
Clonal Variation in the Bark Chemical Properties of Hybrid Aspen: Potential for Added Value Chemicals

 ,
,  ,
,  and
and
Abstract
1. Introduction
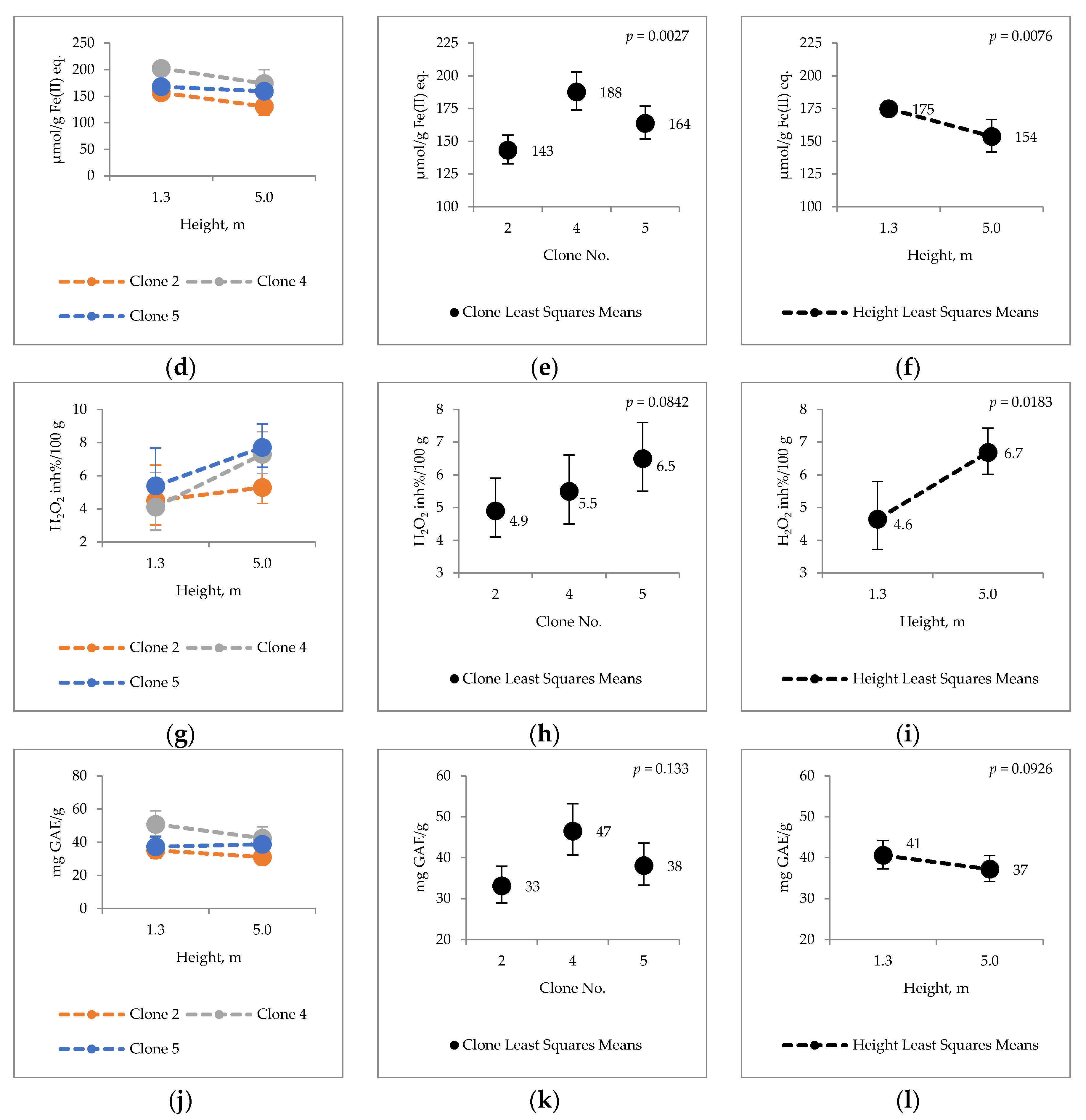
2. Results
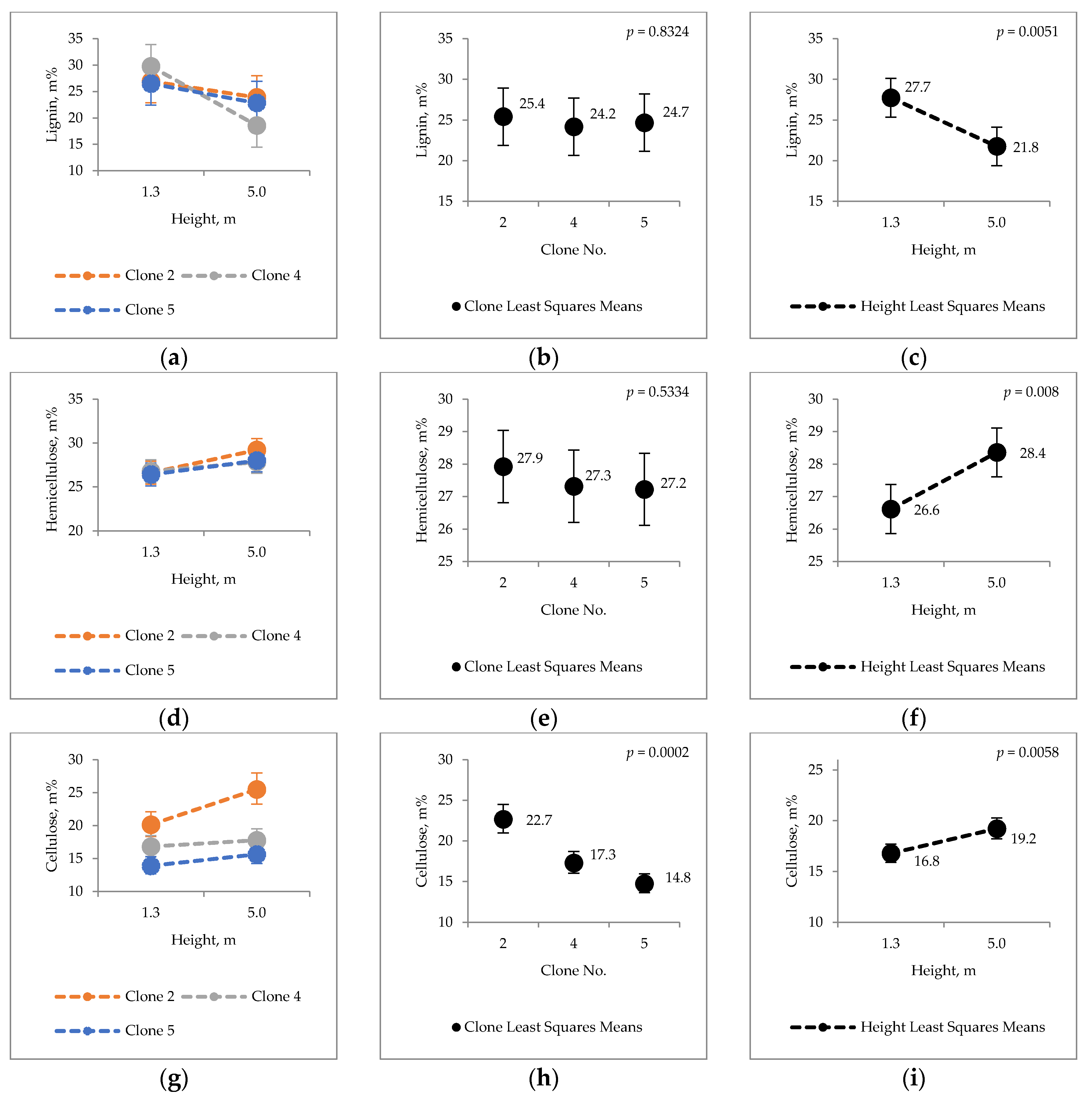
2.1. Clonal Variation in Major Chemical Components
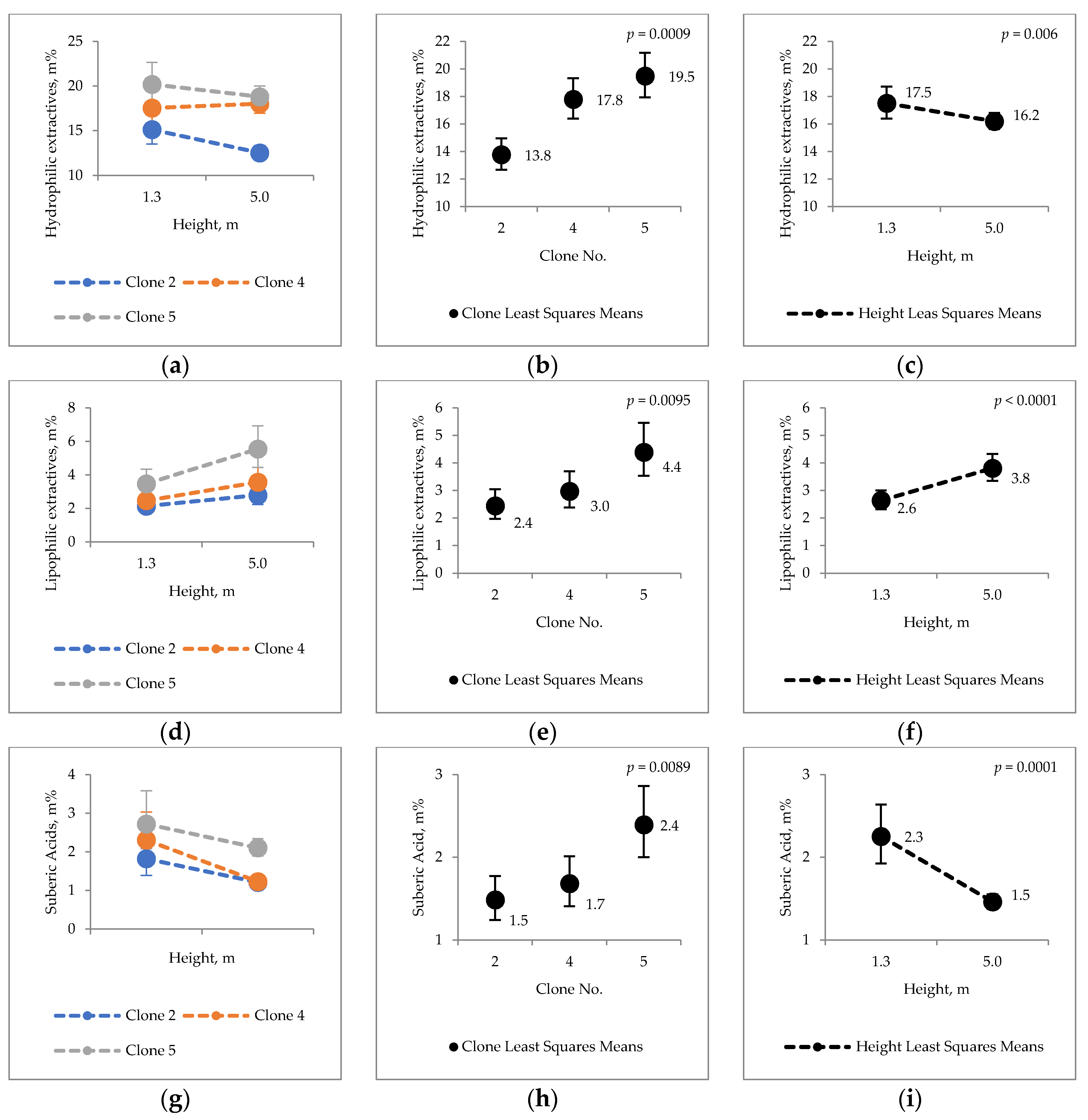
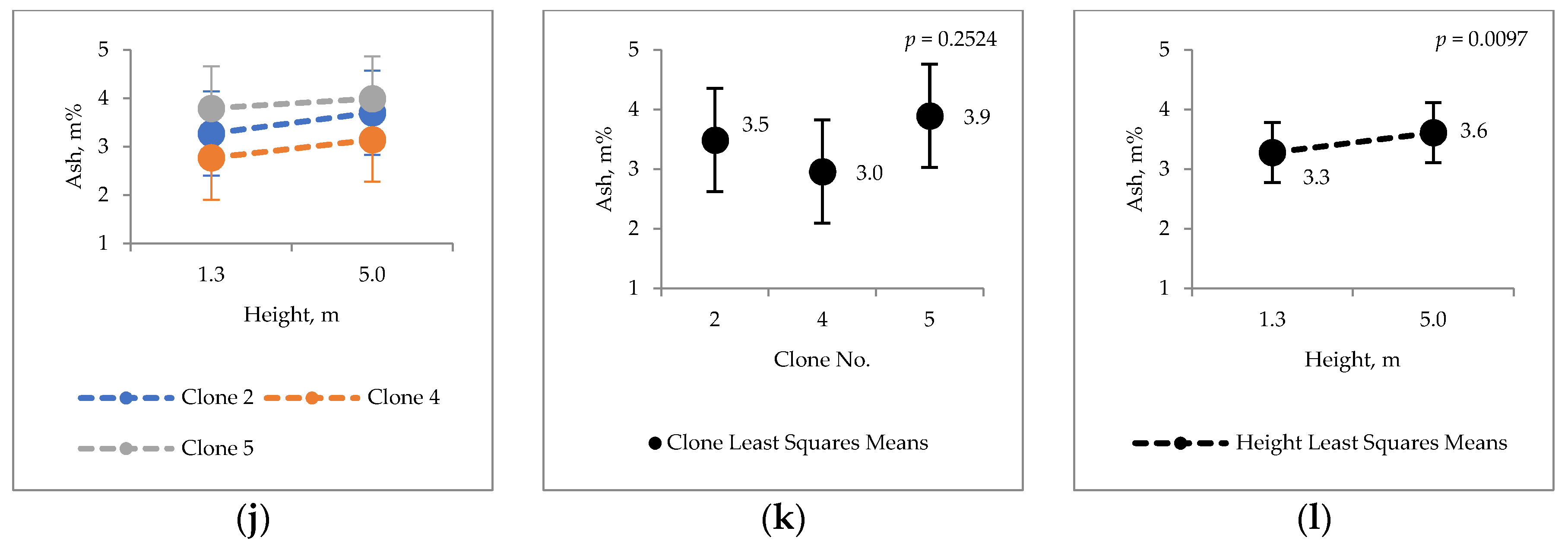
2.2. Extraneous Material
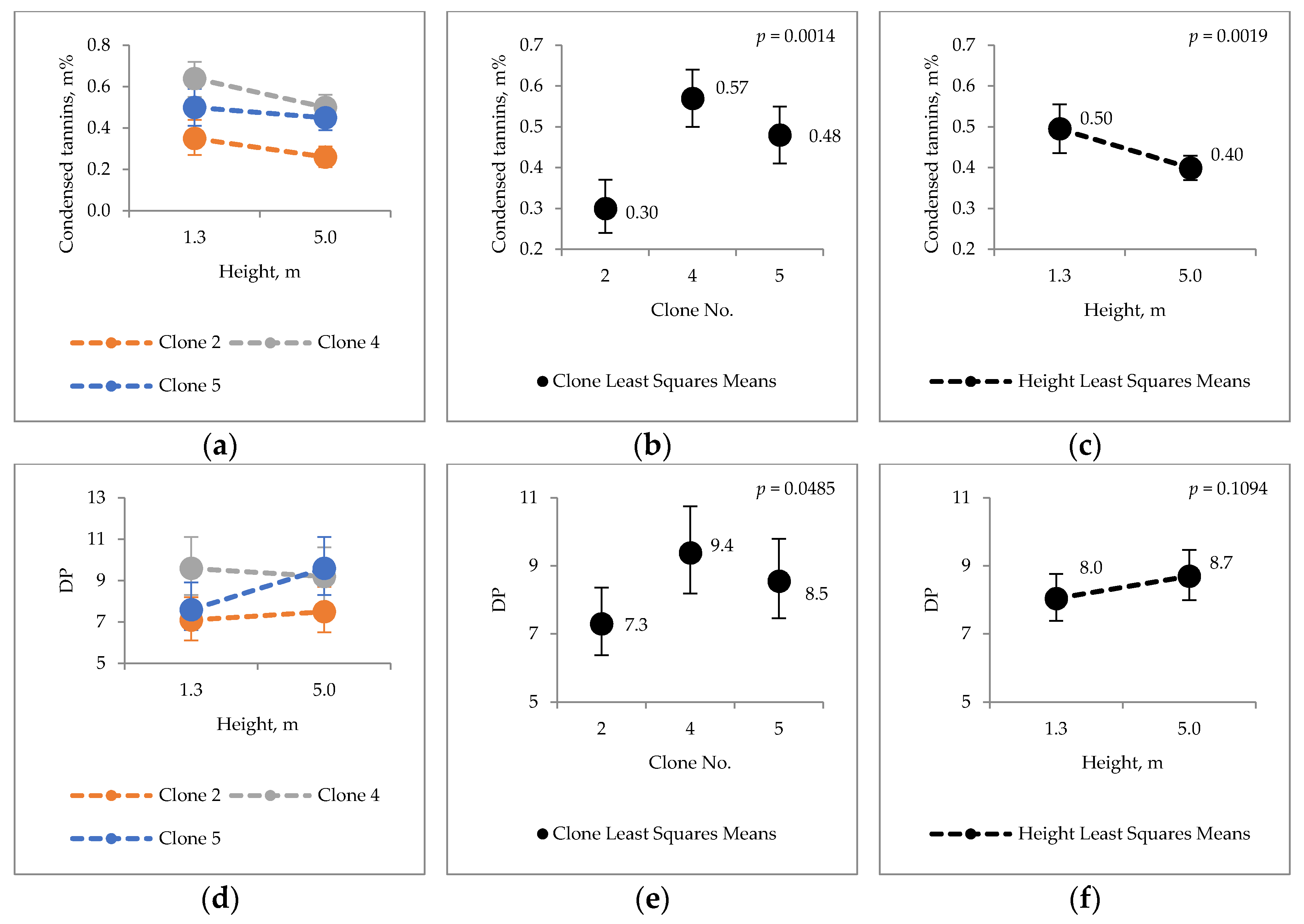
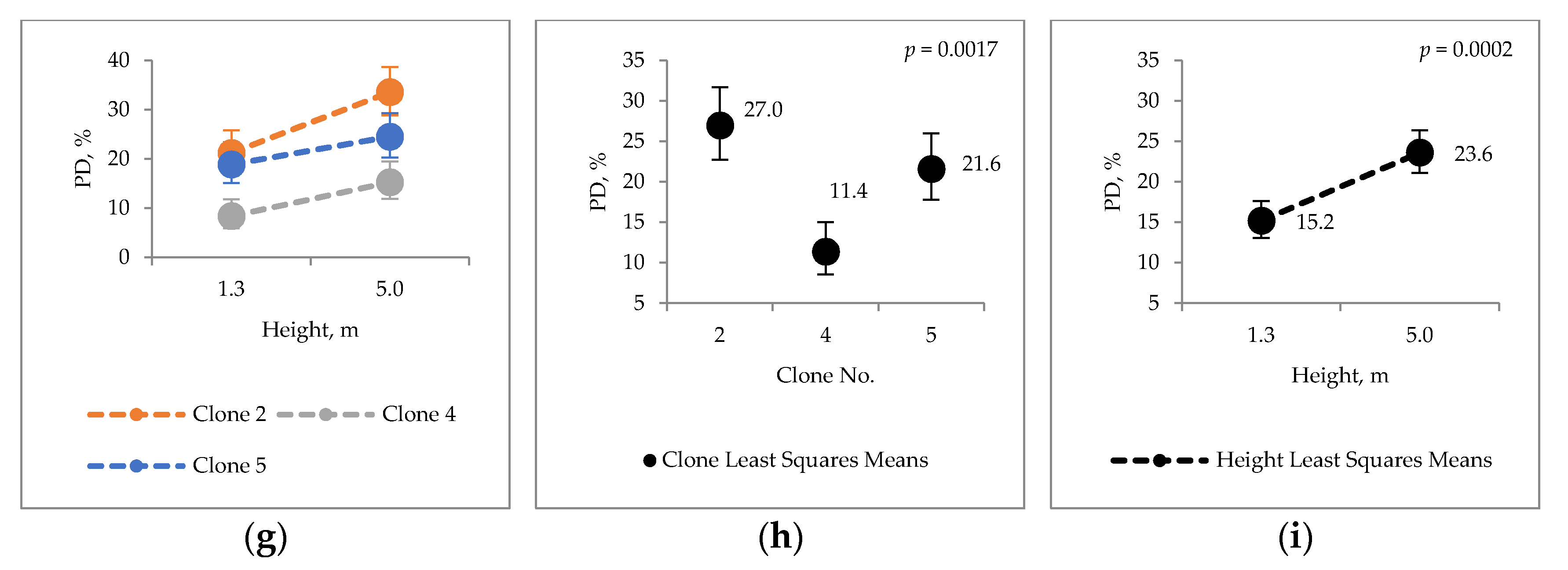
2.3. Condensed Tannins
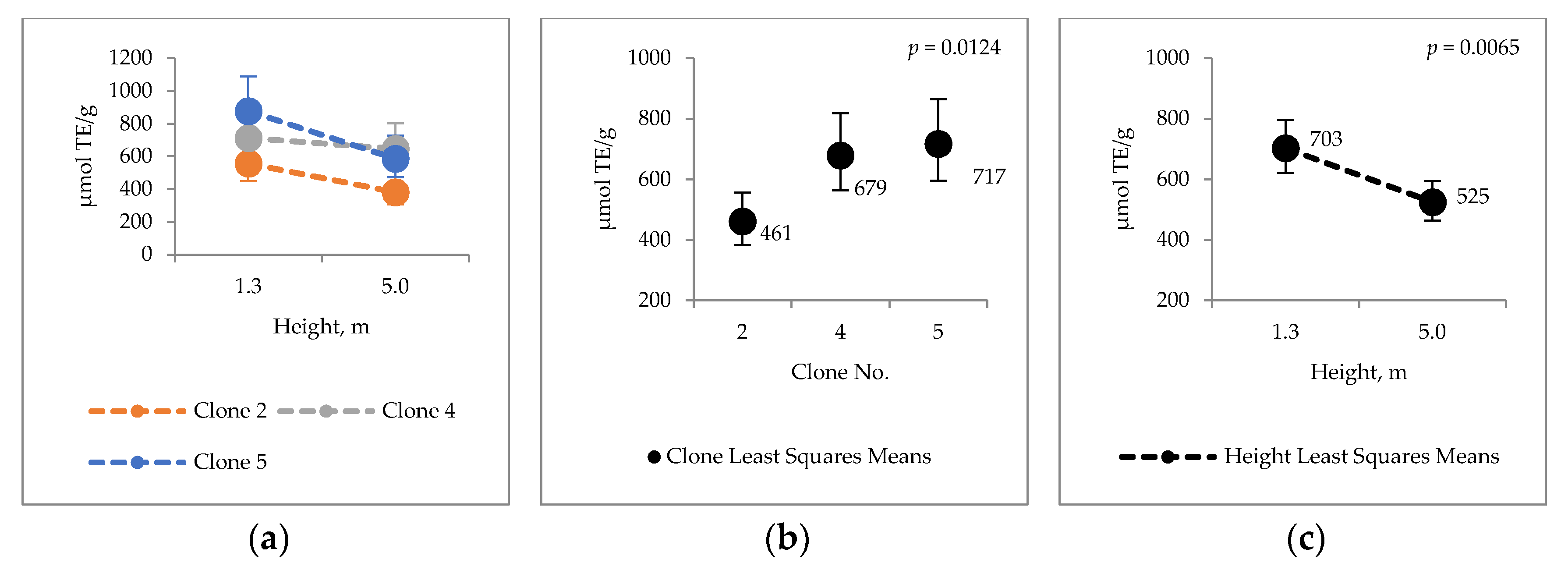
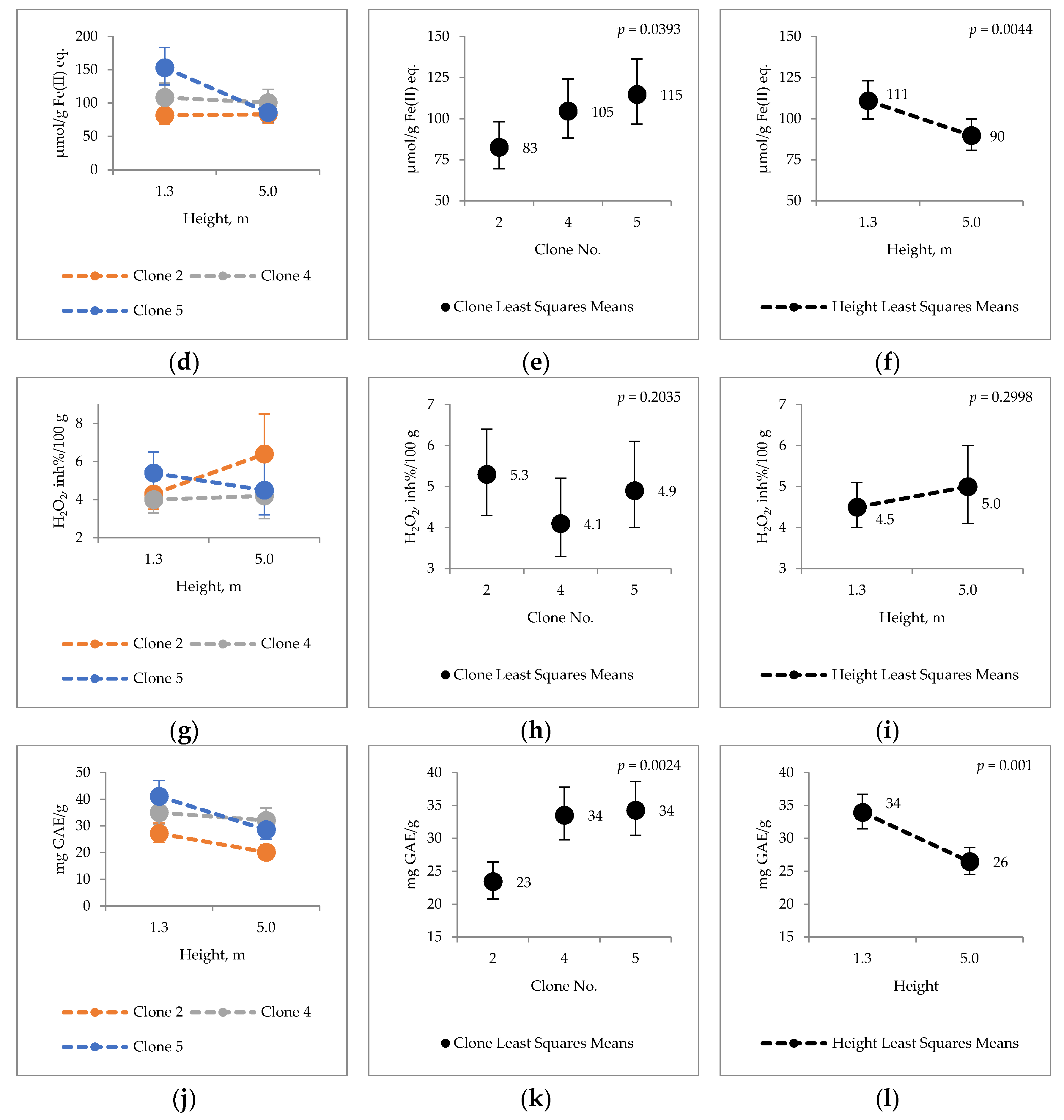
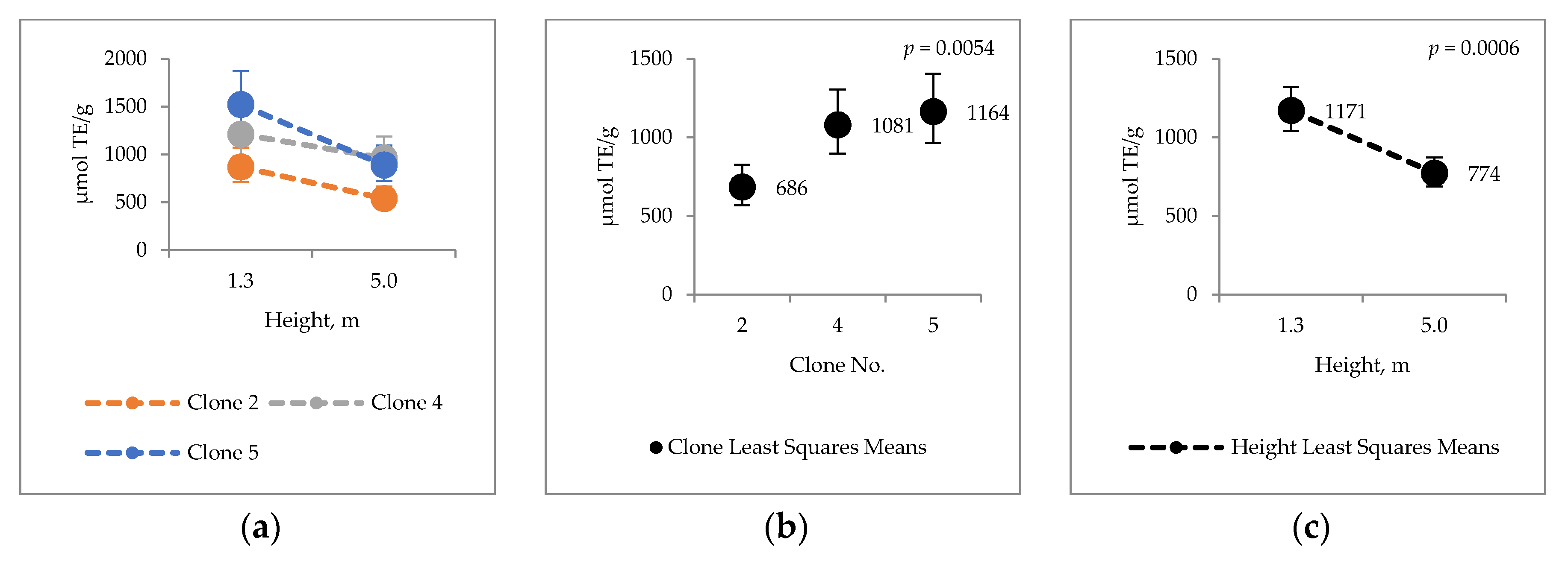
2.4. Bark Hot Water and Ethanol Extracts Antioxidative Capacity and Total Phenol Contents
3. Discussion
3.1. Major Chemical Components of Hybrid Aspen Bark
3.2. Extraneous Material of Hybrid Aspen Bark
3.3. Antioxidative Properties of Hybrid Aspen Bark
4. Materials and Methods
4.1. Sample Trees
4.2. Statistical Analysis
4.3. Wood Analysis
4.3.1. Sample Preparation
4.3.2. Lipohilic and Hydrophilic Extractives
4.3.3. Suberic Acids
4.3.4. Lignin
4.3.5. Cellulose
4.3.6. Hemicellulose
4.4. Condensed Tannins
4.5. Antioxidative Properties and Total Phenol Content
4.5.1. Ferric Reducing Antioxidant Power (FRAP)
4.5.2. Oxygen Radical Absorbance Capacity
4.5.3. H2O2 Scavenging Assay
4.5.4. Folin-Ciocalteu Assay
4.6. Ash Content
