
Caffeic Acid Attenuates Multi-Drug Resistance in Cancer Cells by Inhibiting Efflux Function of Human P-Glycoprotein

 , ,
, , 
Abstract
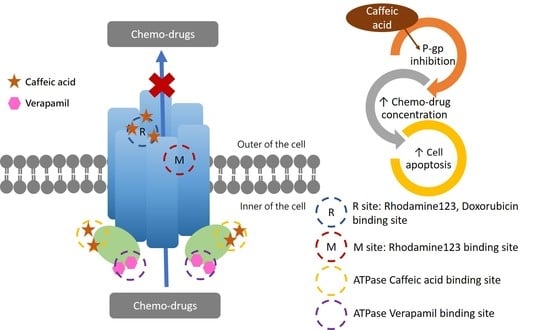
1. Introduction
2. Results
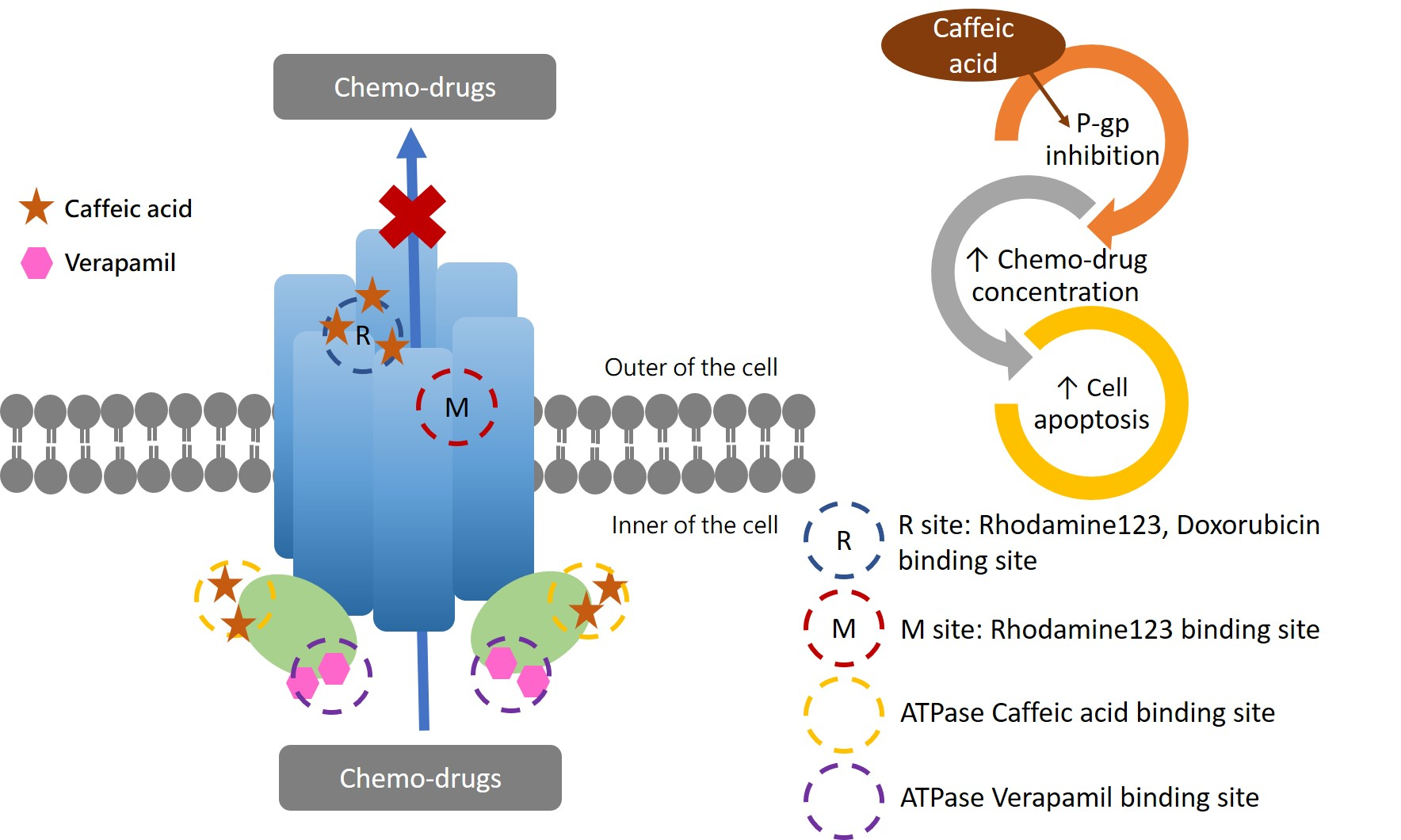
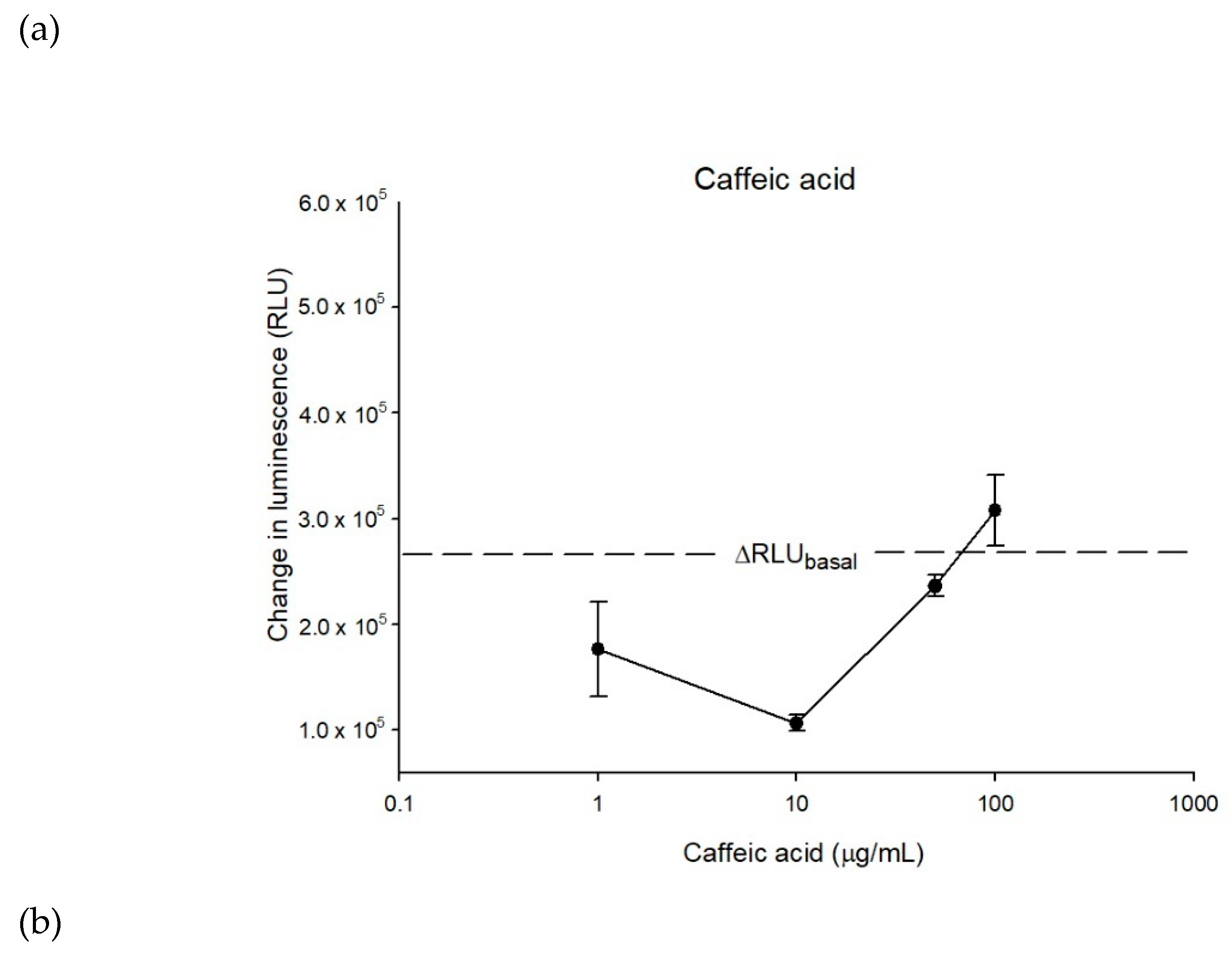
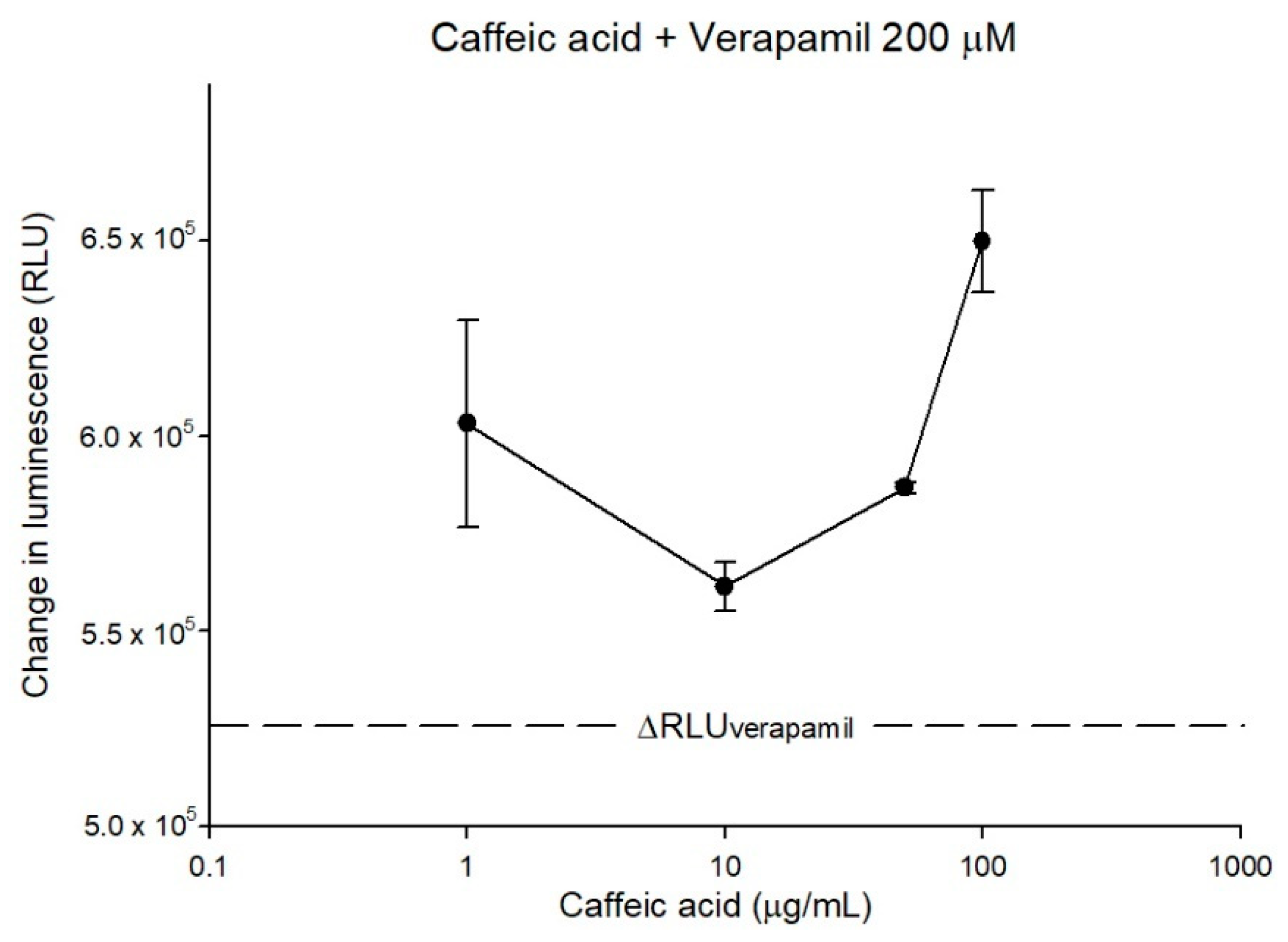
2.1. Caffeic Acid Is Non-Cytotoxic toward Experimental Cell Lines and Is Not a Substrate of P-gp
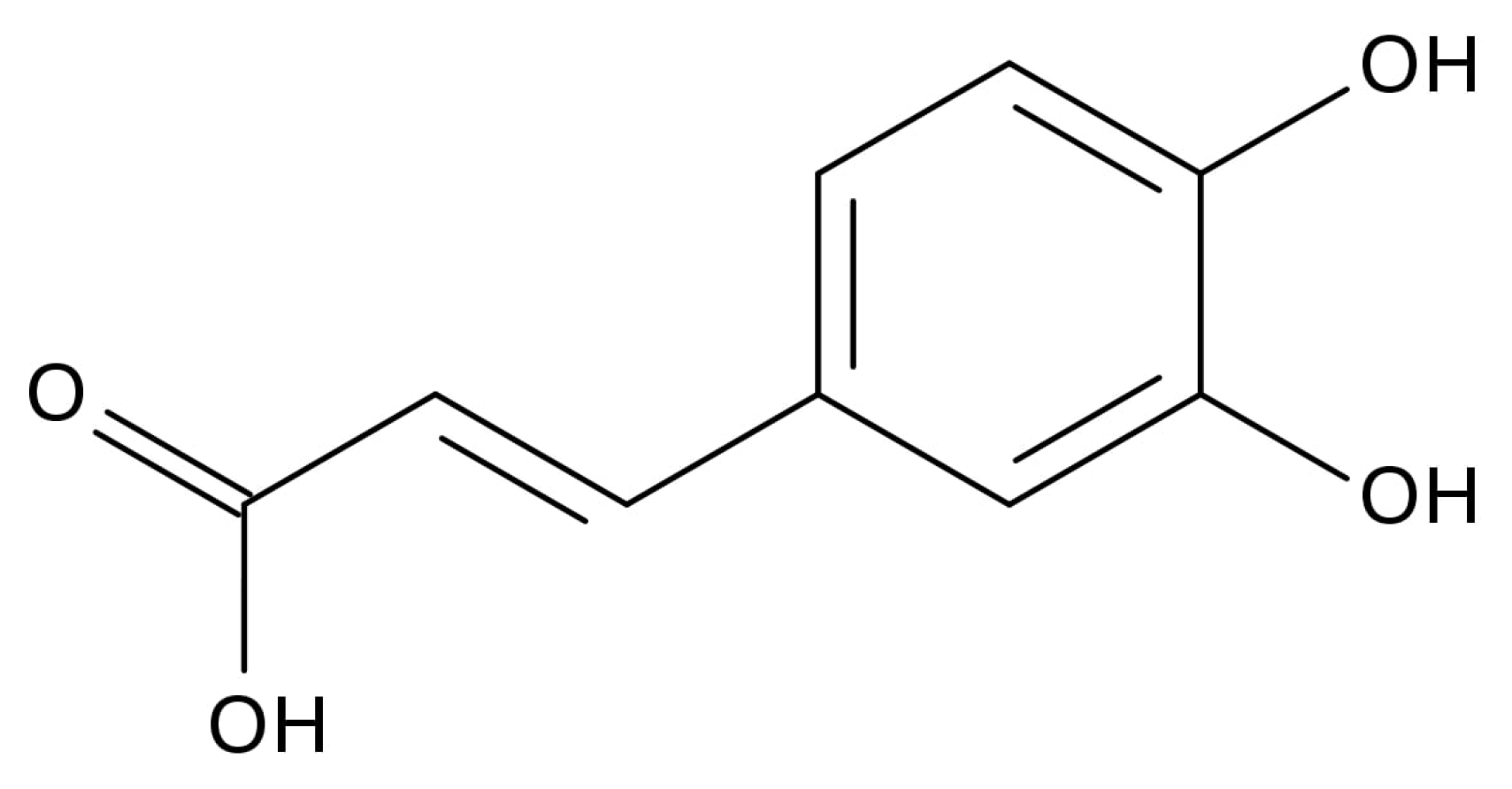
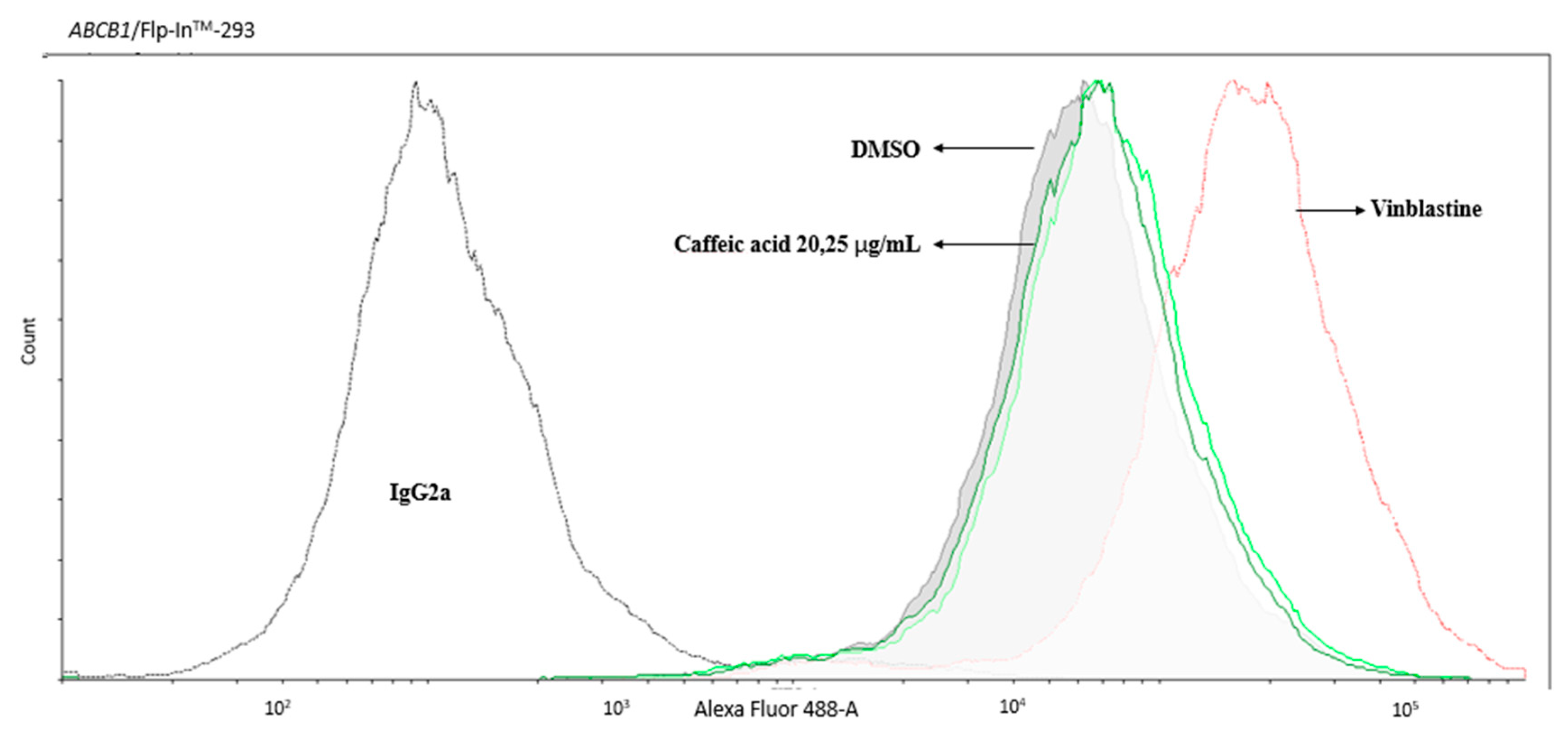
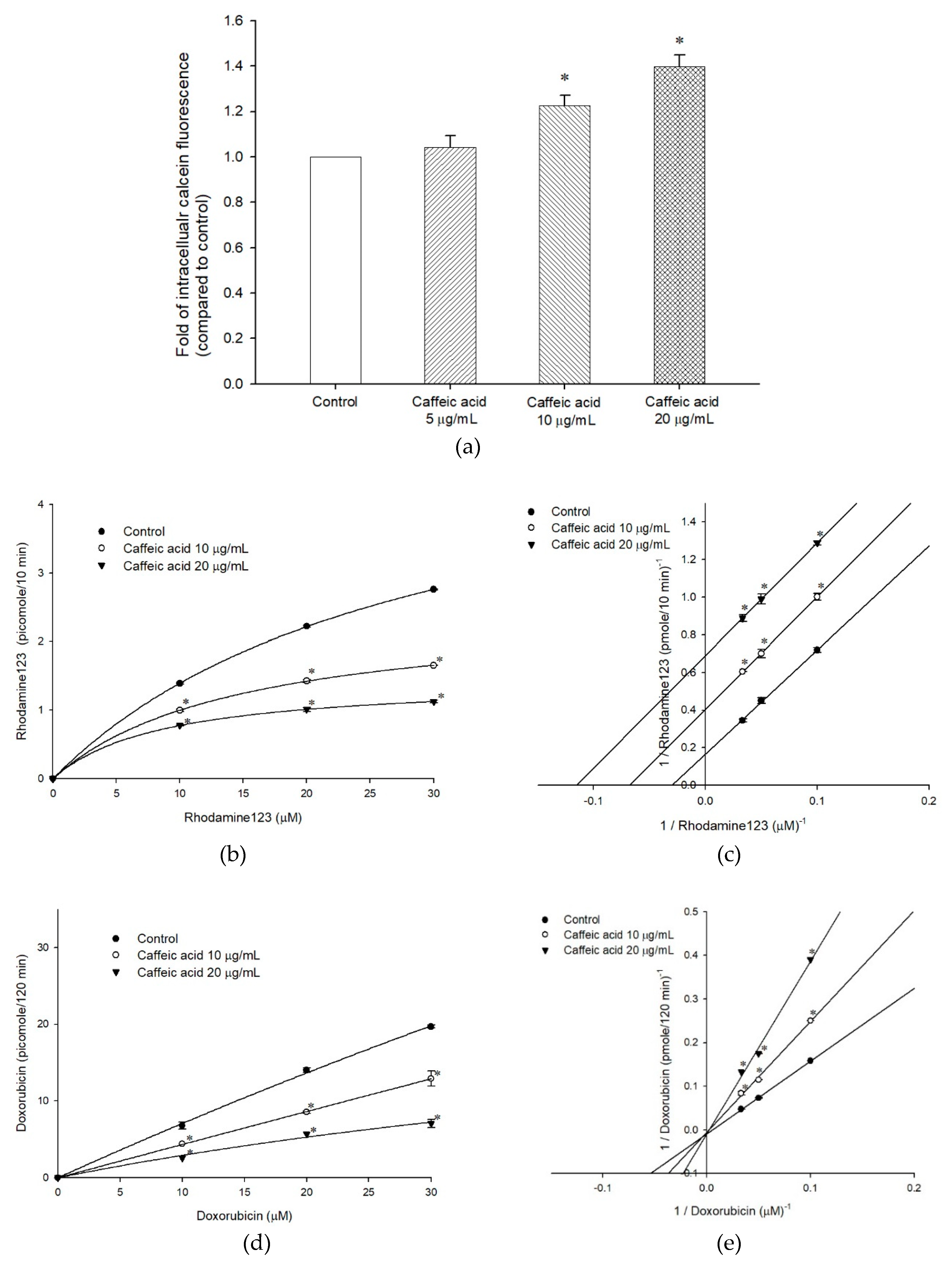
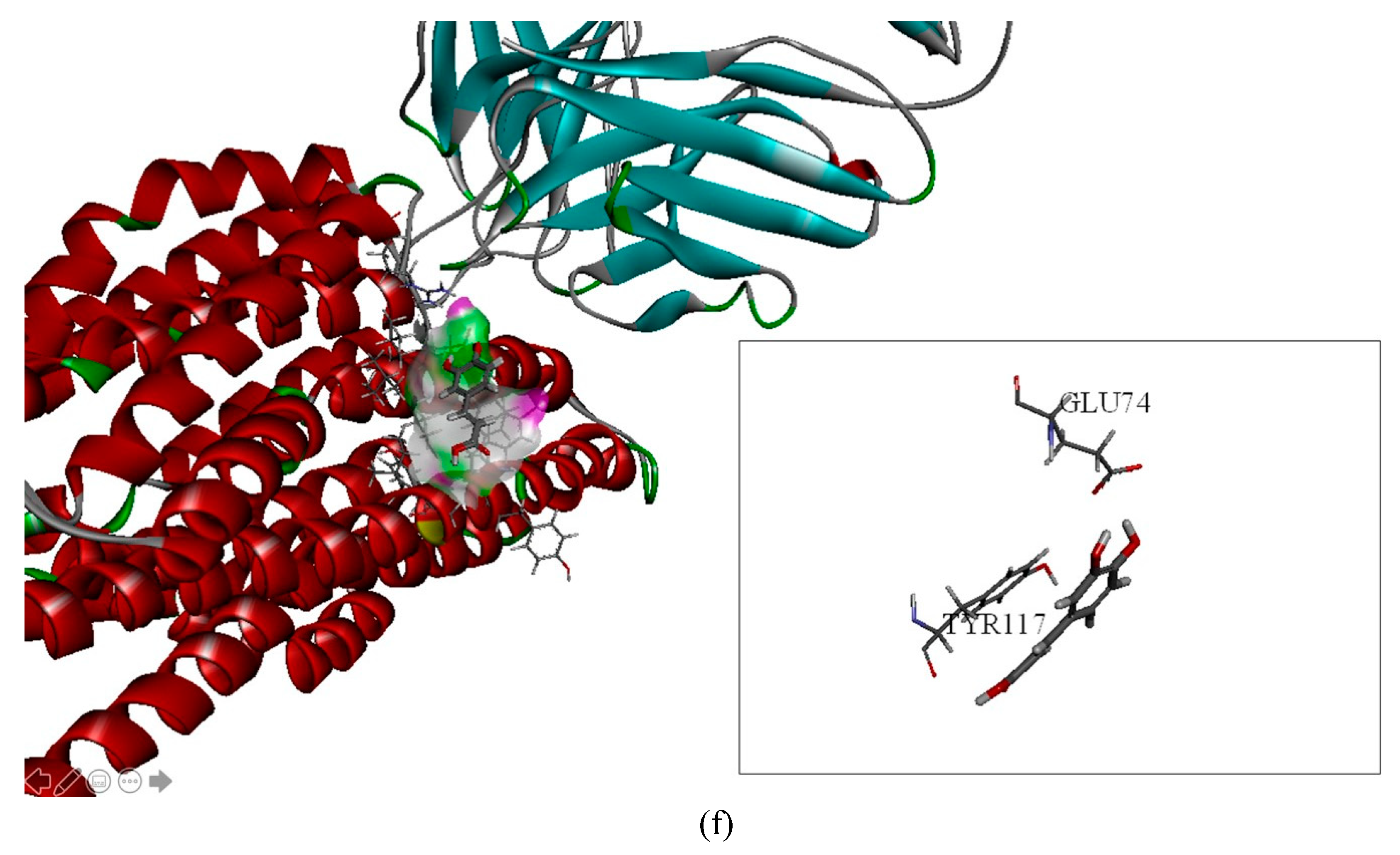
2.2. The Inhibitory Effects, Mechanisms and Binding Modes of Caffeic Acid on Human P-gp Function
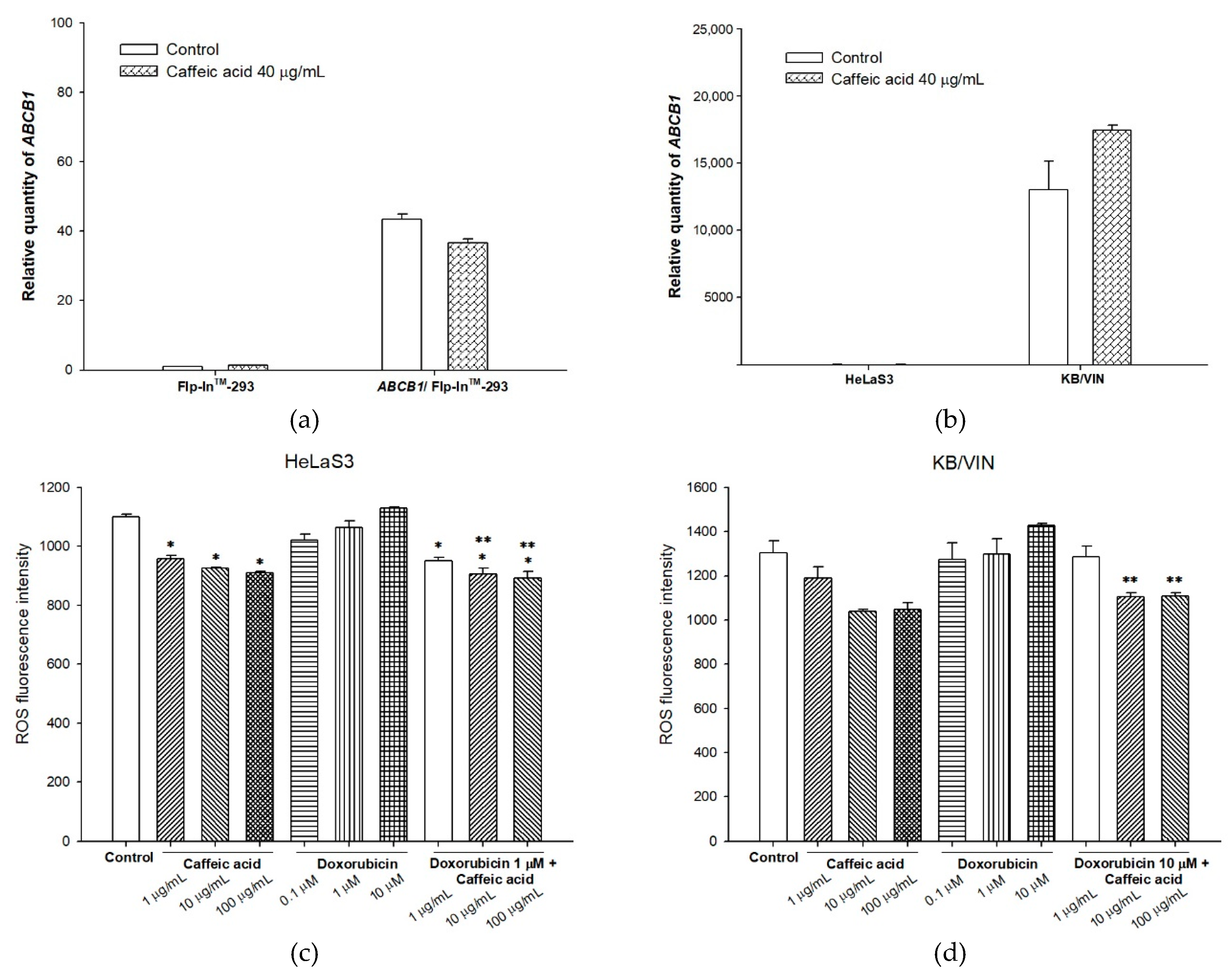
2.3. The Influences of Caffeic Acid on Human P-gp Expression
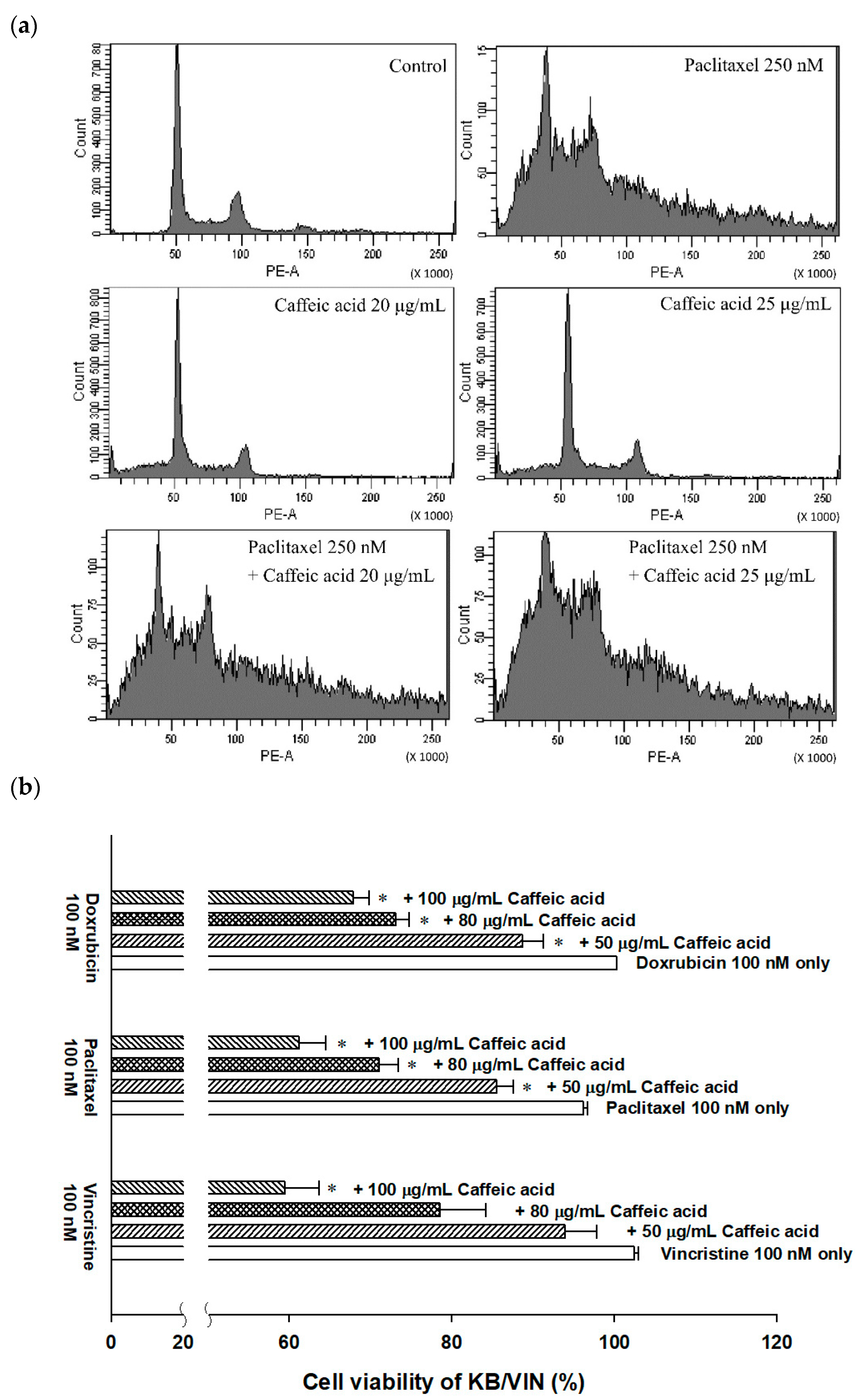
2.4. The Modulating Effects of Caffeic Acid on Cancer Multi-Drug Resistance
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Cell Lines
4.3. Cytotoxicity Determination Assay (SRB Assay)
4.4. MDR1 Shift Assay
4.5. Intracellular Calcein Accumulation Assay
4.6. Rhodamine123 and Doxorubicin Efflux Assay
4.7. P-gp ATPase Activity Assay
4.8. Real-Time Quantitative RT-PCR
4.9. Intracellular Total ROS Activity Assay
4.10. Cell Cycle Analysis
4.11. Molecular Docking
4.12. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Silva, R.; Vilas-Boas, V.; Carmo, H.; Dinis-Oliveira, R.J.; Carvalho, F.; de Lourdes Bastos, M.; Remiao, F. Modulation of P-glycoprotein efflux pump: Induction and activation as a therapeutic strategy. Pharmacol. Ther. 2015, 149, 1–123. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Jaitak, V. Natural products as multidrug resistance modulators in cancer. Eur. J. Med. Chem. 2019, 176, 268–291. [Google Scholar] [CrossRef] [PubMed]
