Synthesis of Elaborate Benzofuran-2-Carboxamide Derivatives through a Combination of 8-Aminoquinoline Directed C–H Arylation and Transamidation Chemistry



Abstract

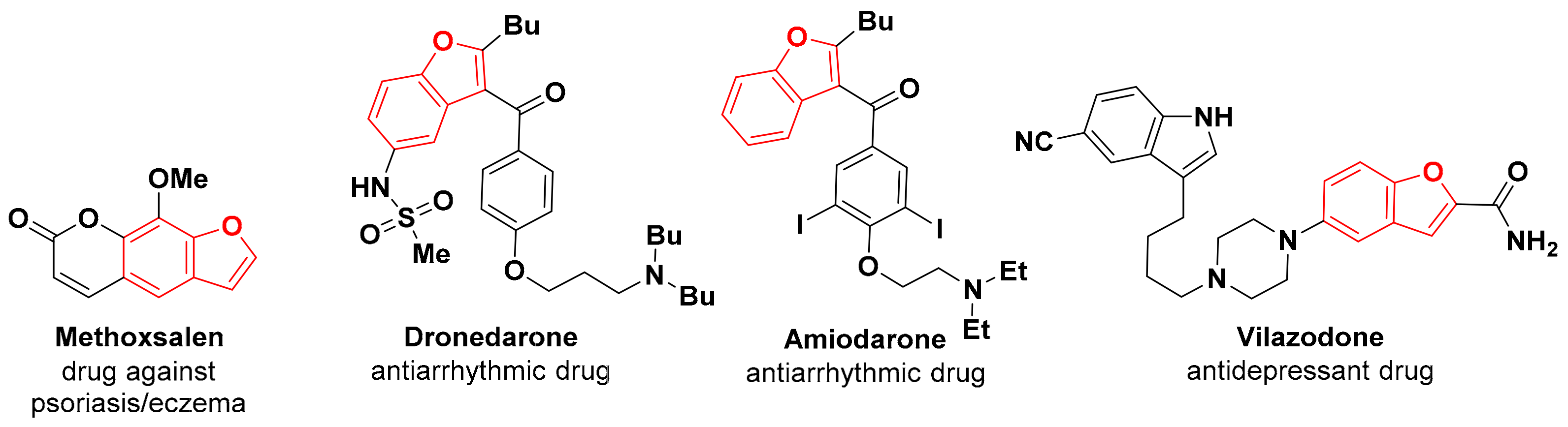
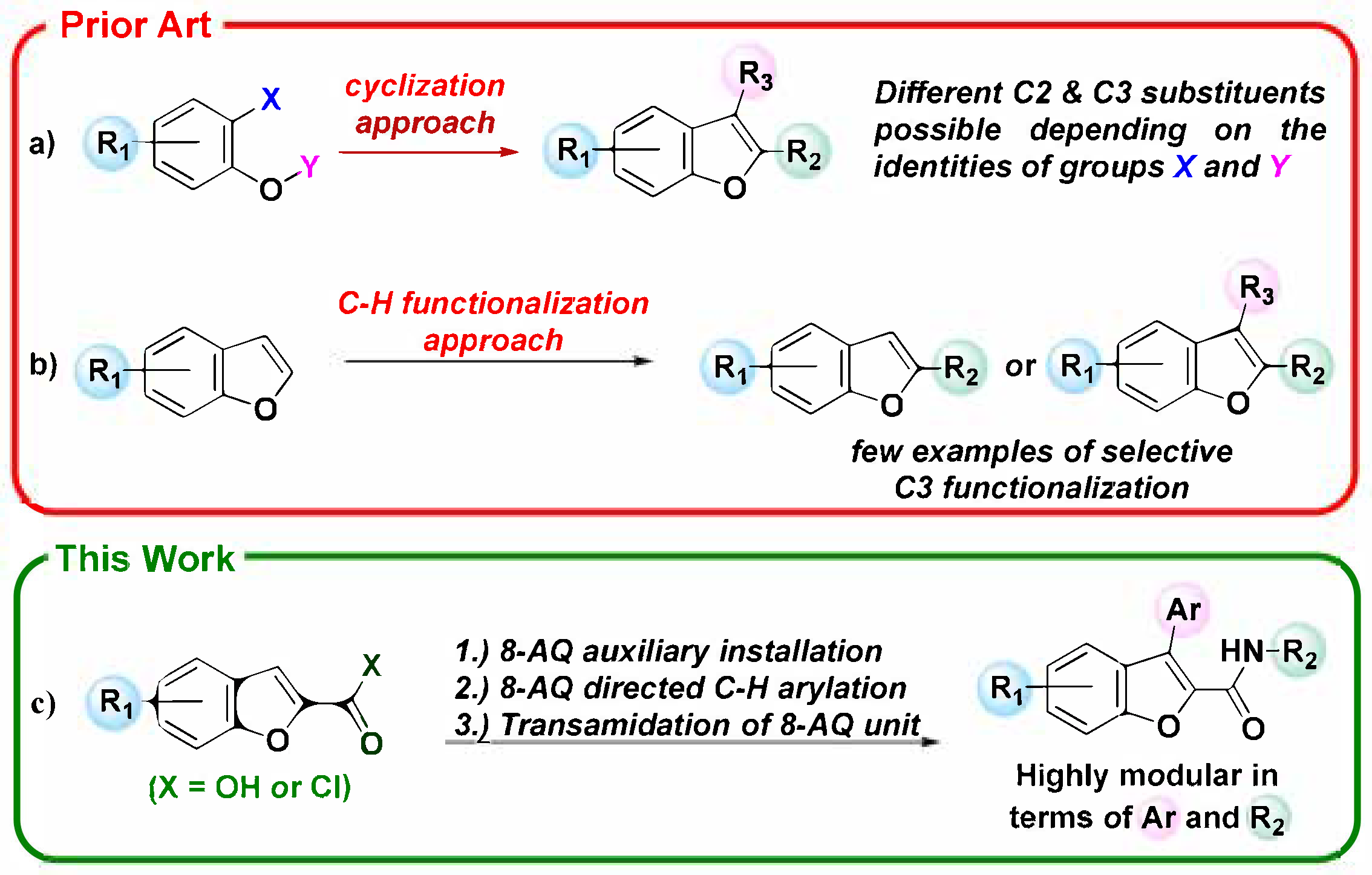
1. Introduction
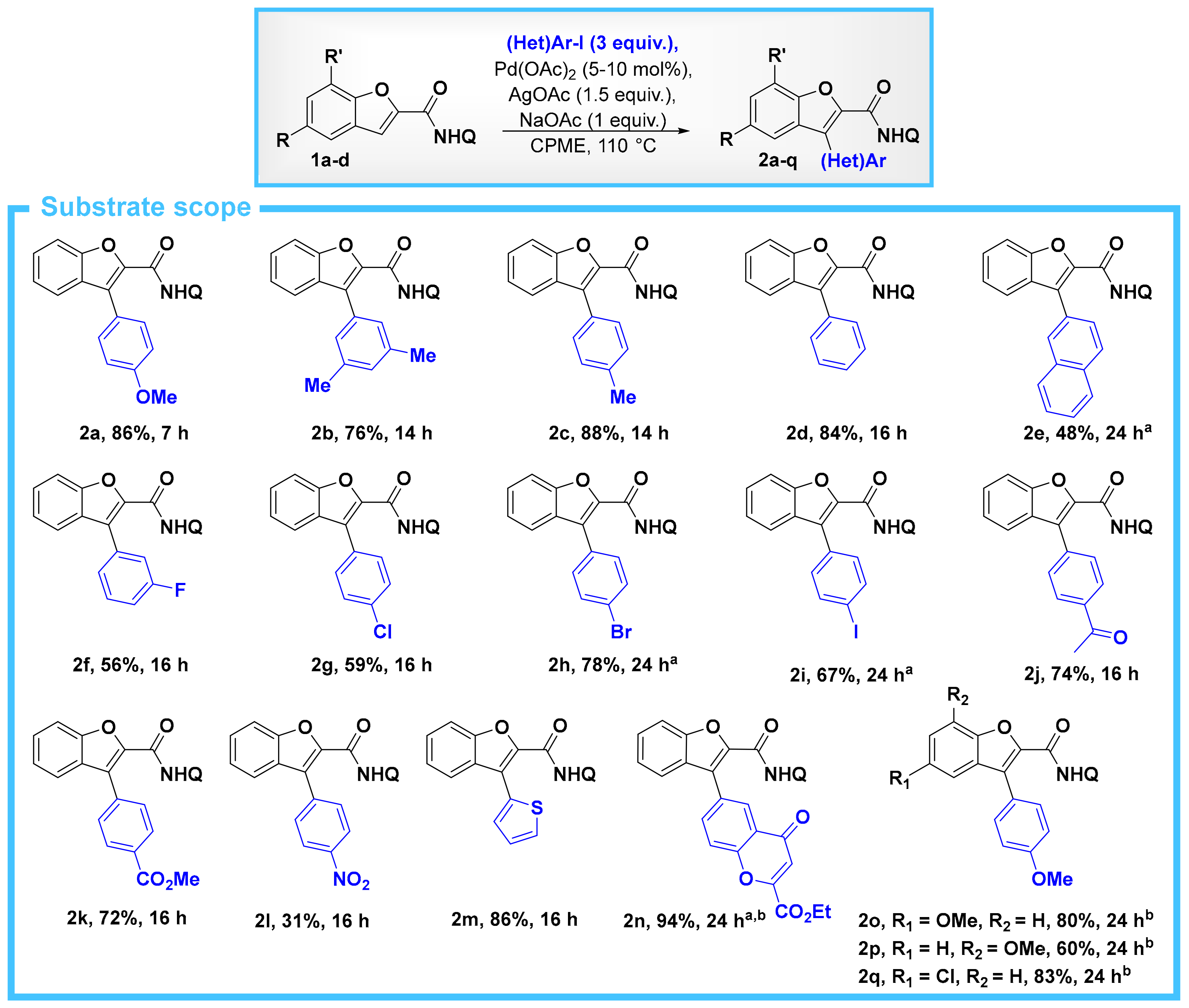
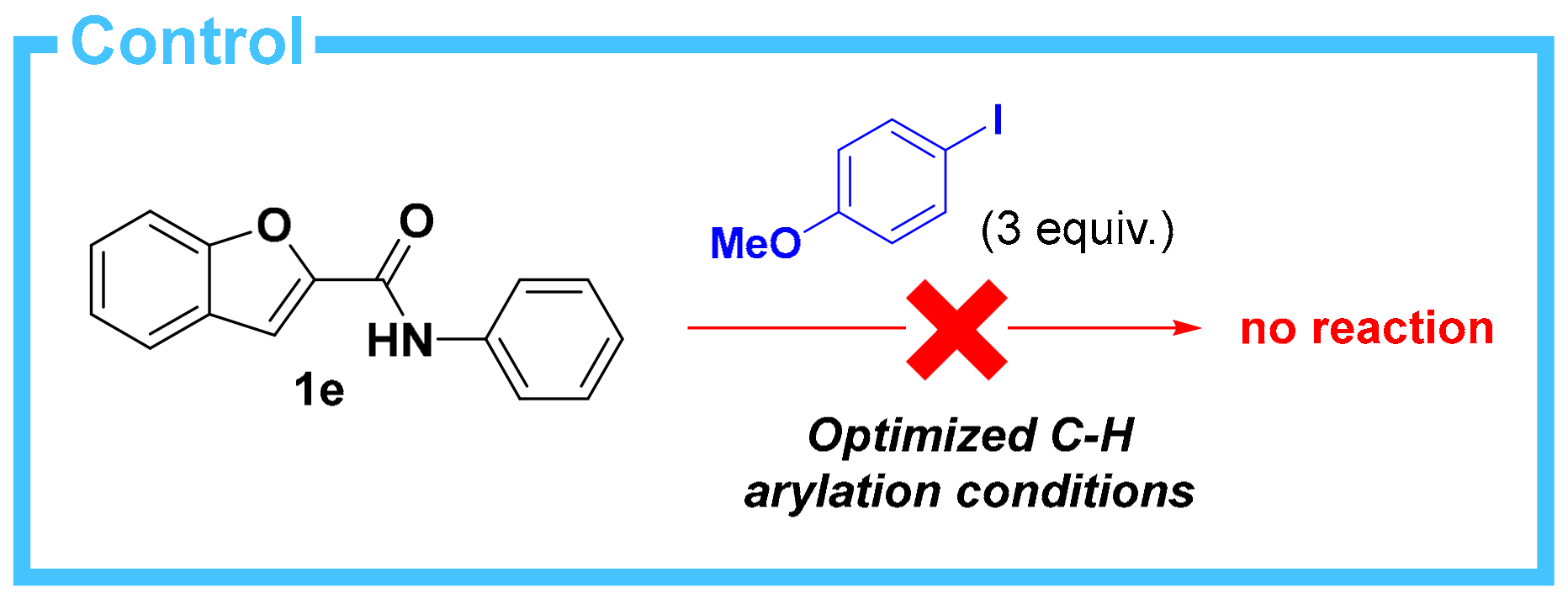
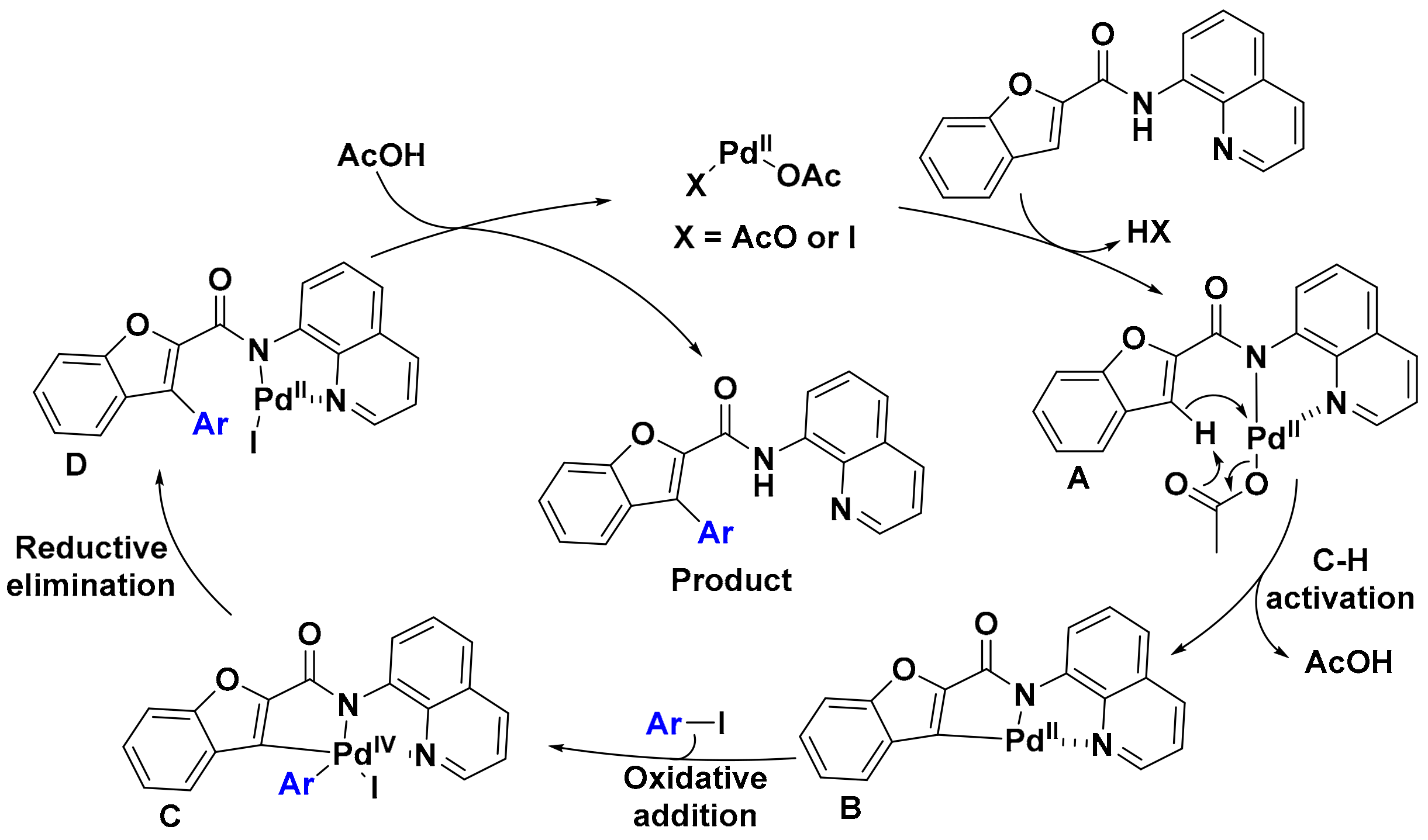
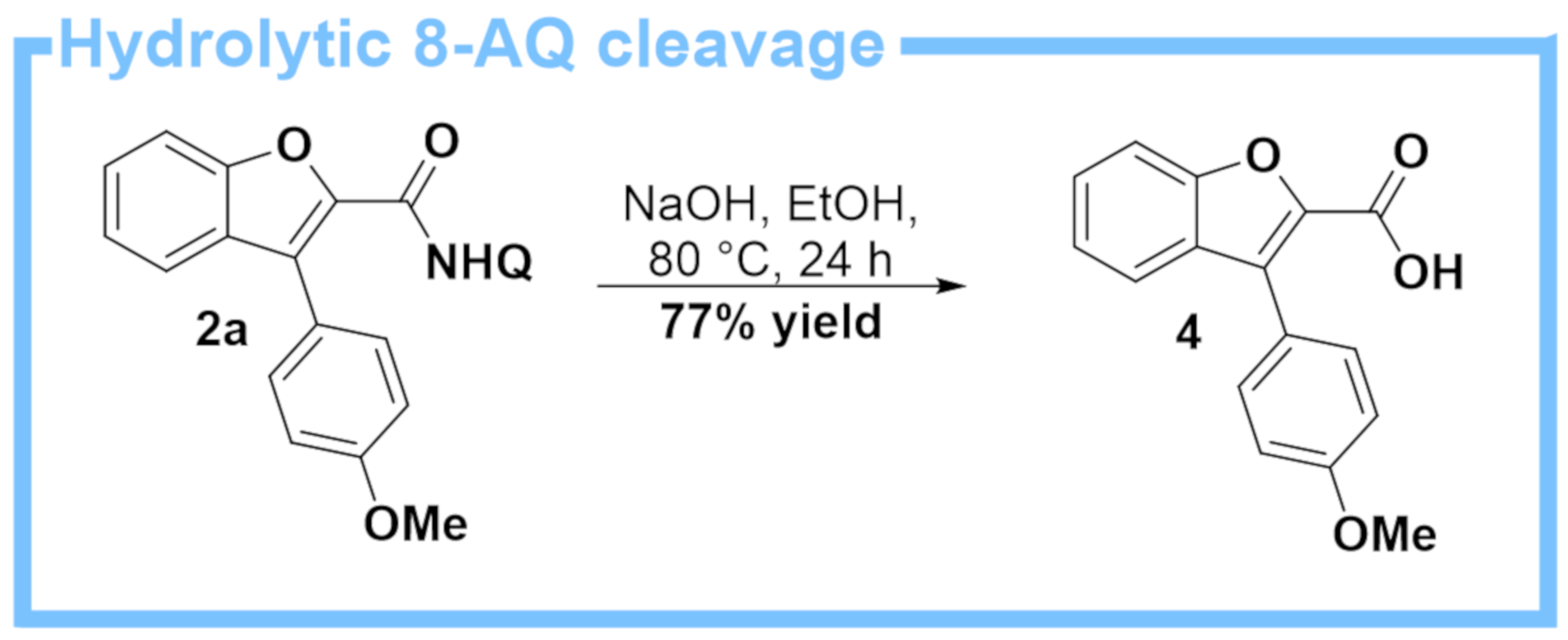
2. Results
3. Discussion
4. Materials and Methods
4.1. General Procedure for the C–H Arylation of Substrates 1a–e
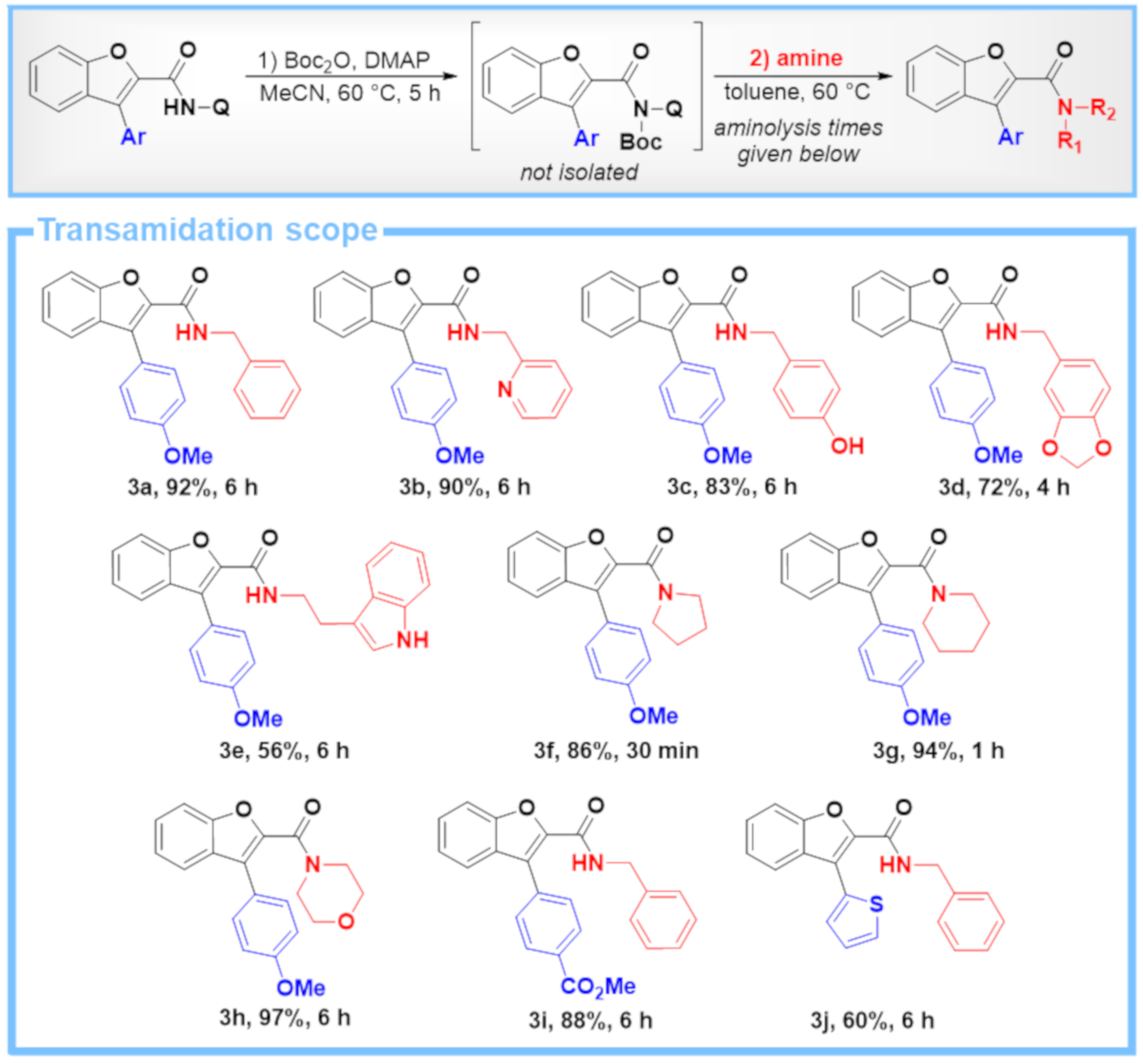
4.2. General Procedure C: Two-Step One-Pot Transamidation of C–H Arylation Products with Different Amines
4.2.1. Boc Activation
4.2.2. Aminolysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References and Notes
- Radadiya, A.; Shah, A. Bioactive benzofuran derivatives: An insight on lead developments, radioligands and advances of the last decade. Eur. J. Med. Chem. 2015, 97, 356–376. [Google Scholar] [CrossRef] [PubMed]
- Hiremathad, A.; Patil, M.R.; Chethana, K.R.; Chand, K.; Amelia Santos, M.; Keri, R.S. Benzofuran: An emerging scaffold for antimicrobial agents. RSC Adv. 2015, 5, 96809–96828. [Google Scholar] [CrossRef]
- Khanam, H. Shamsuzzaman Bioactive Benzofuran derivatives: A review. Eur. J. Med. Chem. 2015, 97, 483–504. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.D.; MacCoss, M.; Lawson, A.D. Rings in drugs. J. Med. Chem. 2014, 57, 5845–5859. [Google Scholar] [CrossRef] [PubMed]
- Khodarahmi, G.; Asadi, P.; Hazzanzadeh, F.; Khodarahmi, E. Benzofuran as a promising scaffold for the synthesis of antimicrobial and antibreast cancer agents: A review. J. Res. Med. Sci. 2015, 20, 1094–1104. [Google Scholar] [CrossRef] [PubMed]
- Salomé, C.; Ribeiro, N.; Chavagnan, T.; Thuaud, F.; Serova, M.; de Gramont, A.; Faivre, S.; Raymond, E.; Désaubry, L. Benzofuran derivatives as anticancer inhibitors of mTOR signaling. Eur. J. Med. Chem. 2014, 81, 181–191. [Google Scholar] [CrossRef]
- Abdelhafez, O.M.; Amin, K.M.; Ali, H.I.; Abdalla, M.M.; Ahmed, E.Y. Design, synthesis and anticancer activity of benzofuran derivatives targeting VEGFR-2 tyrosine kinase. RSC Adv. 2014, 4, 11569–11579. [Google Scholar] [CrossRef]
- More, K.R. Review on Synthetic Routes for Synthesis of Benzofuran-Based Compounds. J. Chem. Pharm. Res. 2017, 9, 210–220. [Google Scholar]
- Heravi, M.M.; Zadsirjan, V. Chapter Five-Recent Advances in the Synthesis of Benzo[b]furans. Adv. Heterocycl. Chem. 2015, 117, 261–376. [Google Scholar]
- Abu-Hashem, A.A.; Hussein, H.A.R.; Aly, A.S.; Gouda, M.A. Synthesis of Benzofuran Derivatives via Different Methods. Synth. Commun. 2014, 44, 2285–2312. [Google Scholar] [CrossRef]