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Beuker, E.; Viherä-Aarnio, A.; Hynynen, J. Growth Potential of First Generation Hybrid Aspen Plantations in Southern Finland. Poplars and Other Fast-Growing Trees—Renewable Resources for Future Green Economies. In Proceedings of the International Poplar Commission, 25th Session, Berlin, Germany, 13–16 September 2016; Working Paper IPC 14. FAO: Rome, Italy, 2016; p. 118. [Google Scholar]
- Hytönen, J. Biomass, nutrient content and energy yield of short-rotation hybrid aspen (P. tremula × P. tremuloides) coppice. For. Ecol. Manag. 2018, 413, 21–31. [Google Scholar] [CrossRef]
- Rytter, L.; Stener, L. Productivity and thinning effects in hybrid aspen (Populus tremula L. × P. tremuloides Michx.) stands in southern Sweden. Int. J. For. Res. 2005, 78, 285–295. [Google Scholar] [CrossRef]
- Hytönen, J.; Beuker, E.; Viherä-Aarnio, A. Clonal variation in basic density, moisture content and heating value of hybrid aspen wood. Silva Fenn. 2018, 52, 1–15. [Google Scholar] [CrossRef]
- Honkaniemi, J.; Ahtikoski, A.; Piri, T. Financial incentives to perform stump treatment against Heterobasidionroot rot in Norway spruce dominated forests, the case of Finland. For. Policy Econ. 2019, 105, 1–9. [Google Scholar] [CrossRef]