- Kathawala, R.J.; Wang, Y.J.; Ashby, C.R., Jr.; Chen, Z.S. Recent advances regarding the role of ABC subfamily C member 10 (ABCC10) in the efflux of antitumor drugs. Chin. J. Cancer 2014, 33, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Wang, C.; Fu, N.; Liu, L.; Zhu, D.; Wei, Z.; Zhang, H.; Xing, J.; Wang, Y. Downregulation of cytokeratin 18 enhances BCRP-mediated multidrug resistance through induction of epithelial-mesenchymal transition and predicts poor prognosis in breast cancer. Oncol. Rep. 2019, 41, 3015–3026. [Google Scholar] [CrossRef]
- Das, M.; Law, S. Role of tumor microenvironment in cancer stem cell chemoresistance and recurrence. Int. J. Biochem. Cell Biol. 2018, 103, 115–124. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar]
- Uddin, M.B.; Roy, K.R.; Hosain, S.B.; Khiste, S.K.; Hill, R.A.; Jois, S.D.; Zhao, Y.; Tackett, A.J.; Liu, Y.Y. An N(6)-methyladenosine at the transited codon 273 of p53 pre-mRNA promotes the expression of R273H mutant protein and drug resistance of cancer cells. Biochem. Pharmacol. 2019, 160, 134–145. [Google Scholar] [CrossRef]
- Bedi, A.; Barber, J.P.; Bedi, G.C.; el-Deiry, W.S.; Sidransky, D.; Vala, M.S.; Akhtar, A.J.; Hilton, J.; Jones, R.J. BCR-ABL-mediated inhibition of apoptosis with delay of G2/M transition after DNA damage: A mechanism of resistance to multiple anticancer agents. Blood 1995, 86, 1148–1158. [Google Scholar] [CrossRef]
- Wilson, C.S.; Medeiros, L.J.; Lai, R.; Butch, A.W.; McCourty, A.; Kelly, K.; Brynes, R.K. DNA topoisomerase IIalpha in multiple myeloma: A marker of cell proliferation and not drug resistance. Mod. Pathol. 2001, 14, 886–891. [Google Scholar] [CrossRef]
- Filomeni, G.; Turella, P.; Dupuis, M.L.; Forini, O.; Ciriolo, M.R.; Cianfriglia, M.; Pezzola, S.; Federici, G.; Caccuri, A.M. 6-(7-Nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol, a specific glutathione S-transferase inhibitor, overcomes the multidrug resistance (MDR)-associated protein 1-mediated MDR in small cell lung cancer. Mol. Cancer Ther. 2008, 7, 371–379. [Google Scholar] [CrossRef]