- Huang, W.; Xu, J.; Liu, C.; Chen, Z.; Gu, Y. Lewis Acid-Catalyzed Synthesis of Benzofurans and 4,5,6,7-Tetrahydrobenzofurans from Acrolein Dimer and 1,3-Dicarbonyl Compounds. J. Org. Chem. 2019, 84, 2941–2950. [Google Scholar] [CrossRef] [PubMed]
- Merkushev, A.A.; Strelnikov, V.N.; Uchuskin, M.G.; Trushnov, I.V. A simple synthesis of benzofurans by acid-catalyzed domino reaction of salicyl alcohols with N-tosylfurfurylamine. Tetrahedron 2017, 73, 6523–6529. [Google Scholar] [CrossRef]
- Warner, A.J.; Churn, A.; McGough, J.S.; Ingleson, M.J. BCl3-Induced Annulative Oxo- and Thioboration for the Formation of C3-Borylated Benzofurans and Benzothiophenes. Angew. Chem. Int. Ed. 2017, 56, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Bankar, S.K.; Mathew, J.; Ramasastry, S.S.V. Synthesis of benzofurans via an acid catalysed transacetalisation/Fries-type O → C rearrangement/Michael addition/ring-opening aromatisation cascade of β-pyrones. Chem. Commun. 2016, 52, 5569–5572. [Google Scholar] [CrossRef] [PubMed]
- Contiero, F.; Jones, K.M.; Matts, E.A.; Porzelle, A.; Tomkinson, N.C.O. Direct Preparation of Benzofurans from O-Arylhydroxylamines. Synlett 2009, 3003–3006. [Google Scholar]
- Witczak, M.; Kwiecien, H. Efficient Cyclization of 2-Phenoxyalkanals to 2-Alkylbenzo[b]furans. Synth. Commun. 2005, 35, 2223–2230. [Google Scholar] [CrossRef]
- Ishibashi, K.; Nakajima, K.; Sugioka, Y.; Sugiyama, M.; Hamata, T.; Horikoshi, H.; Nishi, T. Synthesis of 2-Phenylbenzofuran Derivatives as Testosterone 5α-Reductase Inhibitor. Chem. Pharm. Bull. 1999, 47, 226–240. [Google Scholar] [CrossRef][Green Version]
- Bruneau, A.; Gustafson, K.P.J.; Yuan, N.; Tai, C.-W.; Persson, I.; Zou, X.; Backvall, J.E. Synthesis of Benzofurans and Indoles from Terminal Alkynes and Iodoaromatics Catalyzed by Recyclable Palladium Nanoparticles Immobilized on Siliceous Mesocellular Foam. Chem. Eur. J. 2017, 23, 12886–12891. [Google Scholar] [CrossRef]
- Bosiak, M.J. A Convenient Synthesis of 2-Arylbenzo[b]furans from Aryl Halides and 2-Halophenols by Catalytic One-Pot Cascade Method. ACS Catal. 2016, 6, 2429–2434. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Akiyama, T.; Sasou, H.; Katsumata, H.; Manabe, K. One-Pot Synthesis of Substituted Benzo[b]furans and Indoles from Dichlorophenols/Dichloroanilines Using a Palladium-Dihydroxyterphenylphosphine Catalyst. J. Org. Chem. 2016, 81, 5450–5463. [Google Scholar] [CrossRef]
- Bochicchio, A.; Chiummiento, L.; Funicello, M.; Lopardo, M.T.; Lupattelli, P. Efficient synthesis of 5-nitro-benzo[b]furans via 2-bromo-4-nitro-phenyl acetates. Tetrahedron Lett. 2010, 51, 2824–2827. [Google Scholar] [CrossRef]
- Lingam, V.S.P.R.; Vinodkumar, R.; Mukkanti, K.; Thomas, A.; Gopalan, B. A simple approach to highly functionalized benzo[b]furans from phenols and aryl iodides via aryl propargyl ethers. Tetrahedron Lett. 2008, 49, 4260–4264. [Google Scholar] [CrossRef]
