
The Key Role of IP6K: A Novel Target for Anticancer Treatments?




{kind=link}
{kind=link}
Abstract
1. Introduction
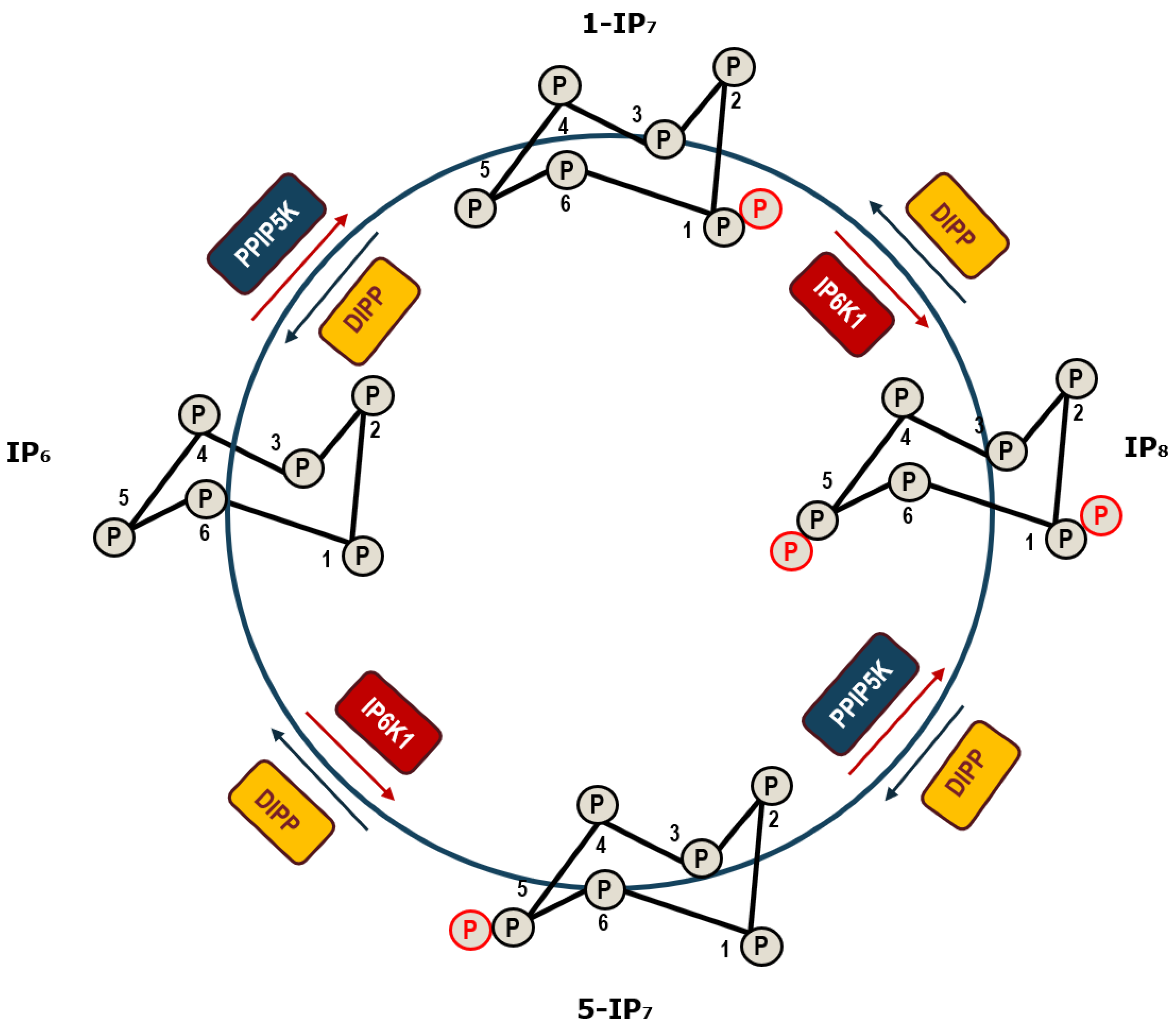
2. Inositol Pyrophosphates
3. IP6Ks: Balance, Activity, and Regulation in Physiological Homeostasis and Cancer
3.1. IP6K1
3.2. IP6K2
3.3. IP6K3
4. Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thota, S.G.; Bhandari, R. The emerging roles of inositol pyrophosphates in eukaryotic cell physiology. J. Biosci. 2015, 40, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, M.; Fuso, A.; Dinicola, S.; Cucina, A.; Bevilacqua, A. Pharmacodynamics and pharmacokinetics of inositol(s) in health and disease. Expert Opin. Drug Metab. Toxicol. 2016, 14, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.G. Phosphoinositides in constitutive membrane traffic. Physiol. Rev. 2004, 84, 699–730. [Google Scholar] [CrossRef]
- Martin, T.F. Phosphoinositide lipids as signaling molecules: Common themes for signal transduction, cytoskeletal regulation, and membrane trafficking. Annu. Rev. Cell Dev. Biol. 1998, 14, 231–264. [Google Scholar] [CrossRef] [PubMed]
- Monserrate, J.P.; York, J.D. Inositol phosphate synthesis and the nuclear processes they affect. Curr. Opin. Cell Biol. 2010, 22, 365–373. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The calcium entry pas de deux. Science 2000, 287, 1604–1605. [Google Scholar] [CrossRef]
- Irvine, R.F. 20 years of Ins(1,4,5)P3, and 40 years before. Nat. Rev. Mol. Cell Biol. 2003, 4, 586–590. [Google Scholar] [CrossRef]
- Michell, R.H. Inositol derivatives: Evolution and functions. Nat. Rev. Mol. Cell Biol. 2008, 9, 151–161. [Google Scholar] [CrossRef]
- Raboy, V. Myo-Inositol-1,2,3,4,5,6-hexakisphosphate. Phytochemistry 2003, 64, 1033–1043. [Google Scholar] [CrossRef]
- Plimmer, R.H.; Page, H.J. An investigation of Phytin. Biochem. J. 1913, 7, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Shears, S.B. Assessing the omnipotence of inositol hexakisphosphate. Cell Signal. 2001, 13, 151–158. [Google Scholar] [CrossRef]
- Hanakahi, L.A.; Bartlet-Jones, M.; Chappell, C.; Pappin, D.; West, S.C. Binding of inositol phosphate to DNA-PK and stimulation of double-strand break repair. Cell 2000, 102, 721–729. [Google Scholar] [CrossRef]
- Macbeth, M.R.; Schubert, H.L.; Vandemark, A.P.; Lingam, A.T.; Hill, C.P.; Bass, B.L. Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science 2005, 309, 1534–1539. [Google Scholar] [CrossRef]
- Stephens, L.; Radenberg, T.; Thiel, U.; Vogel, G.; Khoo, K.H.; Dell, A.; Jackson, T.R.; Hawkins, P.T.; Mayr, G.W. The detection, purification, structural characterization and metabolism of diphosphoinositol pentakisphosphate(s) and bisdiphosphoinositol tetrakisphosphate(s). J. Biol. Chem. 1993, 268, 4009–4015. [Google Scholar]
- Menniti, F.S.; Miller, R.N.; Putney, J.W., Jr.; Shears, S.B. Turnover of inositol polyphosphate pyrophosphates in pancreatoma cells. J. Biol. Chem. 1993, 268, 3850–3856. [Google Scholar]
- Shears, S.B. Inositol pyrophosphates: Why so many phosphates? Adv. Biol. Regul. 2015, 57, 203–216. [Google Scholar] [CrossRef]
- Wilson, M.; Livermore, T.; Saiardi, A. Inositol pyrophosphates: Between signalling and metabolism. Biochem. J. 2013, 452, 369–379. [Google Scholar] [CrossRef]
- Shears, S.B. Diphosphoinositol polyphosphates: Metabolic messengers? Mol. Pharmacol. 2009, 76, 236–252. [Google Scholar] [CrossRef]
- Glennon, M.C.; Shears, S.B. Turnover of inositol pentakisphosphates, inositol hexakisphosphate and diphosphoinositol polyphosphates in primary cultured hepatocytes. Biochem. J. 1993, 293, 583–590. [Google Scholar] [CrossRef]
- Chakraborty, A.; Koldobskiy, M.A.; Sixt, K.M.; Juluri, K.R.; Mustafa, A.K.; Snowman, A.M.; van Rossum, D.B.; Patterson, R.L.; Snyder, S.H. HSP90 regulates cell survival via inositol hexakisphosphate kinase-2. Proc. Natl. Acad. Sci. USA 2008, 105, 1134–1139. [Google Scholar] [CrossRef] [PubMed]
- Koldobskiy, M.A.; Chakraborty, A.; Werner, J.K., Jr.; Snowman, A.M.; Juluri, K.R.; Vandiver, M.S.; Kim, S.; Heletz, S.; Snyder, S.H. p53-mediated apoptosis requires inositol hexakisphosphate kinase-2. Proc. Natl. Acad. Sci. USA 2010, 107, 20947–20951. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Kim, S.; Snyder, S. Inositol pyrophosphates as mammalian cell signals. Sci. Signal. 2011, 4, re1. [Google Scholar] [CrossRef] [PubMed]
- Szijgyarto, Z.; Garedew, A.; Azevedo, C.; Saiardi, A. Influence of inositol pyrophosphates on cellular energy dynamics. Science 2011, 334, 802–805. [Google Scholar] [CrossRef]