- Rodriguez-Antona, C.; Ingelman-Sundberg, M. Cytochrome P450 pharmacogenetics and cancer. Oncogene 2006, 25, 1679–1691. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, R.H. Genetic and epigenetic factors in anticancer drug resistance. J. Natl. Cancer Inst. 2000, 92, 4–5. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, H.; Yang, B.B. Friend or foe: The role of microRNA in chemotherapy resistance. Acta Pharm. Sin 2013, 34, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Pao, W.; Sequist, L.V. Acquired resistance to TKIs in solid tumours: Learning from lung cancer. Nat. Rev. Clin. Oncol. 2014, 11, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Paskeviciute, M.; Petrikaite, V. Overcoming transporter-mediated multidrug resistance in cancer: Failures and achievements of the last decades. Drug Deliv. Transl. Res. 2019, 9, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Yakusheva, E.N.; Titov, D.S. Structure and Function of Multidrug Resistance Protein 1. Biochem. Biokhimiia 2018, 83, 907–929. [Google Scholar] [CrossRef]
- Ling, V.; Thompson, L.H. Reduced permeability in CHO cells as a mechanism of resistance to colchicine. J. Cell. Physiol. 1974, 83, 103–116. [Google Scholar] [CrossRef]
- Mollazadeh, S.; Sahebkar, A.; Hadizadeh, F.; Behravan, J.; Arabzadeh, S. Structural and functional aspects of P-glycoprotein and its inhibitors. Life Sci. 2018, 214, 118–123. [Google Scholar] [CrossRef]
- Leopoldo, M.; Nardulli, P.; Contino, M.; Leonetti, F.; Luurtsema, G.; Colabufo, N.A. An updated patent review on P-glycoprotein inhibitors (2011-2018). Expert Opin. Ther. Pat. 2019, 29, 455–461. [Google Scholar] [CrossRef]
- Joshi, P.; Vishwakarma, R.A.; Bharate, S.B. Natural alkaloids as P-gp inhibitors for multidrug resistance reversal in cancer. Eur. J. Med. Chem. 2017, 138, 273–292. [Google Scholar] [CrossRef]
- O’Brien, M.M.; Lacayo, N.J.; Lum, B.L.; Kshirsagar, S.; Buck, S.; Ravindranath, Y.; Bernstein, M.; Weinstein, H.; Chang, M.N.; Arceci, R.J.; et al. Phase I study of valspodar (PSC-833) with mitoxantrone and etoposide in refractory and relapsed pediatric acute leukemia: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2010, 54, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, H.; Assaraf, Y.G.; Zhao, K.; Xu, X.; Xie, J.; Yang, D.H.; Chen, Z.S. Overcoming ABC transporter-mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist. Updat. 