- Bernini, R.; Cacchi, S.; De Salve, I.; Fabrizi, G. Palladium-Catalyzed Synthesis of Lipophilic Benzo[b]furans from Cardanol. Synthesis 2007, 6, 873–882. [Google Scholar] [CrossRef]
- Kabalka, G.W.; Wang, L.; Pagni, R.M. Sonogashira coupling and cyclization reactions on alumina: A route to aryl alkynes, 2-substituted-benzo[b]furans and 2-substituted-indoles. Tetrahedron 2001, 57, 8017–8028. [Google Scholar] [CrossRef]
- More, K.R.; Mali, R.S. An alternate method for the synthesis of 2-aryl/alkyl-5-bromo-7methoxy benzofurans; application to the synthesis of Egonol, Homoegonol, and analogs via Heck reaction. Tetrahedron 2016, 72, 7496–7504. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar Gangwar, M.; Prakasham, A.P.; Mhatre, D.; Kalita, A.C.; Ghosh, P. Accessing a Biologically Relevant Benzofuran Skeleton by a One-Pot Tandem Heck Alkynylation/Cyclization Reaction Using Well-Defined Palladium N-Heterocyclic Carbene Complexes. Inorg. Chem. 2016, 55, 2882–2893. [Google Scholar] [CrossRef]
- Gao, Y.; Xiong, W.; Chen, H.; Wu, W.; Peng, J.; Gao, Y.; Jiang, H. Pd-Catalyzed Highly Regio- and Stereoselective Formation of C-C Double Bonds: An Efficient Method for the Synthesis of Benzofuran-, Dihydrobenzofuran-, and Indoline-Containing Alkenes. J. Org. Chem. 2015, 80, 7456–7467. [Google Scholar] [CrossRef]
- Yuan, H.; Bi, K.J.; Li, B.; Yue, R.C.; Ye, J.; Shen, Y.H.; Shan, L.; Jin, H.Z.; Sun, Q.Y.; Zhang, W.D. Construction of 2-Substituted-3-Functionalized Benzofurans via Intramolecular Heck Coupling: Application to Enantioselective Total Synthesis of Daphnodorin. B. Org. Lett. 2013, 15, 4742–4745. [Google Scholar] [CrossRef]
- Heravi, M.M.; Fazeli, A. Recent Advances in the Application of the Heck Reaction in the Synthesis of Heterocyclic Compounds. Heterocycles 2010, 81, 1979–2026. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.; Wang, T.; Yao, X.; Wang, P.; Wang, P.; Jing, S.; Liang, Y.; Zhang, Z. Oxidant and Transition-Metal-Free Photoinduced Direct Oxidative Annulation of 1-Aryl-2-(furan/thiophen-2-yl)butane-1,3-diones. J. Org. Chem. 2017, 82, 12097–12105. [Google Scholar] [CrossRef]
- Xia, Z.; Khaled, O.; Mouries-Mansuy, V.; Ollivier, C.; Fensterbank, L. Dual Photoredox/Gold Catalysis Arylative Cyclization of o-Alkynylphenols with Aryldiazonium Salts: A Flexible Synthesis of Benzofurans. J. Org. Chem. 2016, 81, 7182–7190. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Das, J. A novel photochemical Wittig reaction for the synthesis of 2-aryl/alkylbenzofurans. Tetrahedron Lett. 2011, 52, 1112–1116. [Google Scholar] [CrossRef]
- Sumanthi, T.; Balasubramanian, K.K. A photochemical route to 2-alkenyl- and 2-ethynylbenzofurans. Tetrahedron Lett. 1990, 31, 3775–3778. [Google Scholar] [CrossRef]
