
Huisgen [3 + 2] Dipolar Cycloadditions of Phthalazinium Ylides to Activated Symmetric and Non-Symmetric Alkynes


 , , and
, , and 
Abstract
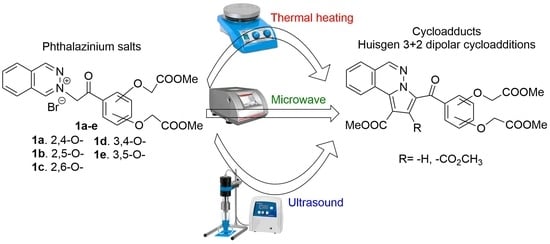
1. Introduction
2. Results and Discussion
3. Materials and Methods
General Procedure
General Procedure for Synthesis of Pyrrolophthalazine Derivatives, 5a–e and 6a–e under Conventional TH Conditions, MW and US Irradiation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moldoveanu, C.; Amariucai-Mantu, D.; Mangalagiu, V.; Antoci, V.; Maftei, D.; Mangalagiu, I.I.; Zbancioc, G. Microwave Assisted Reactions of Fluorescent Pyrrolodiazine Building Blocks. Molecules 2019, 24, 3760. [Google Scholar] [CrossRef] [PubMed]
- Zbancioc, G.; Huhn, T.; Groth, U.; Deleanu, C.; Mangalagiu, I.I. Pyrrolodiazine derivatives as blue organic luminophores: Synthesis and properties. Part 3. Tetrahedron 2010, 66, 4298–4306. [Google Scholar] [CrossRef]
- Cheng, Y.; Ma, B.; Wudl, F. Synthesis and Optical Properties of a Series of Pyrrolopyridazine Derivatives: Deep Blue Organic Luminophors for Electroluminescent Devices. J. Mater. Chem. 1999, 9, 2183–2188. [Google Scholar] [CrossRef]
- Popovici, L.; Amarandi, R.M.; Mangalagiu, I.I.; Mangalagiu, V.; Danac, R. Synthesis, molecular modelling and anticancer evaluation of new pyrrolo[1,2-b]pyridazine and pyrrolo[2,1-a]phthalazinederivatives. J. Enz. Inhib. Med. Chem. 2019, 34, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.M.; Jain, V.; Chen, T.L.; Patel, A.S.; Pidugu, H.B.; Lin, Y.W.; Wu, M.H.; Huang, J.R.; Wu, H.C.; Shah, A.; et al. Design and Synthesis of 1,2-Bis(hydroxymethyl)pyrrolo[2,1-a]phthalazine Hybrids as Potent Anticancer Agents that Inhibit Angiogenesis and Induce DNA Interstrand Cross-links. J. Med. Chem. 2019, 62, 2404–2418. [Google Scholar] [CrossRef]
- Sandeep, C.; Basavaraj, P.; Venugopala, K.N.; Rashmi, S.K.; Rashmi, V.; Odhav, B. Efficient synthesis and characterization of ethyl 7-acetyl-2-substituted 3-(substitutedbenzoyl) indolizine-1-carboxylates for in vitro anticancer activity. Asian J. Chem. 2016, 28, 1043–1048. [Google Scholar] [CrossRef]
- Sonnet, P.; Dallemagne, P.; Guillon, J.; Enguehard, C.; Stiebing, S.; Tanguy, J.; Bureau, R.; Rault, S.; Auvray, P.; Moslemi, S.; et al. New aromatase inhibitors. Synthesis and biological activity of aryl–substituted pyrrolizine and indolizine derivatives. Bioorg. Med. Chem. 2000, 8, 945–955. [Google Scholar] [CrossRef]
- Al Matarneh, C.; Ciobanu, C.I.; Mangalagiu, V.; Zbancioc, G.; Danac, R. Microwave assisted synthesis of six member ring azaheterocycles and their antimycobacterial and anticancer evaluation. Rev. Chim. 2020, 71, 287–293. [Google Scholar] [CrossRef]
- Mantu, D.; Luca, M.C.; Moldoveanu, C.; Zbancioc, G.; Mangalagiu, I.I. Synthesis and antituberculosis activity of some new pyridazine derivatives. Part II. Eur. J. Med. Chem. 2010, 45, 5164–5168. [Google Scholar] [CrossRef]