- Saiardi, A. How inositol pyrophosphates control cellular phosphate homeostasis? Adv. Biol. Regul. 2012, 52, 351–359. [Google Scholar] [CrossRef]
- Chakraborty, A.; Koldobskiy, M.A.; Bello, N.T.; Maxwell, M.; Potter, J.J.; Juluri, K.R.; Maag, D.; Kim, S.; Huang, A.S.; Dailey, M.J.; et al. Inositol pyrophosphates inhibit Akt signaling, thereby regulating insulin sensitivity and weight gain. Cell 2010, 143, 897–910. [Google Scholar] [CrossRef]
- Rao, F.; Cha, J.; Xu, J.; Xu, R.; Vandiver, M.S.; Tyagi, R.; Tokhunts, R.; Koldobskiy, M.A.; Fu, C.; Barrow, R.; et al. Inositol pyrophosphates mediate the DNA-PK/ATM-p53 cell death pathway by regulating CK2 phosphorylation of Tti1/Tel2. Mol. Cell 2014, 54, 119–132. [Google Scholar]
- Bizzarri, M.; Dinicola, S.; Bevilacqua, A.; Cucina, A. Broad spectrum anticancer activity of myo-inositol and inositol hexakisphosphate. Int. J. Endocrinol. 2016, 2016, 5616807. [Google Scholar] [CrossRef]
- Mukherjee, S.; Haubner, J.; Chakraborty, A. Targeting the inositol pyrophosphate biosynthetic enzymes in metabolic diseases. Molecules 2020, 25, 1403. [Google Scholar] [CrossRef]
- Li, X.; Gu, C.; Hostachy, S.; Sahu, S.; Wittwer, C.; Jessen, H.J.; Fiedler, D.; Wang, H.; Shears, S.B. Control of XPR1-dependent cellular phosphate efflux by InsP8 is an exemplar for functionally-exclusive inositol pyrophosphate signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 3568–3574. [Google Scholar] [CrossRef]
- Bizzarri, M.; Dinicola, S.; Cucina, A. Myoinositol and inositol hexakisphosphate in the treatment of breast cancer: Molecular mechanisms. Pre-Menopause Menopause Beyond 2018, 233–241. [Google Scholar] [CrossRef]
- Barker, C.J.; Illies, C.; Gaboardi, G.C.; Berggren, P.O. Inositol pyrophosphates: Structure, enzymology and function. Cell. Mol. Life Sci. 2009, 66, 3851–3871. [Google Scholar] [CrossRef] [PubMed]
- Saiardi, A.; Bhandari, R.; Resnick, A.C.; Snowman, A.M.; Snyder, S.H. Phosphorylation of proteins by inositol pyrophosphates. Science 2004, 306, 2101–2105. [Google Scholar] [CrossRef] [PubMed]
- Mulugu, S.; Bai, W.; Fridy, P.C.; Bastidas, R.J.; Otto, J.C.; Dollins, D.E.; Haystead, T.A.; Ribeiro, A.A.; York, J.D. Conserved family of enzymes that phosphorylate inositol hexakisphosphate. Science 2007, 316, 106–109. [Google Scholar] [CrossRef]
- Draskovic, P.; Saiardi, A.; Bhandari, R.; Burton, A.; Ilc, G.; Kovacevic, M.; Snyder, S.H.; Podobnik, M. Inositol hexakisphosphate kinase products contain diphosphate and triphosphate groups. Chem. Biol. 2008, 15, 274–286. [Google Scholar] [CrossRef]
- Lin, H.; Fridy, P.C.; Ribeiro, A.A.; Choi, J.H.; Barma, D.K.; Vogel, G.; Falck, J.R.; Shears, S.B.; York, J.D.; Mayr, G.W. Structural analysis and detection of biological inositol pyrophosphates reveal that the family of VIP/ diphosphoinositol pentakisphosphate kinases are 1/3-kinases. J. Biol. Chem. 2009, 284, 1863–1872. [Google Scholar] [CrossRef]
- Voglmaier, S.M.; Bembenek, M.E.; Kaplin, A.I.; Dorman, G.; Olszewski, J.D.; Prestwich, G.D.; Snyder, S.H. Purified inositol hexakisphosphate kinase is an ATP synthase: Diphosphoinositol pentakisphosphate as a high-energy phosphate donor. Proc. Natl. Acad. Sci. USA 1996, 93, 4305–4310. [Google Scholar] [CrossRef]
- Saiardi, A.; Erdjument-Bromage, H.; Snowman, A.M.; Tempst, P.; Snyder, S.H. Synthesis of diphosphoinositol pentakisphosphate by a newly identified family of higher inositol polyphosphate kinases. Curr. Biol. 1999, 9, 1323–1326. [Google Scholar] [CrossRef]
- Caffrey, J.J.; Safrany, S.T.; Yang, X.; Shears, S.B. Discovery of molecular and catalytic diversity among human diphosphoinositolpolyphosphate phosphohydrolases. An expanding Nudt family. J. Biol. Chem. 2000, 275, 12730–12736. [Google Scholar] [CrossRef]
- Topolski, B.; Jakopec, V.; Künzel, N.A.; Fleig, U. Inositol pyrophosphate kinase Asp1 modulates chromosome segregation fidelity and spindle function in Schizosaccharomyces pombe. Mol. Cell Biol. 2016, 36, 3128–3140. [Google Scholar] [CrossRef]
- Dollins, D.E.; Bai, W.; Fridy, P.C.; Otto, J.C.; Neubauer, J.L.; Gattis, S.G.; Mehta, K.P.M.; York, J.D. Vip1 is a kinase and pyrophosphatase switch that regulates inositol diphosphate signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 9356–9364. [Google Scholar] [CrossRef] [PubMed]
- Harmel, R.K.; Puschmann, R.; Nguyen Trung, M.; Saiardi, A.; Schmieder, P.; Fiedler, D. Harnessing 13C-labeled myo-inositol to interrogate inositol phosphate messengers by NMR. Chem. Sci. 2019, 10, 5267–5274. [Google Scholar] [CrossRef] [PubMed]