- Blomqvist, M.; Kosunen, M.; Starr, M.; Kantila, T.; Holopainen, M.; Lyytikäinen-saarenmaa, P. Modelling the predisposition of Norway spruce to Ips typographus L. infestation by means of environmental factors in southern Finland. Eur. J. For. Res. 2018, 137, 675–691. [Google Scholar] [CrossRef]
- Forest Industries’ Wood Consumption. 2019. Available online: https://stat.luke.fi/en/forest-industries-wood-consumption-2019_en (accessed on 30 August 2020).
- Devappa, R.K.; Rakshit, S.K.; Dekker, R.F.H. Forest biorefinery: Potential of poplar phytochemicals as value-added co-products. Biotechnol. Adv. 2015, 33, 681–716. [Google Scholar] [CrossRef]
- Doiuf, P.N.; Stevanovic, T.; Cloutier, A. Antioxidant properties and polyphenol contents of trembling aspen bark extracts. Wood Sci. Technol. 2009, 43, 457–470. [Google Scholar] [CrossRef]
- Voipio, R.; Laakso, T. Pienikokoisten puiden maanpäällisen biomassan kemiallinen koostumus. Summary: Chemical composition of the above ground biomass of small-sized trees. Metsäntutkimuslaitos 1992, 789, 22. [Google Scholar]
- Alekseeva, E.A.; Agranat, A.L.; Solodkij, F.T. Chemical composition of lipids of Aspen bark. Gidroliz. Lesokhimicheskaya Promyshlennost 1970, 23, 13–14. [Google Scholar]
- Holloway, P.J. Some variations in the composition of suberin from the cork layers of higher plants. Phytochemistry 1983, 22, 495–502. [Google Scholar] [CrossRef]