- Pandey, G.; Krishna, A.; Bhalerao, U.T. A one step synthesis of 2-substituted benzofurans from 2-aryl-1-substituted ethane-1-ones by photoinduced set reactions. Tetrahedron Lett. 1989, 30, 1867–1870. [Google Scholar] [CrossRef]
- Deng, G.; Li, M.; Yu, K.; Liu, C.; Liu, Z.; Duan, S.; Chen, W.; Yang, X.; Zhang, H.; Walsh, P.J. Synthesis of Benzofuran Derivatives through Cascade Radical Cyclization/Intermolecular Coupling of 2-Azaallyls. Angew. Chem. Int. Ed. 2019, 58, 2826–2830. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cheng, S.; Cai, Z.; Liu, P.; Sun, P. Radical Addition Cascade Cyclization of 1,6-Enynes with DMSO To Access Methylsulfonylated and Carbonylated Benzofurans under Transition-Metal-Free Conditions. J. Org. Chem. 2018, 83, 9344–9352. [Google Scholar] [CrossRef]
- Zheng, H.X.; Shan, X.H.; Qu, J.P.; Kang, Y.B. Strategy for Overcoming Full Reversibility of Intermolecular Radical Addition to Aldehydes: Tandem C-H and C-O Bonds Cleaving Cyclization of (Phenoxymethyl)arenes with Carbonyls to Benzofurans. Org. Lett. 2018, 20, 3310–3313. [Google Scholar] [CrossRef]
- Rueping, M.; Leiendecker, M.; Das, A.; Poisson, T.; Bui, L. Potassium tert-butoxide mediated Heck-type cyclization/isomerization–benzofurans from organocatalytic radical cross-coupling reactions. Chem. Commun. 2011, 47, 10629–10631. [Google Scholar] [CrossRef]
- Beckwith, A.L.J.; Meijs, G.F. Formation of dihydrobenzofurans by radical cyclization. J. Chem. Soc. Chem. Comm. 1981, 136–137. [Google Scholar] [CrossRef]
- Yi, W.; Chen, W.; Liu, F.X.; Zhong, Y.; Wu, D.; Zhou, Z.; Gao, H. Rh(III)-Catalyzed and Solvent-Controlled Chemoselective Synthesis of Chalcone and Benzofuran Frameworks via Synergistic Dual Directing Groups Enabled Regioselective C–H Functionalization: A Combined Experimental and Computational Study. ACS Catal. 2018, 8, 9508–9519. [Google Scholar] [CrossRef]
- Ichake, S.S.; Konala, A.; Kavala, V.; Kuo, C.W.; Yao, C.F. Palladium-Catalyzed Tandem C-H Functionalization/Cyclization Strategy for the Synthesis of 5-Hydroxybenzofuran Derivatives. Org. Lett. 2017, 19, 54–57. [Google Scholar] [CrossRef]
- Agasti, S.; Sharma, U.; Naveen, T.; Maiti, D. Orthogonal selectivity with cinnamic acids in 3-substituted benzofuran synthesis through C–H olefination of phenols. Chem. Commun. 2015, 51, 5375–5378. [Google Scholar] [CrossRef] [PubMed]
- Agasti, S.; Maity, S.; Szabo, K.J.; Maiti, D. Palladium-Catalyzed Synthesis of 2,3-Disubstituted Benzofurans: An Approach Towards the Synthesis of Deuterium Labeled Compounds. Adv. Synth. Catal. 2015, 357, 2331–2338. [Google Scholar] [CrossRef] [PubMed]
- Rajesh, M.; Thirupathi, N.; Reddy, T.J.; Kanojiya, S.; Sridhar, M. Pd-Catalyzed Isocyanide Assisted Reductive Cyclization of 1-(2-Hydroxyphenyl)-propargyl Alcohols for 2-Alkyl/Benzyl Benzofurans and Their Useful Oxidative Derivatization. J. Org. Chem. 2015, 80, 12311–12320. [Google Scholar] [CrossRef] [PubMed]