2016, 27, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Cripe, L.D.; Uno, H.; Paietta, E.M.; Litzow, M.R.; Ketterling, R.P.; Bennett, J.M.; Rowe, J.M.; Lazarus, H.M.; Luger, S.; Tallman, M.S. Zosuquidar, a novel modulator of P-glycoprotein, does not improve the outcome of older patients with newly diagnosed acute myeloid leukemia: A randomized, placebo-controlled trial of the Eastern Cooperative Oncology Group 3999. Blood 2010, 116, 4077–4085. [Google Scholar] [CrossRef] [PubMed]
- Dash, R.P.; Jayachandra Babu, R.; Srinivas, N.R. Therapeutic Potential and Utility of Elacridar with Respect to P-glycoprotein Inhibition: An Insight from the Published In Vitro, Preclinical and Clinical Studies. Eur. J. Drug Metab. Pharmacokinet. 2017, 42, 915–933. [Google Scholar] [CrossRef]
- Han, R.M.; Zhang, J.P.; Skibsted, L.H. Reaction dynamics of flavonoids and carotenoids as antioxidants. Molecules 2012, 17, 2140–2160. [Google Scholar] [CrossRef]
- Kumar, S.; Pandey, A.K. Chemistry and biological activities of flavonoids: An overview. Sci. World J. 2013, 2013, 162750. [Google Scholar] [CrossRef]
- Meyer, A.S.; Heinonen, M.; Frankel, E.N. Antioxidant interactions of catechin, cyanidin, caffeic acid, quercetin, and ellagic acid on human LDL oxidation. Food Chem. 1998, 61, 71–75. [Google Scholar] [CrossRef]
- Jaman, M.S.; Sayeed, M.A. Ellagic acid, sulforaphane, and ursolic acid in the prevention and therapy of breast cancer: Current evidence and future perspectives. Breast Cancer (TokyoJpn.) 2018, 25, 517–528. [Google Scholar] [CrossRef]
- Xing, Y.; Peng, H.Y.; Zhang, M.X.; Li, X.; Zeng, W.W.; Yang, X.E. Caffeic acid product from the highly copper-tolerant plant Elsholtzia splendens post-phytoremediation: Its extraction, purification, and identification. J. Zhejiang Univ. Sci. B 2012, 13, 487–493. [Google Scholar] [CrossRef]