- Yemele, M.C.N.; Koubaa, A.; Cloutier, A.; Soulounganga, P.; Wolcott, M. Effect of bark fiber content and size on the mechanical properties of bark/HDPE composites. Compos. Part A Appl. Sci. Manuf. 2010, 41, 131–137. [Google Scholar] [CrossRef]
- Yemelea, M.C.N.; Koubaab, A.; Cloutierc, A.; Souloungangac, P.; Stevanovic, T.; Wolcottd, M.P. Effects of hot water treatment of raw bark, coupling agent, and lubricants on properties of bark/HDPE composites. Ind. Crops Prod. 2013, 42, 50–56. [Google Scholar] [CrossRef]
- Kumar, A.; Jyske, T.; Möttönen, V. Properties of Injection Molded Biocomposites Reinforced with Wood Particles of Short-Rotation Aspen and Willow. Polymers 2020, 12, 257. [Google Scholar] [CrossRef]
- Sjöström, E. Wood Chemistry, Fundamentals and Applications, 2nd ed.; Academic Press: San Diego, CA, USA, 1993; pp. 54, 63, 72. [Google Scholar]
- Brinchi, L.; Cotana, F.; Fortunati, E.; Kenny, J.M. Production of nanocrystalline cellulose from lignocellulosic biomass: Technology and applications. Carbohydr. Polym. 2013, 94, 154–169. [Google Scholar] [CrossRef]
- Lora, J.H.; Glasser, W.G. Recent Industrial Applications of Lignin: A Sustainable Alternative to Nonrenewable Materials. J. Polym. Environ. 2002, 10, 39–47. [Google Scholar] [CrossRef]
- Stewart, D. Lignin as a base material for materials applications: Chemistry, application and economics. Ind. Crops Prod. 2008, 27, 202–207. [Google Scholar] [CrossRef]