- Sharma, U.; Naveen, T.; Maji, A.; Manna, S.; Maiti, D. Palladium-Catalyzed Synthesis of Benzofurans and Coumarins from Phenols and Olefins. Angew. Chem. Int. Ed. 2013, 52, 12669–12673. [Google Scholar] [CrossRef]
- Xu, G.; Liu, K.; Sun, J. Gold-Catalyzed Controllable C2-Functionalization of Benzofurans with Aryl Diazoesters. Org. Lett. 2018, 20, 72–75. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Jie, K.; Huang, L.; Guo, S.; Cai, H. Copper-Catalyzed C2 and C3 Phosphonation of Benzofuran and Benzothiophene with Trialkyl Phosphites. ChemCatChem 2018, 10, 716–719. [Google Scholar] [CrossRef]
- Fang, H.; Guo, L.; Zhang, Y.; Yao, W.; Huang, Z. A Pincer Ruthenium Complex for Regioselective C–H Silylation of Heteroarenes. Org. Lett. 2016, 18, 5624–5627. [Google Scholar] [CrossRef]
- Paymode, D.J.; Ramana, C.V. Ruthenium(II)-Catalyzed C3 Arylation of 2-Aroylbenzofurans with Arylboronic Acids/Aryltrifluoroborates via Carbonyl-Directed C–H Bond Activation. J. Org. Chem. 2015, 80, 11551–11558. [Google Scholar] [CrossRef]
- Schramm, Y.; Takeuchi, M.; Semba, K.; Nakao, Y.; Hartwig, J.F. Anti-Markovnikov Hydroheteroarylation of Unactivated Alkenes with Indoles, Pyrroles, Benzofurans, and Furans Catalyzed by a Nickel–N-Heterocyclic Carbene System. J. Am. Chem. Soc. 2015, 137, 12215–12218. [Google Scholar] [CrossRef]
- Yin, S.-C.; Zhou, Q.; Zhao, X.-Y.; Shao, L.-X. N-Heterocyclic Carbene-Palladium(II)-1-Methylimidazole Complex Catalyzed Direct C–H Bond Arylation of Benzo[b]furans with Aryl Chlorides. J. Org. Chem. 2015, 80, 8916–8921. [Google Scholar] [CrossRef] [PubMed]
- Loukotova, L.; Yuan, K.; Doucet, H. Regiocontroled Palladium-Catalysed Direct Arylation at Carbon C2 of Benzofurans using Benzenesulfonyl Chlorides as the Coupling Partners. ChemCatChem 2014, 6, 1303–1309. [Google Scholar] [CrossRef]
- Carrer, A.; Brinet, D.; Florent, J.-C.; Rousselle, P.; Bertounesque, E. Palladium-Catalyzed Direct Arylation of Polysubstituted Benzofurans. J. Org. Chem. 2012, 77, 1316–1327. [Google Scholar] [CrossRef]
- Dao-Huy, T.; Haider, M.; Glatz, F.; Schnürch, M.; Mihovilovic, M.D. Direct Arylation of Benzo[b]furan and Other Benzo-Fused Heterocycles. Eur. J. Org. Chem. 2014, 8119–8125. [Google Scholar] [CrossRef] [PubMed]
- Liégault, B.; Petrov, I.; Gorelsky, S.I.; Fagnou, K. Modulating Reactivity and Diverting Selectivity in Palladium-Catalyzed Heteroaromatic Direct Arylation Through the Use of a Chloride Activating/Blocking Group. J. Org. Chem. 2010, 75, 1047–1060. [Google Scholar] [CrossRef] [PubMed]
- Ionita, M.; Roger, J.; Doucet, H. Palladium Catalyzed Direct 3-Arylation of Benzofurans using Low Catalyst Loadings. ChemSusChem 2010, 3, 367–376. [Google Scholar] [CrossRef]
