Revisiting the Rearrangement of Dewar Thiophenes

 ,
, 
Abstract
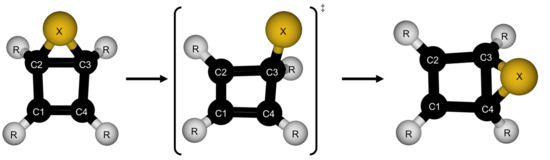
1. Introduction
2. Computational Details
3. Structures
4. Reaction Profiles and Forces
5. Evolution of Bonding
5.1. Bond Orders and Their Derivatives
5.2. Evolution of the Dewar Rearrangement in S-oxide Perfluorotetramethyl Thiophene
5.3. Analysis of the Electron Densities
5.4. NBO and AdNDP Analysis
6. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kobayashi, Y.; Kumadaki, I. Heterocycles Dewar and Related Compounds. In Advances in Heterocyclic Chemistry; Elsevier: New York, NY, USA, 1982; Volume 31, p. 169. [Google Scholar]
- Bushweller, C.H.; Ross, J.A.; Lemal, D.M. Automerization of a Dewar thiophene and its exo-S-oxide. A dramatic contrast. J. Am. Chem. Soc. 1977, 99, 629–631. [Google Scholar] [CrossRef]
- Dorogan, I.V.; Minkin, V.I.; Novikova, L.M. Computer simulation of the mechanisms and energetics of circumambulatory rearrangements of Dewar furan, thiophene and selenophene. Mendeleev Commun. 2003, 13, 205–207. [Google Scholar] [CrossRef]
- Martin Birney, D. Theory, experiment and unusual features of potential energy surfaces of pericyclic and pseudopericyclic reactions with sequential transition structures Curr. Org. Chem. 2010, 14, 1658–1668. [Google Scholar]
- Ross, J.A.; Seiders, R.P.; Lemal, D.M. An extraordinarily facile sulfoxide automerization. J. Am. Chem. Soc. 1976, 98, 4325–4327. [Google Scholar] [CrossRef]
- Schleyer, P.V.R.; Wu, J.I.; Cossío, F.P.; Fernández, I. Aromaticity in transition structures. Chem. Soc. Rev. 2014, 43, 4909–4921. [Google Scholar] [CrossRef]
- Jalife, S.; Martínez-Guajardo, G.; Zavala-Oseguera, C.; Fernández-Herrera, M.A.; Schleyer, P.V.R.; Merino, G. Mechanistic elucidation of the 2-norbornyl to 1,3-dimethylcyclopentenyl cation isomerization. Eur. J. Org. Chem. 2014, 35, 7955–7959. [Google Scholar] [CrossRef]
- Jalife, S.; Judy, I.; Wu, C.; Martínez-Guajardo, G.; Schleyer, P.V.R.; Fernández-Herrera, M.A.; Merino, G. The 9-homocubyl cation rearrangement revisited. Chem. Commun. 2015, 51, 5391–5393. [Google Scholar] [CrossRef]
- Jalife, S.; Mondal, S.; Cabellos, J.L.; Martinez-Guajardo, G.; Fernandez-Herrera, M.A.; Merino, G. The cubyl cation rearrangements. Chem. Commun. 2016, 52, 3403–3405. [Google Scholar] [CrossRef] [PubMed]
- Jalife, S.; Mondal, S.; Osorio, E.; Cabellos, J.L.; Martinez-Guajardo, G.; Fernandez-Herrera, M.A.; Merino, G. Nonclassical 21-homododecahedryl cation rearrangement revisited. Org. Lett. 2016, 18, 1140–1142. [Google Scholar] [CrossRef] [PubMed]
- Barroso, J.; Cabellos, J.L.; Pan, S.; Murillo, F.; Zarate, X.; Fernandez-Herrera, M.A.; Merino, G. Revisiting the racemization mechanism of helicenes. Chem. Commun. 2018, 54, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Toro-Labbé, A. Characterization of chemical reactions from the profiles of energy, chemical Potential, and hardness. J. Phys. Chem. A 1999, 103, 4398–4403. [Google Scholar] [CrossRef]
- Toro-Labbé, A.; Gutiérrez-Oliva, S.; Murray, J.; Politzer, P. A new perspective on chemical and physical processes: The reaction force. Mol. Phys. 2007, 105, 2619–2625. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Jaque, P. Perspectives on the reaction force constant. J. Mol. Model. 2013, 19, 4111. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions—The IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Hargis, J.C.; Vöhringer-Martinez, E.; Woodcock, H.L.; Toro-Labbé, A.; Schaefer III, H.F. Characterizing the mechanism of the double proton transfer in the formamide dimer. J. Phys. Chem. A 2011, 115, 2650–2657. [Google Scholar] [CrossRef]
- Gómez, S.; Guerra, D.; López, J.G.; Toro-Labbé, A.; Restrepo, A. A Detailed Look at the Reaction Mechanisms of Substituted Carbenes with Water. J. Phys. Chem. A 2013, 117, 1991–1999. [Google Scholar] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 (Revision E.01); Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154119. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. Natural Bond Order 6.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2013. [Google Scholar]