- Rajasekaran, S.S.; Kim, J.; Gaboardi, G.C.; Gromada, J.; Shears, S.B.; Dos Santos, K.T.; Nolasco, E.L.; Ferreira, S.S.; Illies, C.; Kohler, M.; et al. Inositol hexakisphosphate kinase 1 is a metabolic sensor in pancreatic beta-cells. Cell. Signal. 2018, 46, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Nagel, A.; Barker, C.J.; Berggren, P.O.; Illies, C. Diphosphosinositol polyphosphates and energy metabolism: Assay for ATP/ADP ratio. Methods Mol. Biol. 2010, 645, 123–131. [Google Scholar]
- Boer, V.M.; Crutchfield, C.A.; Bradley, P.H.; Botstein, D.; Rabinowitz, J.D. Growth-limiting intracellular metabolites in yeast growing under diverse nutrient limitations. Mol. Biol. Cell. 2010, 21, 198–211. [Google Scholar] [CrossRef]
- Lonetti, A.; Szijgyarto, Z.; Bosch, D.; Loss, O.; Azevedo, C.; Saiardi, A. Identification of an evolutionarily conserved family of inorganic polyphosphate endopolyphosphatases. J. Biol. Chem. 2011, 286, 31966–31974. [Google Scholar] [CrossRef]
- Choi, K.; Mollapour, E.; Shears, S.B. Signal transduction during environmental stress: InsP(8) operates within highly restricted contexts. Cell Signal. 2005, 17, 533–1541. [Google Scholar] [CrossRef]
- Pesesse, X.; Choi, K.; Zhang, T.; Shears, S.B. Signaling by higher inositol polyphosphates. Synthesis of bisdiphosphoinositol tetrakisphosphate (“InsP8”) is selectively activated by hyperosmotic stress. J. Biol. Chem. 2004, 279, 43378–43381. [Google Scholar] [CrossRef]
- Choi, K.; Mollapour, E.; Choi, J.H.; Shears, S.B. Cellular energetic status supervises the synthesis of bis-diphosphoinositol tetrakisphosphate independently of AMP-activated protein kinase. Mol. Pharmacol. 2008, 74, 527–536. [Google Scholar] [CrossRef]
- Laussmann, T.; Eujen, R.; Weisshuhn, C.M.; Thiel, U.; Vogel, G. Structures of diphospho-myo-inositol pentakisphosphate and bisdiphospho-myo-inositol tetrakisphosphate from Dictyostelium resolved by NMR analysis. Biochem. J. 1996, 315, 715–720. [Google Scholar] [CrossRef]
- Wu, M.; Dul, B.E.; Trevisan, A.J.; Fiedler, D. Synthesis and characterization of non-hydrolysable diphosphoinositol polyphosphate second messengers. Chem. Sci. 2013, 4, 405–410. [Google Scholar] [CrossRef]
- Gokhale, N.A.; Zaremba, A.; Janoshazi, A.K.; Weaver, J.D.; Shears, S.B. PPIP5K1 modulates ligand competition between diphosphoinositol polyphosphates and PtdIns(3,4,5)P3 for polyphosphoinositide-binding domains. Biochem. J. 2013, 453, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Roig, C.; Viéitez, C.; Posas, F.; de Nadal, E. The Rpd3L HDAC complex is essential for the heat stress response in yeast. Mol. Microbiol. 2010, 76, 1049–1062. [Google Scholar] [CrossRef] [PubMed]
- Worley, J.; Luo, X.; Capaldi, A.P. Inositol pyrophosphates regulate cell growth and the environmental stress response by activating the HDAC Rpd3L. Cell. Rep. 2013, 3, 1476–1482. [Google Scholar] [CrossRef] [PubMed]
- Blind, R.D. Structural analyses of inositol phosphate second messengers bound to signaling effector proteins. Adv. Biol. Regul. 2020, 75, 100667. [Google Scholar] [CrossRef]
- Saiardi, A.; Caffrey, J.J.; Snyder, S.H.; Shears, S.B. The inositol hexakisphosphate kinase family: Catalytic flexibility and function in yeast vacuole biogenesis. J. Biol. Chem. 2000, 275, 24686–24692. [Google Scholar] [CrossRef]
- Luo, H.R.; Huang, Y.E.; Chen, J.C.; Saiardi, A.; Iijima, M.; Ye, K.; Huang, Y.; Nagata, E.; Devreotes, P.; Snyder, S.H. Inositol pyrophosphates mediate chemotaxis in dictyostelium via pleckstrin homology domain-ptdIns(3,4,5)P3 interactions. Cell 2003, 114, 559–572. [Google Scholar] [CrossRef]
- Saiardi, A.; Nagata, E.; Luo, H.R.; Snowman, A.M.; Snyder, S.H. Identification and characterization of a novel inositol hexakisphosphate kinase. J. Biol. Chem. 2001, 276, 39179–39185. [Google Scholar] [CrossRef]
- Chakraborty, A. The inositol pyrophosphate pathway in health and diseases. Biol. Rev. Camb. Philos. Soc. 2018, 93, 1203–1227. [Google Scholar] [CrossRef]
- Takazawa, K.; Perret, J.; Dumont, J.E.; Erneux, C. Molecular cloning and expression of a human brain inositol 1,4,5-trisphosphate 3-kinase. Biochem. Biophys. Res. Commun. 1991, 174, 529–535. [Google Scholar] [CrossRef]
- Fridy, P.C.; Otto, J.C.; Dollins, D.E.; York, J.D. Cloning and characterization of two human VIP1-like inositol hexakisphosphate and diphosphoinositol pentakisphosphate kinases. J. Biol. Chem. 2007, 282, 30754–30762. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, C.; Szijgyarto, Z.; Saiardi, A. The signaling role of inositol hexakisphosphate kinases (IP6Ks). Adv. Enzym. Regul. 2011, 51, 74–82. [Google Scholar] [CrossRef]