- Larbi, K.S.; Djebbar, S.; Soulé, J.-F.; Doucet, H. Reactivity of benzofuran and benzothiophene in palladium-catalysed direct C2,C3-diarylations. J. Organomet. Chem. 2017, 843, 32–39. [Google Scholar] [CrossRef]
- Shen, K.; Fu, Y.; Li, J.-N.; Liu, L.; Guo, Q.-X. What are the pKa values of C–H bonds in aromatic heterocyclic compounds in DMSO? Tetrahedron 2007, 63, 1568–1576. [Google Scholar] [CrossRef]
- Verho, O.; Pourghasemi Lati, M.; Oschmann, M. A Two-Step Procedure for the Overall Transamidation of 8-Aminoquinoline Amides Proceeding via the Intermediate N-Acyl-Boc-Carbamates. J. Org. Chem. 2018, 83, 4464–4476. [Google Scholar] [CrossRef]
- Zaitsev, V.G.; Shabashov, D.; Daugulis, O. Highly Regioselective Arylation of sp3 C-H Bonds Catalyzed by Palladium Acetate. J. Am. Chem. Soc. 2005, 127, 13154–13155. [Google Scholar] [CrossRef]
- Shabashov, D.; Daugulis, O. Auxiliary-Assisted Palladium-Catalyzed Arylation and Alkylation of sp2 and sp3 Carbon-Hydrogen Bonds. J. Am. Chem. Soc. 2010, 132, 3965–3972. [Google Scholar] [CrossRef] [PubMed]
- Daugulis, O.; Roane, J.; Tran, L.D. Bidentate, Monoanionic Auxiliary-Directed Functionalization of Carbon-Hydrogen Bonds. Acc. Chem. Res. 2015, 48, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, A.J.; Ricke, A.; Oschmann, M.; Verho, O. Convenient Access to Chiral Cyclobutanes with Three Contiguous Stereocenters from Verbenone by Directed C(sp3)−H arylation. Chem. Eur. J. 2019, 25, 5154–5157. [Google Scholar] [CrossRef] [PubMed]
- Antermite, D.; Affron, D.P.; Bull, J.A. Regio- and Stereoselective Palladium-Catalyzed C(sp3)–H Arylation of Pyrrolidines and Piperidines with C(3) Directing Groups. Org. Lett. 2018, 20, 3948–3952. [Google Scholar] [CrossRef] [PubMed]
- Melillo, B.; Zoller, J.; Hua, B.K.; Verho, O.; Borghs, J.C.; Nelson, S.D., Jr.; Maetani, M.; Wawer, M.J.; Clemons, P.A.; Schreiber, S.L. Synergistic Effects of Stereochemistry and Appendages on the Performance Diversity of a Collection of Synthetic Compounds. J. Am. Chem. Soc. 2018, 140, 11784–11790. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-J.; Sun, W.-W.; Yu, Q.-Y.; Cao, P.; Dong, X.-P.; Wu, B. Stereoselective synthesis of (−)-3-PPP through palladium-catalysed unactivated C(sp3)–H arylation at the C-3 position of l-pipecolinic acid. Tetrahedron Lett. 2017, 58, 606–609. [Google Scholar]
- Maetani, M.; Zoller, J.; Melillo, B.; Verho, O.; Kato, N.; Pu, J.; Comer, E.; Schreiber, S.L. Synthesis of a Bicyclic Azetidine with In Vivo Antimalarial Activity Enabled by Stereospecific, Directed C(sp3)–H Arylation. J. Am. Chem. Soc. 2017, 139, 11300–11306. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, Y.; Wang, Z.; Zeng, T.; Liu, P.; Engle, K.M. Catalytic Intermolecular Carboamination of Unactivated Alkenes via Directed Aminopalladation. J. Am. Chem. Soc. 2017, 139, 11261–11270. [Google Scholar] [CrossRef]