- Zubarev, D.Y.; Boldyrev, A.I. Developing paradigms of chemical bonding: Adaptive natural density partitioning. Phys. Chem. Chem. Phys. 2008, 10, 5207–5217. [Google Scholar] [CrossRef] [PubMed]
- Zubarev, D.Y.; Boldyrev, A.I. Revealing intuitively assessable chemical bonding patterns in organic aromatic molecules via adaptive natural density partitioning. J. Org. Chem. 2008, 73, 9251–9258. [Google Scholar] [CrossRef] [PubMed]
- Duarte, F.; Toro-Labbé, A. The mechanism of H2 activation by (amino)carbenes. J. Phys. Chem. A 2011, 115, 3050–3059. [Google Scholar] [CrossRef]
- Giraldo, C.; Gómez, S.; Weinhold, F.; Restrepo, A. Insight into the mechanism of the Michael reaction. ChemPhysChem 2016, 17, 2022–2034. [Google Scholar] [CrossRef]
- Bader, R. Atoms in Molecules. A Quantum Theory; Oxford University Press: Oxford, UK, 1994. [Google Scholar]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Popelier, P.L.A. On the full topology of the Laplacian of the electron density. Coord. Chem. Rev. 2000, 197, 169–189. [Google Scholar] [CrossRef]
- Popelier, P.L.A. Atoms in Molecules: An Introduction; Prentice Hall: London, UK, 2000. [Google Scholar]
- Bader, R.F.W. The quantum mechanical basis of conceptual chemistry. Monatsh. Chem. 2005, 136, 819–854. [Google Scholar] [CrossRef]
- Becke, A. The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Merino, G.; Vela, A.; Heine, T. Description of electron delocalization via the analysis of molecular fields. Chem. Rev. 2005, 105, 3812–3841. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From weak to strong interactions: A comprehensive analysis of the topological and energetic properties of the electron density distribution involving X–H⋯F–Y systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural bond orbital methods. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 1–42. [Google Scholar] [CrossRef]
- Wiberg, K.B. Application of the Pople-Santry-Segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]
Sample Availability: Not available. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Reactant | TS | Product |
|---|---|---|---|
| Bond Distance | |||
| C1-C2 | 1.48 | 1.40 | 1.35 |
| C1-C4 | 1.35 | 1.40 | 1.48 |
| C2-C3 | 1.50 | 1.49 | 1.49 |
| C3-C4 | 1.49 | 1.49 | 1.5 |
| C1-S9 | 2.62 | 2.69 | 2.62 |
| C2-S9 | 1.89 | 2.31 | 2.61 |
| C3-S9 | 1.88 | 1.81 | 1.88 |
| C4-S9 | 2.61 | 2.31 | 1.89 |
| S9-O10 | 1.45 | 1.45 | 1.45 |
| Dihedral Angles | |||
| dr | 0 | 0 | 0 |
| F3C-C1-C2-C3 | −170 | −171 | −169 |
| F3C-C2-C3-C4 | 143 | 179 | −169 |
| F3C-C3-C4-C1 | 142 | 136 | 140 |
| F3C-C4-C1-C2 | −168 | 179 | 144 |
| X | R | Ea | W1 | W2 | ||
|---|---|---|---|---|---|---|
| Electronic | Gibbs at 157 °C | Gibbs at −135.8 °C | ||||
| S = O | H | 8.26 | 8.25 | 7.79 | 3.58 | 4.68 |
| CF3 | 7.23 | 7.57 | 6.86 | 4.29 | 2.94 | |
| (6.7 ± 0.1) | ||||||
| S | Ha | 22.14 | 21.16 | 21.24 | 8.76 | 13.38 |
| CF3 | 26.46 | 25.06 | 25.35 | 15.64 | 10.82 | |
| (22.1 ± 0.1) | ||||||
| CH2 | H | 35.06 | 32.98 | 33.16 | 15.46 | 19.6 |
| CF3 | 38.13 | 35.73 | 36.4 | 20.42 | 17.71 |
| R = CF3 | R = H | |||
|---|---|---|---|---|
| Atoms | ON | Atoms | ON | |
| X = SO | C2-S9 | 1.16 | C2-S9 | 1.21 |
| C4-S9 | 1.16 | C4-S9 | 1.21 | |
| X = S | C2-S9 | 1.58 | C2-S9 | 1.66 |
| C4-S9 | 1.58 | C4-S9 | 1.66 | |
| X = CH2 | C2-S9 | 1.37 | C2-S9 | 1.48 |
| C4-S9 | 1.37 | C4-S9 | 1.48 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez, S.; Osorio, E.; Dzib, E.; Islas, R.; Restrepo, A.; Merino, G. Revisiting the Rearrangement of Dewar Thiophenes. Molecules 2020, 25, 284. https://doi.org/10.3390/molecules25020284
Gómez S, Osorio E, Dzib E, Islas R, Restrepo A, Merino G. Revisiting the Rearrangement of Dewar Thiophenes. Molecules. 2020; 25(2):284. https://doi.org/10.3390/molecules25020284
Chicago/Turabian StyleGómez, Sara, Edison Osorio, Eugenia Dzib, Rafael Islas, Albeiro Restrepo, and Gabriel Merino. 2020. "Revisiting the Rearrangement of Dewar Thiophenes" Molecules 25, no. 2: 284. https://doi.org/10.3390/molecules25020284
APA StyleGómez, S., Osorio, E., Dzib, E., Islas, R., Restrepo, A., & Merino, G. (2020). Revisiting the Rearrangement of Dewar Thiophenes. Molecules, 25(2), 284. https://doi.org/10.3390/molecules25020284