- Safrany, S.T.; Caffrey, J.J.; Yang, X.; Bembenek, M.E.; Moyer, M.B.; Burkhart, W.A.; Shears, S.B. A novel context for the ‘MutT’ module, a guardian of cell integrity, in a diphosphoinositol polyphosphate phosphohydrolase. EMBO J. 1998, 17, 6599–6607. [Google Scholar] [CrossRef] [PubMed]
- Onnebo, S.M.; Saiardi, A. Inositol pyrophosphates modulate hydrogen peroxide signalling. Biochem. J. 2009, 423, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Barker, C.J.; Wright, J.; Hughes, P.J.; Kirk, C.J.; Michell, R.H. Complex changes in cellular inositol phosphate complement accompany transit through the cell cycle. Biochem. J. 2004, 380, 465–473. [Google Scholar] [CrossRef]
- Wundenberg, T.; Grabinski, N.; Lin, H.; Mayr, G.W. Discovery of InsP6-kinases as InsP6-dephosphorylating enzymes provides a new mechanism of cytosolic InsP6 degradation driven by the cellular ATP/ADP ratio. Biochem. J. 2014, 462, 173–184. [Google Scholar] [CrossRef]
- Nair, V.S.; Gu, C.; Janoshazi, A.K.; Jessen, H.J.; Wang, H.; Shears, S.B. Inositol pyrophosphate synthesis by diphosphoinositol pentakisphosphate kinase-1 is regulated by phosphatidylinositol(4,5)bisphosphate. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef]
- Wilson, M.S.; Jessen, H.J.; Saiardi, A. The inositol hexakisphosphate kinases IP6K1 and -2 regulate human cellular phosphate homeostasis, including XPR1-mediated phosphate export. J. Biol. Chem. 2019, 294, 11597–11608. [Google Scholar] [CrossRef]
- Gu, C.; Nguyen, H.N.; Ganini, D.; Chen, Z.; Jessen, H.J.; Gu, Z.; Wang, H.; Shears, S.B. KO of 5-InsP7 kinase activity transforms the HCT116 colon cancer cell line into a hypermetabolic, growth-inhibited phenotype. Proc. Natl. Acad. Sci. USA 2017, 114, 11968–11973. [Google Scholar] [CrossRef]
- Wundenberg, T.; Mayr, G.W. Synthesis and biological actions of diphosphoinositol phosphates (inositol pyrophosphates), regulators of cell homeostasis. Biol. Chem. 2012, 393, 979–998. [Google Scholar] [CrossRef]
- Otto, J.C.; Kelly, P.; Chiou, S.T.; York, J.D. Alterations in an inositol phosphate code through synergistic activation of a G protein and inositol phosphate kinases. Proc. Natl. Acad. Sci. USA 2007, 104, 15653–15658. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.; Onnebo, S.M.N.; Azevedo, C.; Saiardi, A. Inositol pyrophosphates: Metabolism and signaling. Cell. Mol. Life. Sci. 2006, 63, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Desfougères, Y.; Wilson, M.S.C.; Laha, D.; Miller, G.J.; Saiardi, A. ITPK1 mediates the lipid-independent synthesis of inositol phosphates controlled by metabolism. Proc. Natl. Acad. Sci. USA 2019, 116, 24551–24561. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Latapy, C.; Xu, J.; Snyder, S.H.; Beaulieu, J.M. Inositol hexakisphosphate kinase-1 regulates behavioral responses via GSK3 signaling pathways. Mol. Psychiatry 2014, 19, 284–293. [Google Scholar]
- Morrison, B.H.; Bauer, J.A.; Lupica, J.A.; Tang, Z.; Schmidt, H.; Didonato, J.A.; Lindner, D.J. Effect of inositol hexakisphosphate kinase 2 on transforming growth factor beta-activated kinase 1 and NF-kappaB activation. J. Biol. Chem. 2007, 282, 15349–15356. [Google Scholar] [CrossRef]
- Fu, C.; Xu, J.; Li, R.J.; Crawford, J.A.; Khan, A.B.; Ma, T.M.; Cha, J.Y.; Snowman, A.M.; Pletnikov, M.V.; Snyder, S.H. Inositol hexakisphosphate kinase-3 regulates the morphology and synapse formation of cerebellar Purkinje cells via spectrin/adducin. J. Neurosci. 2015, 35, 11056–11067. [Google Scholar] [CrossRef]
- Zhu, Q.; Ghoshal, S.; Tyagi, R.; Chakraborty, A. Global IP6K1 deletion enhances temperature modulated energy expenditure which reduces carbohydrate and fat induced weight gain. Mol. Metab. 2017, 6, 73–85. [Google Scholar] [CrossRef]
- Long, Y.C.; Cheng, Z.; Copps, K.D.; White, M.F. 2011. Insulin receptor substrates Irs1 and Irs2 coordinate skeletal muscle growth and metabolism via the Akt and AMPK pathways. Mol. Cell. Biol. 2011, 31, 430–441. [Google Scholar] [CrossRef]
- Osaki, M.; Oshimura, M.; Ito, H. The PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676. [Google Scholar] [CrossRef]