- Verho, O.; Maetani, M.; Melillo, B.; Zoller, J.; Schreiber, S.L. Stereospecific Palladium-Catalyzed C–H Arylation of Pyroglutamic Acid Derivatives at the C3 Position Enabled by 8-Aminoquinoline as a Directing Group. Org. Lett. 2017, 19, 4424–4427. [Google Scholar] [CrossRef]
- Affron, D.P.; Davis, O.A.; Bull, J.A. Regio- and Stereospecific Synthesis of C-3 Functionalized Proline Derivatives by Palladium Catalyzed Directed C(sp3)–H Arylation. Org. Lett. 2014, 16, 4956–4959. [Google Scholar] [CrossRef]
- Beck, J.C.; Lacker, C.R.; Chapman, L.M.; Reisman, S.E. A modular approach to prepare enantioenriched cyclobutanes: Synthesis of (+)-rumphellaone A. Chem. Sci. 2019, 10, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Chapman, L.M.; Beck, J.C.; Lacker, C.R.; Wu, L.; Reisman, S.E. Evolution of a Strategy for the Enantioselective Total Synthesis of (+)-Psiguadial, B. J. Org. Chem. 2018, 83, 6066–6085. [Google Scholar] [CrossRef] [PubMed]
- Chapman, L.M.; Beck, J.C.; Wu, L.; Reisman, S.E. Enantioselective Total Synthesis of (+)-Psiguadial, B. J. Am. Chem. Soc. 2016, 138, 9803–9806. [Google Scholar] [CrossRef] [PubMed]
- Gutekunst, W.R.; Baran, P.S. Applications of C–H Functionalization Logic to Cyclobutane Synthesis. J. Org. Chem. 2014, 79, 2430–2452. [Google Scholar] [CrossRef]
- Gutekunst, W.R.; Gianatassio, R.; Baran, P.S. Sequential Csp3–H Arylation and Olefination: Total Synthesis of the Proposed Structure of Pipercyclobutanamide A. Angew. Chem. Int. Ed. 2012, 51, 7507–7510. [Google Scholar] [CrossRef]
- Frébault, F.; Maulide, N. Total Synthesis and Structural Revision of the Piperarborenines: When Photochemistry Meets C–H Activation. Angew. Chem. Int. Ed. 2012, 51, 2815–2817. [Google Scholar] [CrossRef]
- Gutekunst, W.R.; Baran, P.S. Total Synthesis and Structural Revision of the Piperarborenines via Sequential Cyclobutane C–H Arylation. J. Am. Chem. Soc. 2011, 133, 19076–19079. [Google Scholar] [CrossRef]
- Wu, W.; Yi, J.; Xu, H.; Li, S.; Yuan, R. An Efficient, One-Pot Transamidation of 8-Aminoquinoline Amides Activated by Tertiary-Butyloxycarbonyl. Molecules 2019, 24, 1234. [Google Scholar] [CrossRef]
- Li, G.; Szostak, M. Highly selective transition-metal-free transamidation of amides and amidation of esters at room temperature. Nat. Commun. 2018, 9, 4165. [Google Scholar] [CrossRef]
- Liu, Y.; Shi, S.; Achtenhagen, M.; Liu, R.; Szostak, M. Metal-Free Transamidation of Secondary Amides via Selective N–C Cleavage under Mild Conditions. Org. Lett. 2017, 19, 1614–1617. [Google Scholar] [CrossRef]
- The corresponding acid chloride, benzofuran-2-carbonyl chloride, is also commercially-available to a reasonable cost, and of course it is also possible to use this for the synthesis of 1a. Starting from this acid chloride, we were able to prepare 1a in 97% yield (see SM for experimental details).
