Design of Disruptors of the Hsp90–Cdc37 Interface

 , , ,
, , , 
Abstract
1. Introduction
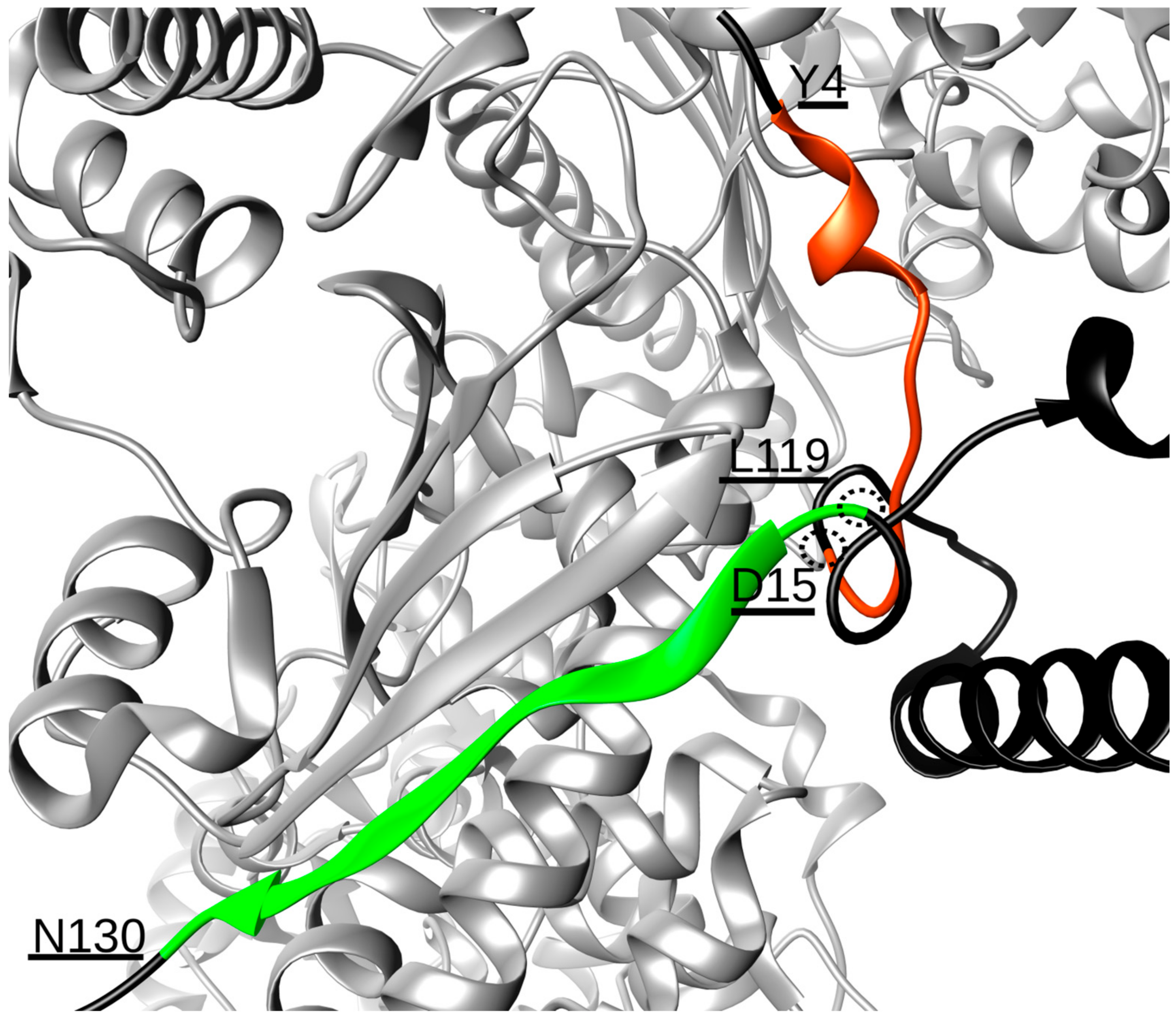
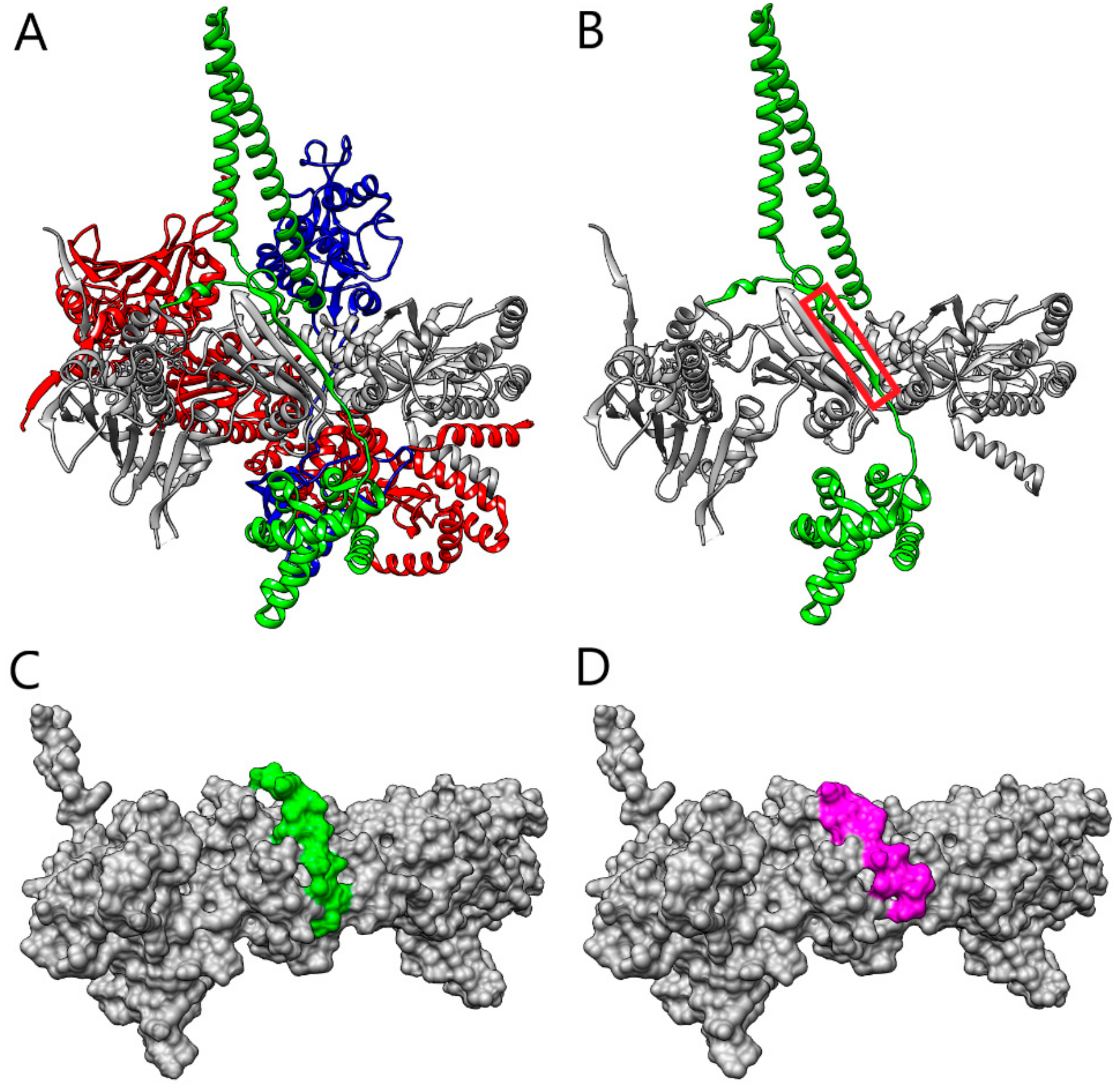
2. Results and Discussion
3. Materials and Methods
3.1. Computational Protocol
3.2. Peptide Synthesis
3.3. Peptide Treatment
3.4. Protein Extraction, Immunoprecipitation and Immunoblotting
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Krykbaeva, I.; Koeva, M.; Kayatekin, C.; Westover, K.D.; Karras, G.I.; Lindquist, S. Quantitative analysis of hsp90-client interactions reveals principles of substrate recognition. Cell 2012, 150, 987–1001. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tan, M.S.; Lu, R.C.; Yu, J.T.; Tan, L. Heat shock proteins at the crossroads between cancer and Alzheimer’s disease. Biomed. Res. Int. 2014, 2014, 239164. [Google Scholar] [CrossRef]
- Wang, T.; Rodina, A.; Dunphy, M.P.; Corben, A.; Modi, S.; Guzman, M.L.; Gewirth, D.T.; Chiosis, G. Chaperome heterogeneity and its implications for cancer study and treatment. J. Biol. Chem. 2018, REV118.002811. [Google Scholar] [CrossRef]
- Ebrahimi-Fakhari, D.; Saidi, L.J.; Eahster, L. Molecular chaperones and protein folding as therapeutic targets in Parkinson’s disease and other synucleinopathies. Acta Neuropathol. Commun. 2013, 1, 79. [Google Scholar] [CrossRef]
- Krukenberg, K.A.; Street, T.O.; Lavery, L.A.; Agard, D.A. Conformational dynamics of the molecular chaperone Hsp90. Q. Rev. Biophys. 2011, 44, 229–255. [Google Scholar] [CrossRef]
- Morra, G.; Potestio, R.; Micheletti, C.; Colombo, G. Corresponding Functional Dynamics across the Hsp90 Chaperone Family: Insights from a Multiscale Analysis of MD Simulations. PLoS Comput. Biol. 2012, 8, e1002433. [Google Scholar] [CrossRef]
- Vettoretti, G.; Moroni, E.; Sattin, S.; Tao, J.; Agard, D.A.A.; Bernardi, A.; Colombo, G. Molecular Dynamics Simulations Reveal the Mechanisms of Allosteric Activation of Hsp90 by Designed Ligands. Sci. Rep. 2016, 6, 23830. [Google Scholar] [CrossRef]