- Currie, R.A.; Walker, K.S.; Gray, A.; Deak, M.; Casamayor, A.; Downes, C.P.; Cohen, P.; Alessi, D.R.; Lucocq, J. Role of phosphatidylinositol 3,4,5-trisphosphate in regulating the activity and localization of 3-phosphoinositide- dependent protein kinase-1. Biochem. J. 1999, 337, 575–583. [Google Scholar] [CrossRef]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, R.W.; Elliott, B.T. Akt/PKB activation and insulin signaling: A novel insulin signaling pathway in the treatment of type 2 diabetes. Diabetes Metab. Syndr. Obes. 2014, 7, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, H.; Liu, J. Akt activation: A potential strategy to ameliorate insulin resistance. Diabetes Res. Clin. Pract. 2019, 156, 107092. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Dong, Y.; Zhang, J.; Scholz, R.; Neumann, D.; Zou, M.H. Identification of the serine 307 of LKB1 as a novel phosphorylation site essential for its nucleocytoplasmic transport and endothelial cell angiogenesis. Mol. Cell. Biol. 2009, 29, 3582–3596. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Ghoshal, S.; Rodrigues, A.; Gao, S.; Asterian, A.; Kamenecka, T.M.; Barrow, J.C.; Chakraborty, A. Adipocyte-specific deletion of Ip6k1 reduces diet-induced obesity by enhancing AMPK-mediated thermogenesis. J. Clin. Investig. 2016, 126, 4273–4288. [Google Scholar] [CrossRef]
- Kaidanovich, O.; Eldar-Finkelman, H. The role of glycogen synthase kinase-3 in insulin resistance and type 2 diabetes. Expert Opin. Ther. Targets 2002, 6, 555–561. [Google Scholar]
- Izumiya, Y.; Hopkins, T.; Morris, C.; Sato, K.; Zeng, L.; Viereck, J.; Hamilton, J.A.; Ouchi, N.; LeBrasseur, N.K.; Walsh, K. Fast/Glycolytic muscle fiber growth reduces fat mass and improves metabolic parameters in obese mice. Cell Metab. 2008, 7, 159–172. [Google Scholar] [CrossRef]
- Barker, C.J.; Berggren, P.O. New horizons in cellular regulation by inositol polyphosphates: Insights from the pancreatic beta-cell. Pharmacol. Rev. 2013, 65, 641–669. [Google Scholar] [CrossRef]
- Illies, C.; Gromada, J.; Fiume, R.; Leibiger, B.; Yu, J.; Juhl, K.; Yang, S.N.; Barma, D.K.; Falck, J.R.; Saiardi, A.; et al. Requirement of inositol pyrophosphates for full exocytotic capacity in pancreatic beta cells. Science 2007, 318, 1299–1302. [Google Scholar] [CrossRef]
- Bhandari, R.; Juluri, K.R.; Resnick, A.C.; Snyder, S.H. Gene deletion of inositol hexakisphosphate kinase 1 reveals inositol pyrophosphate regulation of insulin secretion, growth, and spermiogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 2349–2353. [Google Scholar] [CrossRef]
- Waselle, L.; Gerona, R.R.L.; Vitale, N.; Martin, T.F.J.; Bader, M.F.; Regazzi, R. Role of phosphoinositide signaling in the control of insulin exocytosis. Mol. Endocrinol. 2005, 19, 3097–3106. [Google Scholar] [CrossRef] [PubMed]
- Utzschneider, K.M.; Prigeon, R.L.; Carr, D.B.; Hull, R.L.; Tong, J.; Shofer, J.B.; Retzlaff, B.M.; Knopp, R.H.; Kahm, S.E. Impact of differences in fasting glucose and glucose tolerance on the hyperbolic relationship between insulin sensitivity and insulin responses. Diabetes Care 2006, 29, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Stump, C.S.; Short, K.R.; Bigelow, M.L.; Schimke, J.M.; Nair, K.S. Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proc. Natl. Acad. Sci. USA 2003, 100, 7996–8001. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Li, S.; Wu, H.; Zhang, M.; Zhang, X.; Wei, L.; Qin, X.; Gao, E. Oncostatin M (OSM) protects against cardiac ischaemia/reperfusion injury in diabetic mice by regulating apoptosis, mitochondrial biogenesis and insulin sensitivity. J. Cell. Mol. Med. 2015, 6, 1296–1307. [Google Scholar] [CrossRef]
- Thota, S.G.; Unnikannan, C.P.; Thampatty, S.R.; Manorama, R.; Bhandari, R. Inositol pyrophosphates regulate RNA polymerase I-mediated rRNA transcription in Saccharomyces cerevisiae. Biochem. J. 2015, 466, 105–114. [Google Scholar] [CrossRef]
- Yu, W.; Ye, C.; Greenberg, M.L. Inositol hexakisphosphate kinase 1 (IP6K1) regulates inositol synthesis in mammalian cells. J. Biol. Chem. 2016, 291, 10437–10444. [Google Scholar] [CrossRef]
- Loewen, C.J.R.; Gaspar, M.L.; Jesch, S.A.; Delon, C.; Ktistakis, N.T.; Henry, S.A.; Levine, T.P. Phospholipid metabolism regulated by a transcription factor sensing phosphatidic acid. Science 2004, 304, 1644–1647. [Google Scholar] [CrossRef]
- Ye, C.; Bandara, W.M.; Greenberg, M.L. Regulation of inositol metabolism is fine-tuned by inositol pyrophosphates in Saccharomyces cerevisiae. J. Biol. Chem. 2013, 288, 24898–24908. [Google Scholar] [CrossRef]
- Burton, A.; Azevedo, C.; Andreassi, C.; Riccio, A.; Saiardi, A. Inositol pyrophosphates regulate JMJD2C-dependent histone demethylation. Proc. Natl. Acad. Sci. USA 2013, 110, 18970–18975. [Google Scholar] [CrossRef]
- Rao, F.; Xu, J.; Khan, A.B.; Gadalla, M.M.; Cha, J.Y.; Xu, R.; Tyagi, R.; Dang, Y.; Chakraborty, A.; Snyder, S.H. Inositol hexakisphosphate kinase-1 mediates assembly/disassembly of the CRL4-signalosome complex to regulate DNA repair and cell death. Proc. Natl. Acad. Sci. USA 2014, 111, 16005–16010. [Google Scholar] [CrossRef]
