem. Asian J. 2011, 6, 2240–2245. [Google Scholar] [CrossRef]
- Lator, A.; Gaillard, Q.G.; Mérel, D.S.; Lohier, J.-F.; Gaillard, S.; Poater, A.; Renaud, J.-L. Room-Temperature Chemoselective Reductive Alkylation of Amines Catalyzed by a Well-Defined Iron(II) Complex Using Hydrogen. J. Org. Chem. 2019, 84, 6813–6829. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | HCO2X | Fe | Temperature (°C) | Time (h) | Conv. (%) b |
| 1 | HCO2H | Fe4 | 100 | 24 | 0 |
| 2 | HCO2H/Et3N (1/1) | Fe4 | 100 | 24 | 100 |
| 3 | HCO2Na | Fe4 | 100 | 24 | 100 |
| 4 | HCO2K | Fe4 | 100 | 24 | 100 |
| 5 | HCO2Cs | Fe4 | 100 | 24 | 100 |
| 6 | - | Fe4 | 100 | 24 | 0 |
| 7 | HCO2Na | - | 100 | 24 | 0 |
| 8 | HCO2Na | Fe4 | 100 | 16 | 83 |
| 9 | HCO2Na | Fe4 | 80 | 24 | 100 (99%) c |
| 10 | HCO2Na | Fe3 | 80 | 24 | 0 |
| 11 | HCO2Na | Fe4 | 80 | 16 | 81 |
| 12 | HCO2Na | Fe4 | 60 | 24 | 75 |
| 13 d | HCO2Na | Fe4 | 80 | 24 | 53 |
| 14 e | HCO2Na | Fe4 | 80 | 24 | 86 |
| 15 | HCO2Na | Fe5 | 80 | 24 | 100 (98%) c |
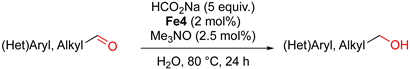 |
 |
 | |||||
|---|---|---|---|---|---|
| Entry | HCO2X (equiv.) | [Fe] | T (°C) | Conv. (%) b | Selectivity (5a)/(5a’) b |
| 1 | HCO2NH4 (5) | Fe4 | 90 | 93 | 77/23 |
| 2 | HCO2NH4 (5) | Fe5 | 90 | 95 | 77/23 |
| 3 | HCO2K (5) | Fe5 | 90 | 94 | 60/40 |
| 4 | HCO2Cs (5) | Fe5 | 90 | 93 | 40/60 |
| 5 | - | Fe5 | 90 | 100 | 0/100 |
| 6 | HCO2NH4 (5) | Fe5 | 85 | 91 | 67/33 |
| 7 | HCO2NH4 (5) | Fe5 | 80 | 83 | 69/31 |
| 8 | HCO2NH4 (5) | Fe5 | 40 | 80 | 52.5/47.5 |
| 9 | HCO2NH4 (6.5) | Fe5 | 90 | 96 | 91/9 (70) c |
 |
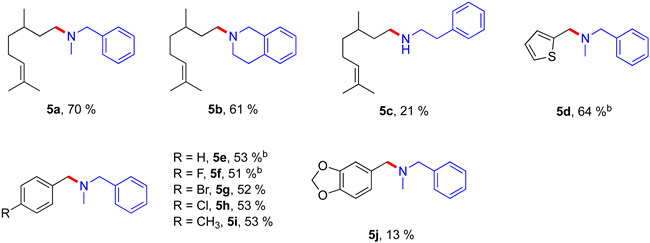 |
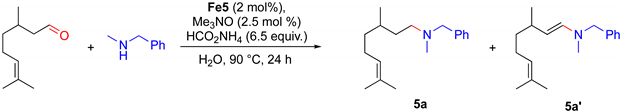 | |||
|---|---|---|---|
| Entry | Run | Conv. (%) c | Selectivity (5a)/(5a’) c |
| 1 | 1 | 96 | 91/9 |
| 2 | 2 | 95 | 96/4 |
| 3 | 3 | 95 | 96/4 |
| 4 | 4 | 96 | 97/3 |
| 5 | 5 | 98 | 96/4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ndiaye, D.; Coufourier, S.; Mbaye, M.D.; Gaillard, S.; Renaud, J.-L. Cyclopentadienone Iron Tricarbonyl Complexes-Catalyzed Hydrogen Transfer in Water. Molecules 2020, 25, 421. https://doi.org/10.3390/molecules25020421
Ndiaye D, Coufourier S, Mbaye MD, Gaillard S, Renaud J-L. Cyclopentadienone Iron Tricarbonyl Complexes-Catalyzed Hydrogen Transfer in Water. Molecules. 2020; 25(2):421. https://doi.org/10.3390/molecules25020421
Chicago/Turabian StyleNdiaye, Daouda, Sébastien Coufourier, Mbaye Diagne Mbaye, Sylvain Gaillard, and Jean-Luc Renaud. 2020. "Cyclopentadienone Iron Tricarbonyl Complexes-Catalyzed Hydrogen Transfer in Water" Molecules 25, no. 2: 421. https://doi.org/10.3390/molecules25020421
APA StyleNdiaye, D., Coufourier, S., Mbaye, M. D., Gaillard, S., & Renaud, J.-L. (2020). Cyclopentadienone Iron Tricarbonyl Complexes-Catalyzed Hydrogen Transfer in Water. Molecules, 25(2), 421. https://doi.org/10.3390/molecules25020421