- Ebringerová, A. Structural Diversity and Application Potential of Hemicelluloses. Macromol. Symp. 2006, 232, 1–12. [Google Scholar] [CrossRef]
- Mamman, A.S.; Lee, J.-M.; Kim, Y.-C.; Hwang, I.T.; Park, N.-J.; Hwang, Y.K.; Chang, J.-S.; Hwang, J.-S. Furfural: Hemicellulose/xylose-derived biochemical. Biofuel Bioprod. Biorefin. 2008, 2, 438–454. [Google Scholar] [CrossRef]
- Rusanen, A.; Lappalainen, K.; Kärkkäinen, J.; Tuuttila, T.; Mikola, M.; Lassi, U. Selective hemicellulose hydrolysis of Scots pine sawdust. Biomass Conv. Bioref. 2019, 9, 283–291. [Google Scholar] [CrossRef]
- Lappalainen, K.; Kuorikoski, E.; Vanvyve, E.; Dong, Y.; Kärkkäinen, J.; Niemelä, M.; Lassi, U. Bronsted and Lewis Acid Catalyzed Conversion of Pulp Industry Waste Biomass to Levulinic Acid. Bioresources 2019, 14, 7025–7040. [Google Scholar]
- Basu, P. Biomass Gasification, Pyrolysis and Torrefaction Practical Design and Theory, 2nd ed.; Elsevier Inc., Academic Press: San Diego, CA, USA, 2013; pp. 87–92. [Google Scholar]
- Hagner, M.; Tiilikkala, K.; Lindqvist, I.; Niemelä, K.; Wikberg, H.; Källi, A.; Rasa, K. Performance of Liquids from Slow Pyrolysis and Hydrothermal Carbonization in Plant Protection. Waste Biomass Valor 2020, 11, 1005–1016. [Google Scholar] [CrossRef]
- Miranda, I.; Gominho, J.; Mirra, I.; Pereira, H. Fractioning and chemical characterization of barks of Betula pendula and Eucalyptus globulus. Ind. Crops. Prod. 2013, 41, 299–305. [Google Scholar] [CrossRef]