- Gundersen, L.L.; Charnock, C.; Negussie, A.H.; Rise, F.; Teklu, S. Synthesis of indolizine derivatives with selective antibacterial activity against Mycobacterium tuberculosis. Eur. J. Pharm. Sci. 2007, 30, 26–35. [Google Scholar] [CrossRef]
- Huang, W.; Zuo, T.; Luo, X.; Jin, H.; Liu, Z.; Yang, Z.; Yu, X.; Zhang, L.; Zhang, L. Indolizine derivatives as HIV–1 VIF–ElonginC interaction inhibitors. Chem. Biol. Drug. Des. 2013, 81, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Narajji, C.; Karvekar, M.D.; Das, A.K. Synthesis and antioxidant activity of 3,3′–diselanediylbis (N, N–disubstituted indolizine–1–carboxamide) and derivatives. S. Afr. J. Chem. 2008, 61, 53–55. [Google Scholar]
- Padwa, A.; Houk, K.N.; Yamaguchi, K. (Eds.) 1,3-Dipolar Cycloaddition Chemistry; John Wiley & Sons: New York, NY, USA, 1984; Volume 2, Chapters 12–13; pp. 277–406, 407–450. [Google Scholar]
- Epiotis, N.D. Theory of Organic Reactions; Springer: Berlin, Germany, 1978; ISBN 978-3-642-66827-2. [Google Scholar]
- Mangalagiu, I.I.; Druta, I.; Constantinescu, M.; Humelnicu, I.; Petrovanu, M. Pyridazinium ylides. Regiochemistry. Tetrahedron 1996, 52, 8853–8862. [Google Scholar] [CrossRef]
- Mangalagiu, I.I.; Petrovanu, M. Pyridazinium ylides. Regiochemistry of addition. Acta Chim. Scand. 1997, 51, 927–931. [Google Scholar] [CrossRef][Green Version]
- Caprosu, M.; Olariu, I.; Mangalagiu, I.I.; Constantinescu, M.; Petrovanu, M. The Regiochemistry of the cycloaddition of 4-R-phenacylpyridazinium ylides to non symmetrical substituted olefins. Eur. J. Org. Chem. 1999, 12, 3501–3504. [Google Scholar] [CrossRef]
- Mangalagiu, I.I.; Mangalagiu, G.; Drochioiu, G.; Deleanu, C.; Petrovanu, M. 4-Methyl pyrimidinium ylides. Part 7: 3+2 Dipolar cycloadditions to non-symmetrical substituted alkenes and alkynes. Tetrahedron 2003, 59, 111–114. [Google Scholar] [CrossRef]
- Dima, S.; Mangalagiu, I.I.; Caprosu, M.; Constantinescu, M.; Humelnicu, I.; Petrovanu, M. The regiochemistry of the cycloaddition of 1-methylphthalazinium ylides to non-symmetrically substituted olefins. J. Serb. Chim. Soc. 1997, 62, 105–111. [Google Scholar]
- Dima, S.; Zbancioc, G.; Mangalagiu, I.I. The 1,3- dipolar cycloaddition of 1-methylphtalazinium ylides to non-symmetrically activated alkenes on a solid support under microwave irradiation. J. Serb. Chim. Soc. 2006, 71, 103–110. [Google Scholar] [CrossRef]
- Dumitrascu, F.; Caproiu, M.T.; Georgescu, F.; Draghici, B.; Popa, M.M.; Georgescu, E. New Pyrrolo[2,1-a]phthalazine Derivatives by One-Pot Three-Component Synthesis. Synlett 2010, 16, 2407–2410. [Google Scholar] [CrossRef]
- Zbancioc, G.; Mangalagiu, I.I. Pyrrolopyridazine Derivatives as Blue Organic Luminophores: Synthesis and Properties. Part 2. Tetrahedron 2010, 66, 278–282. [Google Scholar] [CrossRef]