- Meyer, P.; Prodromou, C.; Hu, B.; Vaughan, C.; Roe, S.M.; Panaretou, B.; Piper, P.W.; Pearl, L.H. Structural and functional analysis of the middle segment of hsp90: Implications for ATP hydrolysis and client protein and cochaperone interactions. Mol. Cell. 2003, 11, 647–658. [Google Scholar] [CrossRef]
- Nemoto, T.; Ohara-Nemoto, Y.; Ota, M.; Takagi, T.; Yokoyama, K. Mechanism of dimer formation of the 90-kDa heat-shock protein. Eur. J. Biochem. FEBS 1995, 233, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Prodromou, C.; Roe, S.M.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 1997, 90, 65–75. [Google Scholar] [CrossRef]
- Neckers, L.; Workman, P. Hsp90 Molecular Chaperone Inhibitors: Are We There Yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Workman, P.; Burrows, F.J.; Neckers, L.; Rosen, N. Drugging the cancer chaperone Hsp90: Combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann. N.Y. Acad. Sci. 2007, 1113, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Bassanini, I.; D’Annessa, I.; Costa, M.; Monti, D.; Colombo, G.; Riva, S. Chemo-enzymatic synthesis of (E)-2,3-diaryl-5-styryl-trans-2,3-dihydrobenzofuran-based scaffolds and their in vitro and in silico evaluation as a novel sub-family of potential allosteric modulators of the 90 kDa heat shock protein (Hsp90). Org. & Biomol. Chem. 2018, 16, 3741–3753. [Google Scholar]
- Ferraro, M.; D’Annessa, I.; Moroni, E.; Morra, G.; Paladino, A.; Rinaldi, S.; Compostella, F.; Colombo, G. Allosteric Modulators of HSP90 and HSP70: Dynamics Meets Function through Structure-Based Drug Design. J. Med. Chem. 2019, 62, 60–87. [Google Scholar] [CrossRef]
- Brandt, G.E.L.; Blagg, B.S.J. Alternate strategies of Hsp90 modulation for the treatment of cancer and other diseases. Curr. Top. Med. Chem. 2009, 9, 1447–1461. [Google Scholar] [CrossRef]