- Pinto, P.C.R.O.; Sousa, A.F.; Silvestre, A.J.D.; Neto, C.P.; Gandini, A.; Eckerman, C.; Holmblom, B. Quercus suber and Betula pendula outer barks as renewable sources of oleochemicals: A comparative study. Ind. Crops. Prod. 2009, 29, 126–132. [Google Scholar] [CrossRef]
- Korpinen, R.I.; Kilpeläinen, P.; Sarjala, T.; Nurmi, M.; Saloranta, P.; Holmbom, T.; Koivula, H.; Mikkonen, K.S.; Willför, S.; Saranpää, P.T. The Hydrophobicity of Lignocellulosic Fiber Network Can Be Enhanced with Suberin Fatty Acids. Molecules 2019, 24, 4391. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Cheng, S.; Yan, Z.; Leitch, M.; Xu, C. Valorization of bark for chemicals and materials: A review. Renew. Sust. Energ. Rev. 2013, 26, 560–578. [Google Scholar] [CrossRef]
- Varila, T.; Brännström, H.; Kilpeläinen, P.; Hellström, J.; Romar, H.; Nurmi, J.; Lassi, U. From Norway spruce bark to carbon foams: Characterization, and applications. Bioresources 2020, 15, 3651–3666. [Google Scholar]
- Lindroth, R.L.; Hwang, S.-Y. Clonal variation in Foliar Chemistry of Quaking Aspen (Populus tremuloides Michx.). Biochem. Syst. Ecol. 1996, 24, 357–364. [Google Scholar] [CrossRef]
- Kähkönen, M.P.; Hopia, A.I.; Vuorela, H.J.; Rauha, J.-P.; Pihlaja, K.; Kujala, T.S.; Heinonen, M. Antioxidant Activity of Plant Extracts Containing Phenolic Compounds. J. Agric. Food Chem. 1999, 47, 3954–3962. [Google Scholar] [CrossRef]