- Ahmed, N.; Escalona, R.; Leung, D.; Chan, E.; Kannourakis, G. Tumour microenvironment and metabolic plasticity in cancer and cancer stem cells: Perspectives on metabolic and immune regulatory signatures in chemoresistant ovarian cancer stem cells. Semin. Cancer Biol. 2018, 53, 265–281. [Google Scholar] [CrossRef]
- Khan, F.A.; Maalik, A.; Murtaza, G. Inhibitory mechanism against oxidative stress of caffeic acid. J. Food Drug Anal. 2016, 24, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Ahn, C.H.; Choi, W.C.; Kong, J.Y. Chemosensitizing activity of caffeic acid in multidrug-resistant MCF-7/Dox human breast carcinoma cells. Anticancer Res. 1997, 17, 1913–1917. [Google Scholar] [PubMed]
- Lin, C.L.; Chen, R.F.; Chen, J.Y.; Chu, Y.C.; Wang, H.M.; Chou, H.L.; Chang, W.C.; Fong, Y.; Chang, W.T.; Wu, C.Y.; et al. Protective effect of caffeic acid on paclitaxel induced anti-proliferation and apoptosis of lung cancer cells involves NF-kappaB pathway. Int. J. Mol. Sci. 2012, 13, 6236–6245. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Omene, C.; Karkoszka, J.; Bosland, M.; Eckard, J.; Klein, C.B.; Frenkel, K. Caffeic acid phenethyl ester (CAPE), derived from a honeybee product propolis, exhibits a diversity of anti-tumor effects in pre-clinical models of human breast cancer. Cancer Lett. 2011, 308, 43–53. [Google Scholar] [CrossRef]
- Nabekura, T.; Kawasaki, T.; Furuta, M.; Kaneko, T.; Uwai, Y. Effects of Natural Polyphenols on the Expression of Drug Efflux Transporter P-Glycoprotein in Human Intestinal Cells. Acs Omega 2018, 3, 1621–1626. [Google Scholar] [CrossRef]
- Takara, K.; Fujita, M.; Matsubara, M.; Minegaki, T.; Kitada, N.; Ohnishi, N.; Yokoyama, T. Effects of propolis extract on sensitivity to chemotherapeutic agents in HeLa and resistant sublines. Phytother. Res. Ptr. 2007, 21, 841–846. [Google Scholar] [CrossRef]
- Montanari, F.; Ecker, G.F. Prediction of drug-ABC-transporter interaction--Recent advances and future challenges. Adv. Drug Deliv. Rev. 2015, 86, 17–26. [Google Scholar] [CrossRef]
