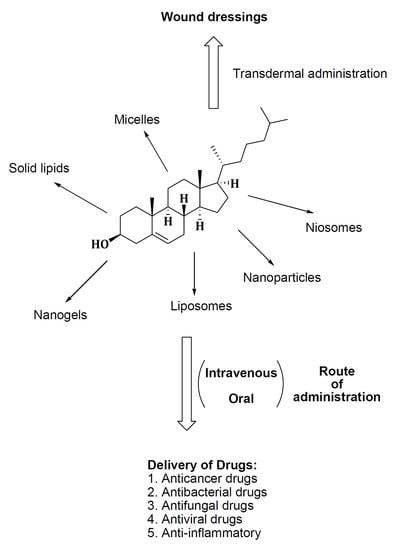
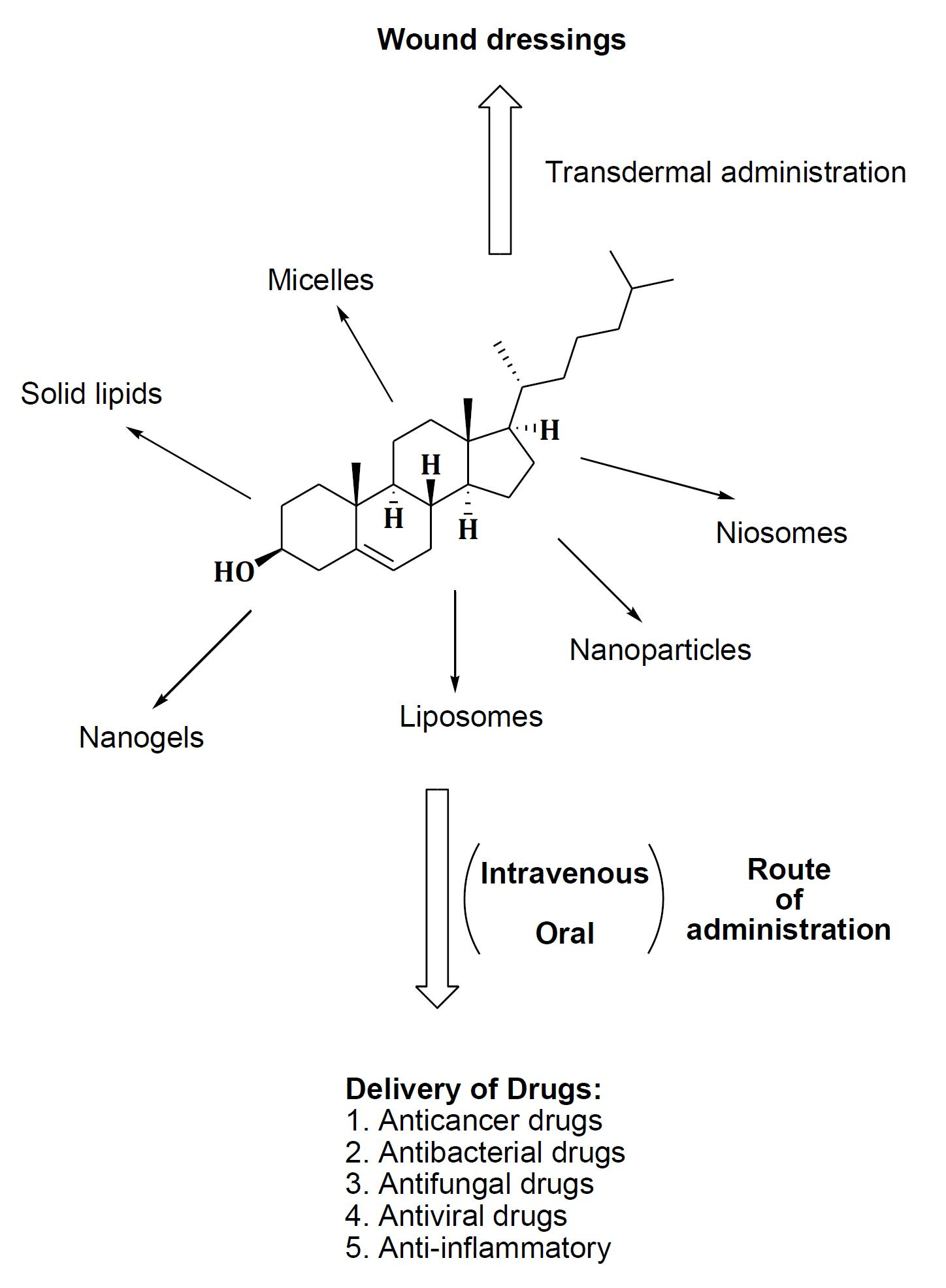
3.1. Micelles
Polymeric micelles are promising drug delivery systems which have attracted considerable interest due to their enhanced solubility in water and prolonged blood circulation [
30,
31]. Incorporation of cholesterol to any drug delivery system improves transportation across the cell membrane. Drugs encapsulated in these micelles can be delivered to specific tissues, prolonging the half-life of the incorporated drug in the blood and achieving a controlled drug release mechanism without exerting severe toxic side effects. Bioactive agents which are incorporated into the core of the polymeric micelles include charged compounds, lipophilic substances and metal complexes. Micelles are structurally stable even at low concentrations [
32].