- Prior, R.L.; Cao, G.; Martin, A.; Sofic, E.; McEwen, J.; O’Brien, C.; Lischner, N.; Ehlenfeldt, M.; Kalt, W.; Krewer, G.; et al. Antioxidant Capacity as Influenced by Total Phenolic and Anthocyanin Content, Maturity, and Variety of Vaccinium Species. J. Agric. Food Chem. 1998, 46, 2686–2693. [Google Scholar] [CrossRef]
- Reuter, J.; Merfort, I.; Schempp, C.M. Botanicals in Dermatology, An Evidence based Review. Am. J. Clin. Dermatol. 2010, 11, 247–267. [Google Scholar]
- Brewer, M.S. Natural Antioxidants: Sources, Compounds, Mechanisms of Action, and Potential Applications. Compr. Rev. Food Sci. Food Saf. 2011, 10, 221–247. [Google Scholar] [CrossRef]
- Ihalainen, M.; Alho, J.; Kolehmainen, O.; Pukkala, T. Expert models for bilberry and cowberry yields in Finnish forests. For. Ecol. Manag. 2002, 157, 15–22. [Google Scholar] [CrossRef]
- Salo, K. Non-timber forest products and their utilization. In Multiple-Use Forestry in the Nordic Countries; Hytönen, M., Ed.; METLA, Finnish Forest Research Institute, Helsinki Research Centre: Helsinki, Finland, 1995; pp. 117–134. [Google Scholar]
- Finnish Food Authority. Available online: https://www.ruokavirasto.fi/en/farmers/plant-production/forest-tree-seed-and-seedling-production/basic-material/approved-basic-material/ (accessed on 29 August 2020).