- Ferreira, R.J.; Ferreira, M.J.; dos Santos, D.J. Molecular docking characterizes substrate-binding sites and efflux modulation mechanisms within P-glycoprotein. J. Chem. Inf. Model. 2013, 53, 1747–1760. [Google Scholar] [CrossRef]
- Martinez, L.; Arnaud, O.; Henin, E.; Tao, H.; Chaptal, V.; Doshi, R.; Andrieu, T.; Dussurgey, S.; Tod, M.; Di Pietro, A.; et al. Understanding polyspecificity within the substrate-binding cavity of the human multidrug resistance P-glycoprotein. FEBS J. 2014, 281, 673–682. [Google Scholar] [CrossRef]
- Ambudkar, S.V.; Dey, S.; Hrycyna, C.A.; Ramachandra, M.; Pastan, I.; Gottesman, M.M. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Ann. Rev. Pharmacol. Toxicol. 1999, 39, 361–398. [Google Scholar] [CrossRef]
- Dey, S.; Ramachandra, M.; Pastan, I.; Gottesman, M.M.; Ambudkar, S.V. Evidence for two nonidentical drug-interaction sites in the human P-glycoprotein. Proc. Natl. Acad. Sci. USA 1997, 94, 10594–10599. [Google Scholar] [CrossRef] [PubMed]
- Sirota, R.; Gibson, D.; Kohen, R. The timing of caffeic acid treatment with cisplatin determines sensitization or resistance of ovarian carcinoma cell lines. Redox Biol. 2017, 11, 170–175. [Google Scholar] [CrossRef]
- Huang, C.; Huang, S.; Li, H.; Li, X.; Li, B.; Zhong, L.; Wang, J.; Zou, M.; He, X.; Zheng, H.; et al. The effects of ultrasound exposure on P-glycoprotein-mediated multidrug resistance in vitro and in vivo. J. Exp. Clin. Cancer Res. 2018, 37, 232. [Google Scholar] [CrossRef] [PubMed]
- Sheu, M.J.; Teng, Y.N.; Chen, Y.Y.; Hung, C.C. The functional influences of common ABCB1 genetic variants on the inhibition of P-glycoprotein by Antrodia cinnamomea extracts. PLoS ONE 2014, 9, e89622. [Google Scholar] [CrossRef]
- Teng, Y.-N.; Wang, Y.-H.; Wu, T.-S.; Hung, H.-Y.; Hung, C.-C. Zhankuic Acids A, B and C from Taiwanofungus camphoratus Act as Cytotoxicity Enhancers by Regulating P-Glycoprotein in Multi-Drug Resistant Cancer Cells. Biomolecules 2019, 9, 759. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.-N.; Hsieh, Y.-W.; Hung, C.-C.; Lin, H.