The use of small interfering RNA (siRNA) is a potential therapeutic approach to treat cancer. It acts by silencing genes that contribute to drug resistance during chemotherapy. Its successful therapy is dependent on the use of delivery vehicles due to its biological instability and inefficient cellular uptake [
33]. Delivering siRNA to tumor sites is a challenge because it degrades rapidly by nucleases and it is poorly translocated due to its high negative charge. Chemical modification of siRNA has been investigated to overcome its limitations, but it is still characterized by increased non-specific binding and high toxicity. On the other hand, curcumin has long been known to be a hydrophobic chemotherapeutic cancer agent [
34]. Muddineti et al. developed a curcumin loaded chitosan-cholesterol micellar system as a potential drug–siRNA carrier for cancer combination therapy [
35].
Cellular uptake studies were done by visualizing A549 cells treated with siRNA and curcumin loaded chitosan-cholesterol micelles (C-CCM/siRNA) under a fluorescence microscope. The uptake of the micelles and their accumulation in the cytosol of A549 cells was rapid within 30 min. Thirty minutes after C-CCM and siRNA treatment of A549 cells, the Geo mean fluorescence of A549 was 14,259 ± 246 and 19,153 ± 357, respectively, and it increased to 2245 ± 142 and 3645 ± 168, respectively, after 120 min [
35]. The results revealed that the drug uptake from the micelle system was dependent on time. Both fluorescence results showed that the drug-loaded micelles exhibited cellular uptake which was time-dependent. The siRNA/curcumin loaded micelles formulation was stable at 4 °C over a period of one month. The results revealed that micelles prepared from chitosan and cholesterol are promising systems for combination therapy in cancer treatment [
35]. However, no cytotoxicity study was investigated to fully understand the micellar system.
α-Tocopherol, a form of vitamin E and α-tocopheryl succinate (a-TOS) have also been recently studied as a drug delivery vehicle resulting from their ability to solubilize hydrophobic drugs [
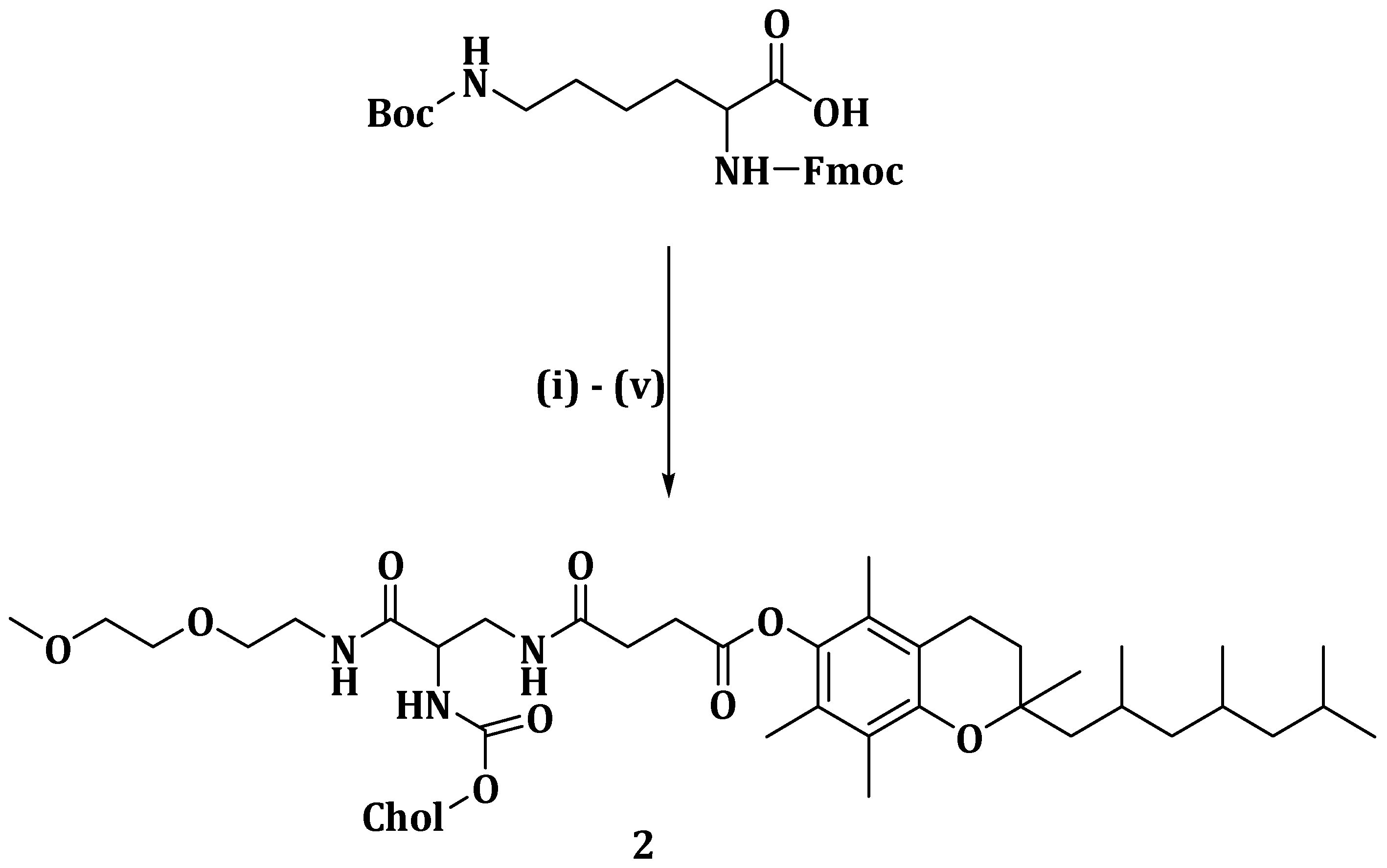
36]. α-TOS induce apoptosis in various cancer cells via membrane destabilization and it is a potential multi-drug resistance inhibitor in cancer therapy. Muddineti et al. developed a drug delivery system composed of vitamin E, a lipophilic core of cholesterol and a hydrophilic corona of polyethylene glycol (PEG), covalently bonded by a trifunctional linker, lysine (