- Burnham, K.; Anderson, D. Model Selection and Multimodel Inference: A Practical Information-Theoretic Approach, 2nd ed.; Springer: New York, NY, USA, 2002. [Google Scholar]
- Gbur, E.; Stroup, W.; McCarter, K.; Durham, S.; Young, L.; Christman, M.; West, M.; Kramer, M. Analysis of Generalized Linear Mixed Models in the Agricultural and Natural Resources Sciences; American Society of Agronomy: Madison, WI, USA, 2012. [Google Scholar]
- Westfall, P. Multiple Testing of General Contrasts Using Logical Constraints and Correlations. J. Am. Stat. Assoc. 1997, 92, 299–306. [Google Scholar] [CrossRef]
- Kenward, M.; Roger, J. An Improved Approximation to the Precision of Fixed Effects from Restricted Maximum Likelihood. Comput. Stat. Data Anal. 2009, 53, 2583–2595. [Google Scholar] [CrossRef]
- Krogell, J.; Holmbom, B.; Pranovich, A.; Hemming, J.; Willför, S. Extraction and chemical characterization of Norway spruce inner and outer bark. Nord. Pulp Pap. Res. J. 2012, 27, 6–17. [Google Scholar] [CrossRef]
- Sluiter, A.; Hames, B.; Ruiz, R.; Scarlata, C.; Sluiter, J.; Templeton, D.; Crocker, D. Determination of structural carbohydrates and lignin in biomass. In Laboratory Analytical Procedure (LAP); NREL: Denver, CO, USA, 2012; pp. 1–15. [Google Scholar]
- Sundherg, A.; Sundherg, K.; Lillandt, C.; Holmbom, B. Determination of hemicelluloses and pectins in wood and pulp fibres by methanolysis and gas chromatography. Nord. Pulp Pap. Res. J. 1996, 11, 216–219. [Google Scholar] [CrossRef]
- Vaario, L.-M.; Asamizu, S.; Sarjala, T.; Matsushita, N.; Onaka, H.; Xia, Y.; Kurokochi, H.; Morinaga, S.-I.; Huang, J.; Zhang, S.; et al. Bioactive properties of streptomyces may affect the dominance of Tricholoma matsutake in shiro. Symbiosis 2020, 81, 1–13. [Google Scholar] [CrossRef]
- Benzie, I.F.F.; Strain, J.J. The Ferric Reducing Ability of Plasma (FRAP) as a Measure of “Antioxidant Power”: The FRAP Assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef]
- Huang, D.; Ou, B.; Hampsch-Woodill, M.; Flanagan, J.A.; Prior, R.L. High-throughput assay of oxygen radical absorbance capacity (ORAC) using a multichannel liquid handling system coupled with a microplate fluorescence reader in 96-well format. J. Agric. Food Chem. 2002, 50, 4437–4444. [Google Scholar] [CrossRef]
- Prior, R.L.; Hoang, H.; Gu, L.; Wu, X.; Bacchiocca, M.; Howard, L.; Hampsch-Woodill, M.; Huang, D.; Ou, B.; Jacob, R. Assays for hydrophilic and lipophilic antioxidant capacity (oxygen radical absorbance capacity (ORACFL)) of plasma and other biological and food samples. J. Agric. Food Chem. 2003, 51, 3273–3279. [Google Scholar] [CrossRef]
- Hazra, B.; Biswas, S.; Mandal, N. Antioxidant and free radical scavenging activity of Spondias pinnata. BMC Complement Altern. Med. 2008, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.-Y.; Woollard, A.C.; Wolff, S.P. Hydrogen peroxide production during experimental protein glycation. FEBS Lett. 1990, 268, 69–71. [Google Scholar] [CrossRef]
- Singleton, V.L.; Rossi, J.A., Jr. Colorimetry of total phenolics with phosphomolybdic-phosphotungstic acid reagents. Am. J. Enol. Viticult. 1965, 16, 144–158. [Google Scholar]
- Singleton, V.L.; Orthofer, R.; Lamuela-Raventós, R.M. Analysis of total phenols and other oxidation substrates and antioxidants by means of Folin-Ciocalteu reagent. Methods Enzymol. 1999, 299, 152–178. [Google Scholar]
- Ainsworth, E.A.; Gillespie, K.M. Estimation of total phenolic content and other oxidation substrates in plant tissues using Folin-Ciocalteu reagent. Nat. Protoc. 2007, 2, 875–877. [Google Scholar] [CrossRef] [PubMed]
- T 211 om-02. Ash in Wood, Pulp, Paper and Paperboard: Combustion at 525 °C; TAPPI: Peachtree Corners, GA, USA, 2002. [Google Scholar]
Sample Availability: Samples are not available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clone No. | National Register Id 1 | H (dm) 2 | LTL (dm) 3 | D 1.3 (mm) 4 | D 5.0 (mm) 5 |
|---|---|---|---|---|---|
| 2 | C05-99-24 | 188 ± 18 | 57 ± 5 | 153 ± 12 | 129 ± 12 |
| 4 | C05-99-34 | 189 ± 21 | 52 ± 19 | 141 ± 12 | 118 ± 13 |
| 5 | C05-99-14 | 180 ± 38 | 52 ± 18 | 124 ± 16 | 104 ± 28 |
| Particle-Size Distribution | |
|---|---|
| <0.063 mm | 11.7% |
| 0.063–0.2 mm | 20.1% |
| 0.2–0.5 mm | 42.3% |
| 0.5–1.25 mm | 25.2% |
| 1.25–2.0 mm | 0.6% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korkalo, P.; Korpinen, R.; Beuker, E.; Sarjala, T.; Hellström, J.; Kaseva, J.; Lassi, U.; Jyske, T. Clonal Variation in the Bark Chemical Properties of Hybrid Aspen: Potential for Added Value Chemicals. Molecules 2020, 25, 4403. https://doi.org/10.3390/molecules25194403
Korkalo P, Korpinen R, Beuker E, Sarjala T, Hellström J, Kaseva J, Lassi U, Jyske T. Clonal Variation in the Bark Chemical Properties of Hybrid Aspen: Potential for Added Value Chemicals. Molecules. 2020; 25(19):4403. https://doi.org/10.3390/molecules25194403
Chicago/Turabian StyleKorkalo, Pasi, Risto Korpinen, Egbert Beuker, Tytti Sarjala, Jarkko Hellström, Janne Kaseva, Ulla Lassi, and Tuula Jyske. 2020. "Clonal Variation in the Bark Chemical Properties of Hybrid Aspen: Potential for Added Value Chemicals" Molecules 25, no. 19: 4403. https://doi.org/10.3390/molecules25194403
APA StyleKorkalo, P., Korpinen, R., Beuker, E., Sarjala, T., Hellström, J., Kaseva, J., Lassi, U., & Jyske, T. (2020). Clonal Variation in the Bark Chemical Properties of Hybrid Aspen: Potential for Added Value Chemicals. Molecules, 25(19), 4403. https://doi.org/10.3390/molecules25194403