- D’Annessa, I.; Sattin, S.; Tao, J.; Pennati, M.; Sànchez-Martìn, C.; Moroni, E.; Rasola, A.; Zaffaroni, N.; Agard, D.A.; Bernardi, A.; et al. Design of allosteric stimulators of the HSP90 ATPase as novel anticancer leads. Chem. A Eur. J. 2017, 23, 5188–5192. [Google Scholar] [CrossRef]
- Verba, K.A.; Wang, R.Y.; Arakawa, A.; Liu, Y.; Shirouzu, M.; Yokoyama, S.; Agard, D.A. Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 2016, 352, 1542–1547. [Google Scholar] [CrossRef]
- Pearl, L.H. Hsp90 and Cdc37-a chaperone cancer conspiracy. Curr. Opin. Genet. & Dev. 2005, 15, 55–61. [Google Scholar]
- Liu, W.; Landgraf, R. Phosphorylated and unphosphorylated serine 13 of CDC37 stabilize distinct interactions between its client and Hsp90 binding domains. Biochemistry 2015, 54, 1493–1504. [Google Scholar] [CrossRef]
- Miyata, Y.; Nishida, E. CK2 binds, phosphorylates and regulatesits pivotal substrate Cdc37, an Hsp90-cochaperone. Mol. Cell. Biochem. 2005, 274, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Trellet, M.; Melquiond, A.S.; Bonvin, A.M. A Unified Conformational Selection and Induced Fit Approach to Protein-Peptide Docking. PLoS ONE 2013, 8, e58769. [Google Scholar] [CrossRef] [PubMed]
- Donsky, E.; Wolfson, H.J. PepCrawler: A fast RRT-based algorithm for high-resolution refinement and binding affinity estimation of peptide inhibitors. Bioinformatics 2001, 27, 2836–2842. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, C.K.; Mollapour, M.; Smith, J.R.; Truman, A.; Hu, B.; Good, V.M.; Panaretou, B.; Neckers, L.; Clarke, P.A.; Workman, P.; et al. Hsp90-dependent activation of protein kinases is regulated by chaperone-targeted dephosphorylation of Cdc37. Mol. Cell 2008, 31, 886–895. [Google Scholar] [CrossRef]