- Luo, H.R.; Saiardi, A.; Nagata, E.; Ye, K.; Yu, H.; Jung, T.S.; Luo, X.; Jain, S.; Sawa, A.; Snyder, S.H. GRAB: A physiologic guanine nucleotide exchange factor for Rab3A, which interacts with inositol hexakisphosphate kinase. Neuron 2001, 31, 439–451. [Google Scholar] [CrossRef]
- Lee, T.S.; Lee, J.Y.; Kyung, J.W.; Yang, Y.; Park, S.J.; Lee, S.; Pavlovic, I.; Kong, B.; Jho, Y.S.; Jessen, H.J.; et al. Inositol pyrophosphates inhibit synaptotagmin-dependent exocytosis. Proc. Natl. Acad. Sci. USA 2016, 113, 8314–8319. [Google Scholar] [CrossRef] [PubMed]
- Mikoshiba, K.; Fukuda, M.; Ibata, K.; Kabayama, H.; Mizutani, A. Role of synaptotagmin, a Ca2+ and inositol polyphosphate binding protein, in neurotransmitter release and neurite outgrowth. Chem. Phys. Lipids 1999, 98, 59–67. [Google Scholar] [CrossRef]
- Chanduri, M.; Rai, A.; Malla, A.B.; Wu, M.; Fiedler, D.; Mallik, R.; Bhandari, R. Inositol hexakisphosphate kinase 1 (IP6K1) activity is required for cytoplasmic dynein-driven transport. Biochem. J. 2016, 473, 3031–3047. [Google Scholar] [CrossRef]
- Soppina, V.; Rai, A.K.; Ramaiya, A.J.; Barak, P.; Mallik, R. Tug-of-war between dissimilar teams of microtubule motors regulates transport and fission of endosomes. Proc. Natl. Acad. Sci. USA 2009, 106, 19381–19386. [Google Scholar] [CrossRef]
- Vallee, R.B.; McKenney, R.J.; Ori-McKenney, K.M. Multiple modes of cytoplasmic dynein regulation. Nat. Cell Biol. 2012, 14, 224–230. [Google Scholar] [CrossRef]
- Azevedo, C.; Burton, A.; Ruiz-Mateos, E.; Marsh, M.; Saiardi, A. Inositol pyrophosphate mediated pyrophosphorylation of AP3B1 regulates HIV-1 Gag release. Proc. Natl. Acad. Sci. USA 2009, 106, 21161–21166. [Google Scholar] [CrossRef]
- Saiardi, A.; Sciambi, C.; McCaffery, J.M.; Wendland, B.; Snyder, S.H. Inositol pyrophosphates regulate endocytic trafficking. Proc. Natl. Acad. Sci. USA 2002, 99, 14206–14211. [Google Scholar] [CrossRef]
- Fu, C.; Xu, J.; Cheng, W.; Rojas, T.; Chin, A.C.; Snowman, A.M.; Harraz, M.M.; Snyder, S.H. Neuronal migration is mediated by inositol hexakisphosphate kinase 1 via alpha-actinin and focal adhesion kinase. Proc. Natl. Acad. Sci. USA 2017, 114, 2036–2041. [Google Scholar] [CrossRef]
- Prasad, A.; Jia, Y.; Chakraborty, A.; Li, Y.; Jain, S.K.; Zhong, J.; Roy, S.G.; Loison, F.; Mondal, S.; Sakai, J.; et al. Inositol hexakisphosphate kinase 1 regulates neutrophil function in innate immunity by inhibiting phosphatidylinositol-(3,4,5)-trisphosphate signaling. Nat. Immunol. 2011, 12, 752–760. [Google Scholar] [CrossRef]
- Barnes, P.J. New concepts in chronic obstructive pulmonary disease. Annu. Rev. Med. 2003, 54, 113–129. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Li, H.; Bajrami, B.; Kwak, H.; Cao, S.; Liu, P.; Zhou, J.; Zhou, Y.; Zhu, H.; Ye, K.; et al. Cigarette smoke (CS) and nicotine delay neutrophil spontaneous death via suppressing production of diphosphoinositol pentakisphosphate. Proc. Natl. Acad. Sci. USA 2013, 110, 7726–7731. [Google Scholar] [CrossRef] [PubMed]
- Jadav, R.S.; Kumar, D.; Buwa, N.; Ganguli, S.; Thampatty, S.R.; Balasubramanian, N.; Bhandari, R. Deletion of inositol hexakisphosphate kinase 1 (IP6K1) reduces cell migration and invasion, conferring protection from aerodigestive tract carcinoma in mice. Cell Signal. 2016, 28, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, M.; Christofori, G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009, 28, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Dinicola, S.; Fabrizi, G.; Masiello, M.G.; Proietti, S.; Palombo, A.; Minini, M.; Harrath, A.H.; Alwasel, S.H.; Ricci, G.; Catizone, A.; et al. Inositol induces mesenchymal-epithelial reversion in breast cancer through cytoskeleton rearrangement. Exp. Cell Res. 2016, 345, 37–50. [Google Scholar] [CrossRef]
- Koguchi, T.; Tanikawa, C.; Mori, J.; Kojima, Y.; Matsuda, K. Regulation of myo-inositol biosynthesis by p53-ISYNA1 pathway. Int. J. Oncol. 2016, 48, 2415–2424. [Google Scholar] [CrossRef]
- Minini, M.; Proietti, S.; Xingkang, H.; Senni, A.; Monti, N.; Fuso, A.; Cucina, A.; Cao, Y.; Bizzarri, M. Myo-Inositol treatment inhibits motility in triple negative breast cancer via miR-125a-5p/IP6K1 axis. In Proceedings of the EMBL Conference, Cancer Genomics, EMBL Advanced Training Centre, Heidelberg, Germany, 4–6 November 2019. [Google Scholar]