-Y. Demethoxycurcumin Modulates Human P-Glycoprotein Function via Uncompetitive Inhibition of ATPase Hydrolysis Activity. J. Agric. Food Chem. 2015, 63, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Robertson, D.H.; Brooks, C.L., 3rd; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER-A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nonlinear Kinetic Parameters | |||
|---|---|---|---|
| ABCB1/Flp-InTM-293 | Vm (pmole/10 min) | Km (µM) | |
| Rhodamine123 only | 9.04 ± 1.00 | 56.52 ± 6.97 | |
| + Caffeic acid, 10 µg/mL | 2.47 ± 0.03* | 15.93 ± 0.18 * | |
| + Caffeic acid, 20 µg/mL | 1.57 ± 0.04 * | 10.72 ± 0.30 * | |
| Ki | 253.44 ± 2.64 | ||
| ABCB1/Flp-InTM-293 | Vm (pmole/120 min) | Km (µM) | |
| Doxorubicin only | 107.52 ± 0.001 | 179.81 ± 0.001 | |
| + Caffeic acid, 10 µg/mL | 108.31 ± 0.68 | 234.76 ± 1.52 * | |
| + Caffeic acid, 20 µg/mL | 107.52 ± 0.001 | 426.16 ± 0.00 * | |
| Ki | 14.13 ± 0.005 | ||
| Cell Line | Flp-InTM293 | ABCB1/Flp-InTM293 | ||
|---|---|---|---|---|
| Compound | IC50 ± S.E. (nM) | RF | IC50 ± S.E. (nM) | RF |
| Vincristine | 9.34 ± 0.43 | 1.00 | 778.11 ± 14.77 | 1.00 |
| + 30 μg/mL Caffeic acid | 3.37 ± 4.30 | 2.77 | 198.04 ± 6.62 | 3.90 |
| + 20 μg/mL Caffeic acid | 7.08 ± 0.09 | 1.31 | 557.46 ± 8.70 | 1.40 |
| + 10 μg/mL Caffeic acid | 9.11 ± 0.32 | 1.02 | 615.03 ± 3.09 | 2.26 |
| Paclitaxel | 89.99 ± 0.50 | 1.00 | 604.09 ± 7.09 | 1.00 |
| + 30 μg/mL Caffeic acid | 40.9 ± 0.50 | 2.20 | 121.55 ± 13.50 | 4.96 * |
| + 20 μg/mL Caffeic acid | 79.3 ± 0.67 | 1.13 | 313.06 ± 37.71 | 1.92 |
| + 10 μg/mL Caffeic acid | 86.9 ± 0.12 | 1.03 | 597.87 ± 11.25 | 1.01 |
| Doxorubicin | 8.55 ± 0.19 | 1.00 | 9023.61 ± 272.90 | 1.00 |
| + 30 μg/mL Caffeic acid | 4.07 ± 4.49 | 2.10 | 596.90 ± 24.18 | 15.11 * |
| + 20 μg/mL Caffeic acid | 7.34 ± 4.67 | 1.20 | 1299.7 ± 37.18 | 6.94 * |
| + 10 μg/mL Caffeic acid | 8.48 ± 2.58 | 1.00 | 2628.1 ± 24.49 | 3.43 |
| ABCB1/Flp-InTM-293 | Percentage of Phase ± SE (%) | |||
| Sub G1 | G0/G1 | S | G2/M | |
| Control | 0.4 ± 0.17 | 35.7 ± 1.5 | 46.2 ± 2.9 | 17.6 ± 4.3 |
| Paclitaxel 250 nM | 11.3 ± 0.2 | 27.4 ± 0.6 | 29.3 ± 1.6 | 31.8 ± 1.3 |