- Padmavathi, R.; Sankar, R.; Gopalakrishnan, B.; Parella, R.; Babu, S.A. Pd(OAc)2/AgOAc Catalytic System Based Bidentate Ligand Directed Regiocontrolled C–H Arylation and Alkylation of the C-3 Position of Thiophene- and Furan-2-carboxamides. Eur. J. Org. Chem. 2015, 3727–3742. [Google Scholar] [CrossRef]
- Attempts to purify these products by column chromatography (with either pentane/EtOAc or pentane/CH2Cl2) resulted in poor material recovery and low yields.
- Rit, R.K.; Yadav, R.; Ghosh, K.; Sahoo, A.K. Reusable directing groups [8-aminoquinoline, picolianamide, sulfoximine] in C(sp3)-H bond activation: Present and future. Tetrahedron 2015, 71, 4450–4459. [Google Scholar] [CrossRef]
- Arroniz, C.; Denis, J.G.; Ironmonger, A.; Rassias, G.; Larrosa, I. An organic cation as a silver(I) analogue for the arylation of sp2 and sp3 C-H bonds with iodoarenes. Chem. Sci. 2014, 5, 3509–3514. [Google Scholar] [CrossRef]
- Daugulis, O.; Zaitsev, V.G. Anilide ortho-arylation by using C-H activation methodology. Angew. Chem. Int. Ed. 2005, 44, 4046–4048. [Google Scholar] [CrossRef]
- Pereira, K.C.; Porter, A.L.; Potavathri, S.; LeBris, A.P.; DeBoef, B. Insight into the palladium-catalyzed oxidative arylation of benzofuran: Heteropoly acid oxidants evoke a Pd(II)/Pd(IV) mechanism. Tetrahedron 2013, 69, 4429–4435. [Google Scholar] [CrossRef]
- Bhaskararao, B.; Singh, S.; Anand, M.; Verma, P.; Prakash, P.; Athira, C.; Malakar, S.; Schaefer, H.F.; Sunoj, R.B. Is silver a mere terminal oxidant in palladium catalyzed C-H bond activation reactions? Chem. Sci. 2020, 11, 208–216. [Google Scholar] [CrossRef]
- Guin, S.; Dolui, P.; Zhang, X.; Paul, S.; Singh, V.K.; Pradhan, S.; Chandrashekar, H.B.; Anjana, S.S.; Paton, R.S.; Maiti, D. Iterative Arylation of Amino Acids and Aliphatic Amines via δ-C(sp3)−H Activation: Experimental and Computational Exploration. Angew. Chem. Int. Ed. 2019, 58, 5633–5638. [Google Scholar] [CrossRef]
- Mudarra, Á.L.; de Salinas, S.M.; Pérez-Temprano, M. Beyond the traditional roles of Ag in catalysis: Transmetalating ability of organosilver(I) species in Pd-catalysed reactions. Org. Biomol. Chem. 2019, 17, 1655–1667. [Google Scholar] [CrossRef]
Sample Availability: Samples of all the reported compounds can be made available from the authors upon request. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Additive (equiv.) | Solvent | Temp (°C) | Time (h) | Yield (%) |
|---|---|---|---|---|---|
| 1 | none | toluene | 110 | 7 | 46 |
| 2 | none | toluene | 110 | 16 | 65 |
| 3 | none | toluene | 120 | 7 | 30 |
| 4 | (BnO)2PO2H (0.2) | toluene | 110 | 7 | 28 |
| 5 | PivOH (0.2) | toluene | 110 | 7 | 61 |
| 6 | PivOH (0.5) | toluene | 110 | 7 | 48 |
| 7 | PivOH (1.0) | toluene | 110 | 7 | 17 |
| 8 | NaOAc (0.5) | toluene | 110 | 7 | 62 |
| 9 | NaOAc (1.0) | toluene | 110 | 7 | 78 |
| 10 | NaOAc (2.0) | toluene | 110 | 7 | 70 |
| 11 | PivOH+NaOAc (0.2 + 1.0) | toluene | 110 | 7 | 56 |
| 12 | NaOAc (1.0) | t-amyl OH | 110 | 7 | 91 |
| 13 | NaOAc (1.0) | MeTHF | 110 | 7 | 81 |
| 14 | NaOAc (1.0) | CPME | 110 | 7 | 93 |
| 15 | NaOAc (1.0) | DCE | 110 | 7 | 69 |
| 16 | NaOAc (1.0) | MeCN | 110 | 7 | 18 |
| 17 [b] | NaOAc (1.0) | CPME | 110 | 15 | 80 |
| 18 [b,c] | NaOAc (1.0) | CPME | 110 | 15 | 73 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oschmann, M.; Johansson Holm, L.; Pourghasemi-Lati, M.; Verho, O. Synthesis of Elaborate Benzofuran-2-Carboxamide Derivatives through a Combination of 8-Aminoquinoline Directed C–H Arylation and Transamidation Chemistry. Molecules 2020, 25, 361. https://doi.org/10.3390/molecules25020361
Oschmann M, Johansson Holm L, Pourghasemi-Lati M, Verho O. Synthesis of Elaborate Benzofuran-2-Carboxamide Derivatives through a Combination of 8-Aminoquinoline Directed C–H Arylation and Transamidation Chemistry. Molecules. 2020; 25(2):361. https://doi.org/10.3390/molecules25020361
Chicago/Turabian StyleOschmann, Michael, Linus Johansson Holm, Monireh Pourghasemi-Lati, and Oscar Verho. 2020. "Synthesis of Elaborate Benzofuran-2-Carboxamide Derivatives through a Combination of 8-Aminoquinoline Directed C–H Arylation and Transamidation Chemistry" Molecules 25, no. 2: 361. https://doi.org/10.3390/molecules25020361
APA StyleOschmann, M., Johansson Holm, L., Pourghasemi-Lati, M., & Verho, O. (2020). Synthesis of Elaborate Benzofuran-2-Carboxamide Derivatives through a Combination of 8-Aminoquinoline Directed C–H Arylation and Transamidation Chemistry. Molecules, 25(2), 361. https://doi.org/10.3390/molecules25020361