- El-Sherbiny, Y.M.; Cox, M.C.; Ismail, Z.A.; Shamsuddin, A.M.; Vucenik, I. G0/G1 arrest and S phase inhibition of human cancer cell lines by inositol hexaphosphate (IP6). Anticancer Res. 2001, 21, 2393–2404. [Google Scholar]
- Jadav, R.S.; Chanduri, M.V.L.; Sengupta, S.; Bhandari, R. Inositol pyrophosphate synthesis by inositol hexakisphosphate kinase 1 is required for homologous recombination repair. J. Biol. Chem. 2013, 288, 3312–3321. [Google Scholar] [CrossRef]
- Wilson, M.S.C.; Bulley, S.J.; Pisani, F.; Irvine, R.F.; Saiardi, A. A novel method for the purification of inositol phosphates from biological samples reveals that no phytate is present in human plasma or urine. Open Biol. 2015, 5, 150014. [Google Scholar] [CrossRef]
- Morrison, B.H.; Bauer, J.A.; Hu, J.; Grane, R.W.; Ozdemir, A.M.; Chawla-Sarkar, M.; Gong, B.; Almasan, A.; Kalvakolanu, D.V.; Lindner, D.J. Inositol hexakisphosphate kinase 2 sensitizes ovarian carcinoma cells to multiple cancer therapeutics. Oncogene 2002, 21, 1882–1889. [Google Scholar] [CrossRef][Green Version]
- Nagata, E.; Luo, H.R.; Saiardi, A.; Bae, B.I.; Suzuki, N.; Snyder, S.H. Inositol hexakisphosphate kinase-2, a physiologic mediator of cell death. J. Biol. Chem. 2005, 280, 1634–1640. [Google Scholar] [CrossRef] [PubMed]
- Nagata, E.; Saiardi, A.; Tsukamoto, H.; Okada, Y.; Itoh, Y.; Satoh, T.; Itoh, J.; Margolis, R.L.; Takizawa, S.; Sawa, A.; et al. Inositol hexakisphosphate kinases induce cell death in Huntington disease. J. Biol. Chem. 2011, 286, 26680–26686. [Google Scholar] [CrossRef] [PubMed]
- Nagata, E.; Nonaka, T.; Moriya, Y.; Fujii, N.; Okada, Y.; Tsukamoto, H.; Itoh, J.; Okada, C.; Satoh, T.; Arai, T.; et al. Inositol hexakisphosphate kinase 2 promotes cell death in cells with cytoplasmic TDP-43 aggregation. Mol. Neurobiol. 2016, 53, 5377–5383. [Google Scholar] [CrossRef]
- Saiardi, A.; Resnick, A.C.; Snowman, A.M.; Wendland, B.; Snyder, S.H. Inositol pyrophosphates regulate cell death and telomere length through phosphoinositide 3-kinaserelated protein kinases. Proc. Natl. Acad. Sci. USA 2005, 102, 1911–1914. [Google Scholar] [CrossRef] [PubMed]
- Morrison, B.H.; Tang, Z.; Jacobs, B.S.; Bauer, J.A.; Lindner, D.J. Apo2L/TRAIL induction and nuclear translocation of inositol hexakisphosphate kinase 2 during IFN-beta-induced apoptosis in ovarian carcinoma. Biochem. J. 2005, 385, 595–603. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rao, F.; Xu, J.; Fu, C.; Cha, J.Y.; Gadalla, M.M.; Xu, R.; Barrow, J.C.; Snyder, S.H. Inositol pyrophosphates promote tumor growth and metastasis by antagonizing liver kinase B1. Proc. Natl. Acad. Sci. USA 2015, 112, 1773–1778. [Google Scholar] [CrossRef]
- Carretero, J.; Shimamura, T.; Rikova, K.; Jackson, A.L.; Wilkerson, M.D.; Borgman, C.L.; Buttarazzi, M.S.; Sanofsky, B.A.; McNamara, K.L.; Brandstetter, K.A.; et al. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell 2010, 17, 547–559. [Google Scholar] [CrossRef]
- Roy, B.C.; Kohno, T.; Iwakawa, R.; Moriguchi, T.; Kiyono, T.; Morishita, K.; Sanchez-Cespedes, M.; Akiyama, T.; Yokota, J. Involvement of LKB1 in epithelial-mesenchymal transition (EMT) of human lung cancer cells. Lung Cancer 2010, 70, 136–145. [Google Scholar] [CrossRef]
- Lizcano, J.M.; Göransson, O.; Toth, R.; Deak, M.; Morrice, N.A.; Boudeau, J.; Hawley, S.A.; Udd, L.; Mäkelä, T.P.; Hardie, D.G.; et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004, 23, 833–843. [Google Scholar] [CrossRef]
- Song, P.; Xie, Z.; Wu, Y. Protein kinase Czeta-dependent LKB1 serine 428 phosphorylation increases LKB1 nucleus export and apoptosis in endothelial cells. J. Biol. Chem. 2008, 283, 12446–12455. [Google Scholar] [CrossRef]
- Alessi, D.R.; Sakamoto, K.; Bayascas, J.R. LKB1-dependent signaling pathways. Annu. Rev. Biochem. 2006, 75, 137–163. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.; Helou, K.; Kovács, A.; Bendahl, P.O.; Bjursell, G.; Fernö, M.; Carlsson, P.; Kannius-Janson, M. Nuclear janus-activated kinase 2/nuclear factor 1-C2 suppresses tumorigenesis and epithelial-to-mesenchymal transition by repressing Forkhead box F1. Cancer Res. 2010, 70, 2020–2029. [Google Scholar] [CrossRef] [PubMed]
- Grudzien-Nogalska, E.; Jiao, X.; Song, M.G.; Hart, R.P.; Kiledjian, M. Nudt3 is an mRNA decapping enzyme that modulates cell migration. RNA 2016, 22, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Morrison, B.H.; Haney, R.; Lamarre, E.; Drazba, J.; Prestwich, G.D.; Lindner, D.J. Gene deletion of inositol hexakisphosphate kinase 2 predisposes to aerodigestive tract carcinoma. Oncogene 2009, 28, 2383–2392. [Google Scholar] [CrossRef]