- Oberoi, J.; Dunn, D.M.; Woodford, M.R.; Mariotti, L.; Schulman, J.; Bourboulia, D.; Mollapour, M.; Vaughan, C.K. Structural and functional basis of protein phosphatase 5 substrate specificity. Proc. Natl. Acad. Sci. USA 2016, 113, 9009–9014. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Case, D.A.B.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham III, T.E.; Darden, T.A.; Duke, R.E.; Gohlke, H.; Goetz, A.W.; et al. Amber14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N-log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Ceriotti, M.; Bussi, G.; Parrinello, M. Langevin equation with colored noise for constant-temperature molecular dynamics simulations. Phys. Rev. Lett. 2009, 102. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.B. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints; Molecular Dynamics of n-Alkanes. J. Comp. Phys. 1977, 23, 327. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. SETTLE: An analytical version of the SHAKE and RATTLE algorithms for rigid water models. J. Comp. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; De Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 webserver: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A protein-protein docking approach based on biochemical and/or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef]
- De Cesare, M.; Sfondrini, L.; Pennati, M.; De Marco, C.; Motta, V.; Tagliabue, E.; Deraco, M.; Balsari, A.; Zaffaroni, N. CpG-oligodeoxynucleotides exert remarkable antitumor activity against diffuse malignant peritoneal mesothelioma orthotopic xenografts. J. Transl. Med. 2016, 14, 25. [Google Scholar] [CrossRef]
Sample Availability: Cdc37p1–5 synthesized peptides are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
| Hsp90:Cdc37 Hydrogen Bonds | |
|---|---|
| VAL318:MET128 | 99.76 |
| HIS320:LYS126 | 90.79 |
| SER322:PHE124 | 88.28 |
| GLU324:LYS121 | 86.99 |
| GLN326:LEU118 | 73.62 |
| LEU327:SER119 | 47.11 |
| LYS399:ASP8 | 51.90 |
| LYS402:ASP14 | 66.62 |
| LYS406:pSER13 | 78.92 |
| THR446:pSER13 | 49.77 |
| ASN389:TYR4 | 68.58 |
| LEU327:SER120 | 63.79 |
| GLU414:SER127 | 92.02 |
| LEU316:ASN130 | 92.05 |