- Rodriquez, M.; Taddei, M. Synthesis of Heterocycles via Microwave-Assisted Cycloadditions and Cyclocondensations. In Microwave-Assisted Synthesis of Heterocycle; Van der Eycken, E., Kappe, C.O., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 213–266. ISBN 9783540309833. [Google Scholar]
- Perreux, L.; Loupy, A. Nonthermal Effects of Microwaves in Organic Synthesis. In Microwaves in Organic Synthesis, 2nd ed.; Loupy, A., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 134–218. ISBN 9783527314522. [Google Scholar]
- Shingare, M.S.; Shingate, B.B. Ultrasound in Synthetic Applications and Organic Chemistry. In Handbook on Applications of Ultrasound: Sonochemistry for Sustainability, 1st ed.; Chen, D., Sharma, S.K., Mudhoo, A., Eds.; CRC Press; Taylor & Francis Group: Boca Raton, FL, USA, 2012; ISBN 9781439842065. [Google Scholar]
- Mason, T.J.; Peters, D. Practical Sonochemistry. Power Ultrasound Uses and Applications, 2nd ed.; Ellis Horwood: Chichester, UK, 2002; ISBN 9781898563839. [Google Scholar]
- Zbancioc, G.; Bejan, V.; Risca, M.; Moldoveanu, C.; Mangalagiu, I.I. Microwave assisted reactions of new azaheterocyles compounds. Molecules 2009, 14, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Moldoveanu, C.; Mangalagiu, I.; Isac, D.L.; Airinei, A.; Zbancioc, G. A New Pathway for the Synthesis of a New Class of Blue Fluorescent Benzofuran Derivatives. Molecules 2018, 23, 1968. [Google Scholar] [CrossRef]
- Xu, J. Microwave Irradiation and Selectivities in Organic Reactions. Prog. Chem. 2007, 19, 700–712. [Google Scholar]
- Xu, J. Selelectivities in microwave-assisted organic reactions. In Advances in Microwave Chemistry; Banik, B.K., Bandyopadhyay, D., Eds.; CRC Press, Taylor & Francis Group: Bosa Roca, FL, USA, 2018; Chapter 6; pp. 257–291. ISBN 9780815375197. [Google Scholar]
- Li, X.; Xu, J. Effects of the Microwave Power on the Microwave-assisted Esterification. Curr. Microw. Chem. 2017, 4, 158–162. [Google Scholar] [CrossRef]
- Zbancioc, G.; Moldoveanu, C.; Zbancioc, A.M.; Mangalagiu, I.I. Microwave Assisted Synthesis of New Pyrrolopyridazine Derivatives with Acetophenone Skeleton. Part V. Curr. Microw. Chem. 2014, 1, 41–46. [Google Scholar] [CrossRef]
- Bejan, V.; Moldoveanu, C.; Mangalagiu, I.I. Ultrasounds assisted reactions of steroid analogous of anticipated biological activities. Ultrason. Sonochem. 2009, 16, 312–315. [Google Scholar] [CrossRef]
- Zbancioc, G.; Zbancioc, A.M.; Mangalagiu, I.I. Ultrasound and microwave assisted synthesis of dihydroxyacetophenone derivatives with or without 1,2-diazine skeleton. Ultrason. Sonochem. 2014, 21, 802–811. [Google Scholar] [CrossRef]
- Zbancioc, G.; Moldoveanu, C.; Mangalagiu, I.I. Ultrasound assisted synthesis of imidazolium salts: An efficient way to ionic liquids. Ultrason. Sonochem. 2015, 23, 376–384. [Google Scholar] [CrossRef]
- Varma, R.S. Green Chemistry with Microwave Energy. In Innovations in Green Chemistry and Green Engineering; Anastas, P.T., Zimmerman, J.B., Eds.; Springer: New York, NY, USA, 2013; ISBN 9781461458166. [Google Scholar]