- Morrison, B.H.; Bauer, J.A.; Kalvakolanu, D.V.; Lindner, D.J. Inositol hexakisphosphate kinase 2 mediates growth suppressive and apoptotic effects of interferon-beta in ovarian carcinoma cells. J. Biol. Chem. 2001, 276, 24965–24970. [Google Scholar] [CrossRef]
- Aoki, M.; Sobek, V.; Maslyar, D.J.; Hecht, A.; Vogt, P.K. Oncogenic transformation by beta-catenin: Deletion analysis and characterization of selected target genes. Oncogene 2002, 21, 6983–6991. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Y.; Yao, R.; Li, J.; Lubet, R.A.; You, M. p53 Transgenic mice are highly susceptible to 4-nitroquinoline-1-oxide-induced oral cancer. Mol. Cancer Res. 2006, 4, 401–410. [Google Scholar] [CrossRef]
- Barcellos-Hoff, M.H.; Cucinotta, F.A. New tricks for an old fox: Impact of TGFβ on the DNA damage response and genomic stability. Sci. Signal. 2014, 2014, 7. [Google Scholar] [CrossRef]
- Moritoh, Y.; Oka, M.; Yasuhara, Y.; Hozumi, H.; Iwachidow, K.; Fuse, H.; Tozawa, R. Inositol hexakisphosphate kinase 3 regulates metabolism and lifespan in mice. Sci. Rep. 2016, 6, 32072. [Google Scholar] [CrossRef]
- Malla, A.B.; Bhandari, R. IP6K1 is essential for chromatoid body formation and temporal regulation of TNP2 and PRM2 expression in mouse spermatids. J. Cell Sci. 2017, 130, 2854–2866. [Google Scholar] [CrossRef]
- Brehm, M.A.; Wundenberg, T.; Williams, J.; Mayr, G.W.; Shears, S.B. A non-catalytic role for inositol 1,3,4,5,6-pentakisphosphate 2-kinase in the synthesis of ribosomal RNA. J. Cell Sci. 2013, 126, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, S.F.; Maag, D.; Maxwell, M.J.; Resnick, A.C.; Juluri, K.R.; Chakraborty, A.; Koldobskiy, M.A.; Cha, S.H.; Barrow, R.; et al. Amino acid signaling to mTOR mediated by inositol polyphosphate multikinase. Cell Metab. 2011, 13, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, U.; Dollins, D.E.; Fridy, P.C.; York, J.D.; Downes, C.P. Characterization of a selective inhibitor of inositol hexakisphosphate kinases: Use in defining biological roles and metabolic relationships of inositol pyrophosphates. J. Biol. Chem. 2009, 284, 10571–10582. [Google Scholar] [CrossRef] [PubMed]
- Sarmah, B.; Wente, S.R. Inositol hexakisphosphate kinase-2 acts as an effector of the vertebrate Hedgehog pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 19921–19926. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, S.; Zhu, Q.; Asteian, A.; Lin, H.; Xu, H.; Ernst, G.; Barrow, J.C.; Xu, B.; Cameron, M.D.; Kamenecka, T.M.; et al. TNP [N2-(m-Trifluorobenzyl), N6-(p-nitrobenzyl)purine] ameliorates diet induced obesity and insulin resistance via inhibition of the IP6K1 pathway. Mol. Metab. 2016, 5, 903–917. [Google Scholar] [CrossRef]
- Chang, Y.T.; Choi, G.; Bae, Y.S.; Burdett, M.; Moon, H.S.; Lee, J.W.; Gray, N.S.; Schultz, P.G.; Meijer, L.; Chung, S.K.; et al. Purine-based inhibitors of inositol-1,4,5-trisphosphate-3-kinase. Chembiochem 2002, 3, 897–901. [Google Scholar] [CrossRef]
- Cheng, J.Q.; Ruggeri, B.; Klein, W.M.; Sonoda, G.; Altomare, D.A.; Watson, D.K.; Testa, J.R. Amplification of AKT2 in human pancreatic cells and inhibition of AKT2 expression and tumorigenicity by antisense RNA. Proc. Natl. Acad. Sci. USA 1996, 93, 3636–3641. [Google Scholar] [CrossRef]
- Vincent, E.E.; Elder, D.J.; Thomas, E.C.; Phillips, L.; Morgan, C.; Pawade, J.; Sohail, M.; May, M.T.; Hetzel, M.R.; Tavare, J.M. Akt phosphorylation on Thr308 but not on Ser473 correlates with Akt protein kinase activity in human non-small cell lung cancer. Br. J. Cancer 2011, 104, 1755–1761. [Google Scholar] [CrossRef]
- Mundi, P.S.; Sachdev, J.; Mccourt, C.; Kalinsky, K. AKT in cancer: New molecular insights and advances in drug development. Br. J. Clin. Pharmacol. 2016, 82, 943–956. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minini, M.; Senni, A.; Unfer, V.; Bizzarri, M. The Key Role of IP6K: A Novel Target for Anticancer Treatments? Molecules 2020, 25, 4401. https://doi.org/10.3390/molecules25194401
Minini M, Senni A, Unfer V, Bizzarri M. The Key Role of IP6K: A Novel Target for Anticancer Treatments? Molecules. 2020; 25(19):4401. https://doi.org/10.3390/molecules25194401
Chicago/Turabian StyleMinini, Mirko, Alice Senni, Vittorio Unfer, and Mariano Bizzarri. 2020. "The Key Role of IP6K: A Novel Target for Anticancer Treatments?" Molecules 25, no. 19: 4401. https://doi.org/10.3390/molecules25194401
APA StyleMinini, M., Senni, A., Unfer, V., & Bizzarri, M. (2020). The Key Role of IP6K: A Novel Target for Anticancer Treatments? Molecules, 25(19), 4401. https://doi.org/10.3390/molecules25194401