| HIS315:ASN130 | 72.82 |
| Peptides Sequence | Peptide Length | Code | Purity |
|---|---|---|---|
| NYSVWDHIEVSDDLSKDGFSKSMVN | 25 | Cdc37p1 | >95% |
| NYSVWDHIEVDDDLSKDGFSKSMVN | 25 | Cdc37p2 | >95% |
| NYSVWDHIEVEDDLSKDGFSKSMVN | 25 | Cdc37p3 | >95% |
| LSKDGFSKSMVN | 12 | Cdc37p4 | >95% |
| PSKDIFLKSMIN | 12 | Cdc37p5 | >95% |
| Peptides Code | Mol. Wt. | ESI-MS Data (m/z) |
|---|---|---|
| Cdc37p1 | 2872.08 | 1436.6 (p1-2H2+); 958.2 (p1-3H3+); 719.1 (p1-4H4+) |
| Cdc37p2 | 2900.09 | 1450.8 (p2-2H2+); 967.6 (p2-3H3+); 726.1 (p2-4H4+) |
| Cdc37p3 | 2913.13 | 1457.6 (p3-2H2+); 972.0 (p3-3H3+); 729.7 (p3-4H4+) |
| Cdc37p4 | 1311.51 | 1311.6 (p4-1H+); 656.5 (p4-2H2+); 438.1 (p4-3H3+) |
| Cdc37p5 | 1391.68 | 1391.7 (p5-1H+); 696.5 (p5-2H2+); 464.7 (p5-3H3+) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Annessa, I.; Hurwitz, N.; Pirota, V.; Beretta, G.L.; Tinelli, S.; Woodford, M.; Freccero, M.; Mollapour, M.; Zaffaroni, N.; Wolfson, H.; et al. Design of Disruptors of the Hsp90–Cdc37 Interface. Molecules 2020, 25, 360. https://doi.org/10.3390/molecules25020360
D’Annessa I, Hurwitz N, Pirota V, Beretta GL, Tinelli S, Woodford M, Freccero M, Mollapour M, Zaffaroni N, Wolfson H, et al. Design of Disruptors of the Hsp90–Cdc37 Interface. Molecules. 2020; 25(2):360. https://doi.org/10.3390/molecules25020360
Chicago/Turabian StyleD’Annessa, Ilda, Naama Hurwitz, Valentina Pirota, Giovanni Luca Beretta, Stella Tinelli, Mark Woodford, Mauro Freccero, Mehdi Mollapour, Nadia Zaffaroni, Haim Wolfson, and et al. 2020. "Design of Disruptors of the Hsp90–Cdc37 Interface" Molecules 25, no. 2: 360. https://doi.org/10.3390/molecules25020360
APA StyleD’Annessa, I., Hurwitz, N., Pirota, V., Beretta, G. L., Tinelli, S., Woodford, M., Freccero, M., Mollapour, M., Zaffaroni, N., Wolfson, H., & Colombo, G. (2020). Design of Disruptors of the Hsp90–Cdc37 Interface. Molecules, 25(2), 360. https://doi.org/10.3390/molecules25020360