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: New York, NY, USA, 1988. [Google Scholar]
- Zbancioc, A.M.; Miron, A.; Tuchilus, C.; Rotinberg, P.; Mihai, C.T.; Mangalagiu, I.I.; Zbancioc, G. Synthesis and In vitro Analysis of Novel Dihydroxyacetophenone Derivatives with Antimicrobial and Antitumor Activities. Med. Chem. 2014, 10, 476–483. [Google Scholar] [CrossRef]
- Mangalagiu, G.; Mangalagiu, I.; Olariu, R.; Petrovanu, M. 4-Methylpyrimidinium ylides II: Selective reactions of pyrimidinium ylides with activated alkyne. Synthesis 2000, 14, 2047–2050. [Google Scholar] [CrossRef]
- Zhou, J.; Hu, Y.; Hu, H. Synthesis of pyrrolo[2,1-a]phthalazines by 1,3-dipolar cycloaddition of phthalazinium N-ylides with alkenes in the presence of tetrakis-pyridine cobalt (II) dichromate. J. Het. Chem. 2000, 37, 1165–1168. [Google Scholar] [CrossRef]
- Dumitrascu, F.; Draghici, C.; Caproiu, M.T.; Dumitrescu, D.; Badoiu, A. New pyrrolo[2,1-a]phthalazine derivatives by 1,2-dipolar cycloaddition reactions. Rev. Roum. Chim. 2006, 51, 643–647. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Microwaves (MW) | Ultrasounds (US) | Conventional (TH) | |||
|---|---|---|---|---|---|---|
| Reaction Time | Yield, % | Reaction Time | Yield, % | Reaction Time | Yield, % | |
| 5a | 10 min | 93 | 20 min | 90 | 360 min | 74 |
| 5b | 10 min | 89 | 20 min | 85 | 360 min | 69 |
| 5c | 10 min | 86 | 20 min | 85 | 360 min | 66 |
| 5d | 10 min | 90 | 20 min | 90 | 360 min | 70 |
| 5e | 10 min | 90 | 20 min | 89 | 360 min | 74 |
| 6a | 40 min | 82 | 60 min | 81 | 1 week | 55 |
| 6b | 40 min | 77 | 60 min | 77 | 1 week | 50 |
| 6c | 40 min | 75 | 60 min | 75 | 1 week | 48 |
| 6d | 40 min | 82 | 60 min | 80 | 1 week | 52 |
| 6e | 40 min | 80 | 60 min | 78 | 1 week | 52 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antoci, V.; Moldoveanu, C.; Danac, R.; Mangalagiu, V.; Zbancioc, G. Huisgen [3 + 2] Dipolar Cycloadditions of Phthalazinium Ylides to Activated Symmetric and Non-Symmetric Alkynes. Molecules 2020, 25, 4416. https://doi.org/10.3390/molecules25194416
Antoci V, Moldoveanu C, Danac R, Mangalagiu V, Zbancioc G. Huisgen [3 + 2] Dipolar Cycloadditions of Phthalazinium Ylides to Activated Symmetric and Non-Symmetric Alkynes. Molecules. 2020; 25(19):4416. https://doi.org/10.3390/molecules25194416
Chicago/Turabian StyleAntoci, Vasilichia, Costel Moldoveanu, Ramona Danac, Violeta Mangalagiu, and Gheorghita Zbancioc. 2020. "Huisgen [3 + 2] Dipolar Cycloadditions of Phthalazinium Ylides to Activated Symmetric and Non-Symmetric Alkynes" Molecules 25, no. 19: 4416. https://doi.org/10.3390/molecules25194416
APA StyleAntoci, V., Moldoveanu, C., Danac, R., Mangalagiu, V., & Zbancioc, G. (2020). Huisgen [3 + 2] Dipolar Cycloadditions of Phthalazinium Ylides to Activated Symmetric and Non-Symmetric Alkynes. Molecules, 25(19), 4416. https://doi.org/10.3390/molecules25194416