| Caffeic acid 20 μg/mL | 3.7 ± 0.6 | 27.4 ± 0.6 | 29.3 ± 1.6 | 31.8 ± 1.3 |
| Caffeic acid 25 μg/mL | 1.2 ± 0.1 | 41.7 ± 0.5 | 37.7 ± 0.2 | 19.2 ± 0.6 |
| Paclitaxel 250 nM + Caffeic acid 20 μg/mL | 24.1 ± 0.3 | 36.6 ± 1.4 | 24.1 ± 1.8 | 15.0 ± 0.5 |
| Paclitaxel 250 nM + Caffeic acid 25 μg/mL | 33.0 ± 9.0 | 27.5 ± 8.5 | 29.7 ± 4.8 | 14.8 ± 0.6 |
| KB/VIN | Percentage of Phase ± SE (%) | |||
| Sub G1 | G0/G1 | S | G2/M | |
| Control | 0.6 ± 0.07 | 37.3 ± 4.0 | 39.2 ± 0.7 | 22.9 ± 3.3 |
| Paclitaxel 250 nM | 12.8 ± 1.5 | 44.6 ± 1.0 | 29.2 ± 0.4 | 13.3 ± 0.1 |
| Caffeic acid 20 μg/mL | 1.5 ± 0.04 | 40.3 ± 0.9 | 45.5 ± 1.3 | 12.8 ± 0.5 |
| Caffeic acid 25 μg/mL | 1.3 ± 0.1 | 37.2 ± 0.4 | 50.1 ± 0.5 | 11.4 ± 0.3 |
| Paclitaxel 250 nM + Caffeic acid 20 μg/mL | 13.2 ± 1.2 | 44.3 ± 0.5 | 31.6 ± 1.0 | 11.0 ± 0.2 |
| Paclitaxel 250 nM + Caffeic acid 25 μg/mL | 12.7 ± 1.3 | 46.9 ± 0.1 | 23.6 ± 1.5 | 16.8 ± 0.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teng, Y.-N.; Wang, C.C.N.; Liao, W.-C.; Lan, Y.-H.; Hung, C.-C. Caffeic Acid Attenuates Multi-Drug Resistance in Cancer Cells by Inhibiting Efflux Function of Human P-Glycoprotein. Molecules 2020, 25, 247. https://doi.org/10.3390/molecules25020247
Teng Y-N, Wang CCN, Liao W-C, Lan Y-H, Hung C-C. Caffeic Acid Attenuates Multi-Drug Resistance in Cancer Cells by Inhibiting Efflux Function of Human P-Glycoprotein. Molecules. 2020; 25(2):247. https://doi.org/10.3390/molecules25020247
Chicago/Turabian StyleTeng, Yu-Ning, Charles C.N. Wang, Wei-Chieh Liao, Yu-Hsuan Lan, and Chin-Chuan Hung. 2020. "Caffeic Acid Attenuates Multi-Drug Resistance in Cancer Cells by Inhibiting Efflux Function of Human P-Glycoprotein" Molecules 25, no. 2: 247. https://doi.org/10.3390/molecules25020247
APA StyleTeng, Y.-N., Wang, C. C. N., Liao, W.-C., Lan, Y.-H., & Hung, C.-C. (2020). Caffeic Acid Attenuates Multi-Drug Resistance in Cancer Cells by Inhibiting Efflux Function of Human P-Glycoprotein. Molecules, 25(2), 247. https://doi.org/10.3390/molecules25020247