Scheme 1) (
2), which was loaded with curcumin [
37]. Micelles
2 loaded with curcumin (C-CVM) had a bigger hydrophobic compartment size varying in the range of 162.2–175.8 nm compared to polyethylene glycol amine phosphatidyl ethanolamine micelles (PPM) (20.4–24.4 nm). Micelles
2 displayed high drug encapsulation efficiencies of between 97.2% and 98.6% [
37].
It exhibited reduced cell viability, good cellular uptake and sustained drug release in B16F10 and MDAMB-231 cell lines. A low hemolysis activity between 1.10% and 2.81%, which is below the acceptable range of less than 5%, was reported in the cholesterol vitamin E-based polymer, indicating the safe use of the formulation for in vivo studies. Cellular uptake studies showed that the micelles,
2 and C-PPM were taken up in dose- and time-dependent manner as the fluorescence was brighter after 4 h when compared to an hour after administration of the formulation [
37]. Both micelles,
2 and C-PPM, showed no significant difference in the cellular uptake which is due to the presence of PEG-based polymeric corona. In vitro cytotoxicity evaluation on the free curcumin, CVM, PPM and micelles
2 and C-PPM in B16F10 and MDA-MB-231 cell lines revealed that PPM showed no cytotoxic effect on both cell lines with nearly 100% cell survival. However, the presence of α-TOS led to CVM showing significant cytotoxic effect [
37].
Cytotoxic study profiles of C-PPM, free curcumin and micelles
2 for 6 h followed by incubation for 24 h were 48.7%, 58.2% and 42.8, respectively, in B16F10 cells and 74.2%, 59.6% and 58.2%, respectively, in MDA-MB-231 cells. The cell viabilities demonstrated by CVM and PPM (at a concentration of 50 µg/mL) were 42.4% and 91.7%, respectively, in B16F10, 45.2% and 92.3% in MDA-MB-231 cells, respectively. The results showed significant cytotoxicity induced by micelles
2 and this was due to the synergism between α-TOS-conjugated CVM and curcumin. Growth inhibition results showed that the cellular uptake of micelles
2 into the tumor spheroids inhibited cell proliferation resulting in an enhanced therapeutic effect. The developed micelles
2 are potential therapeutics for the treatment of drug-resistant tumors [
37].
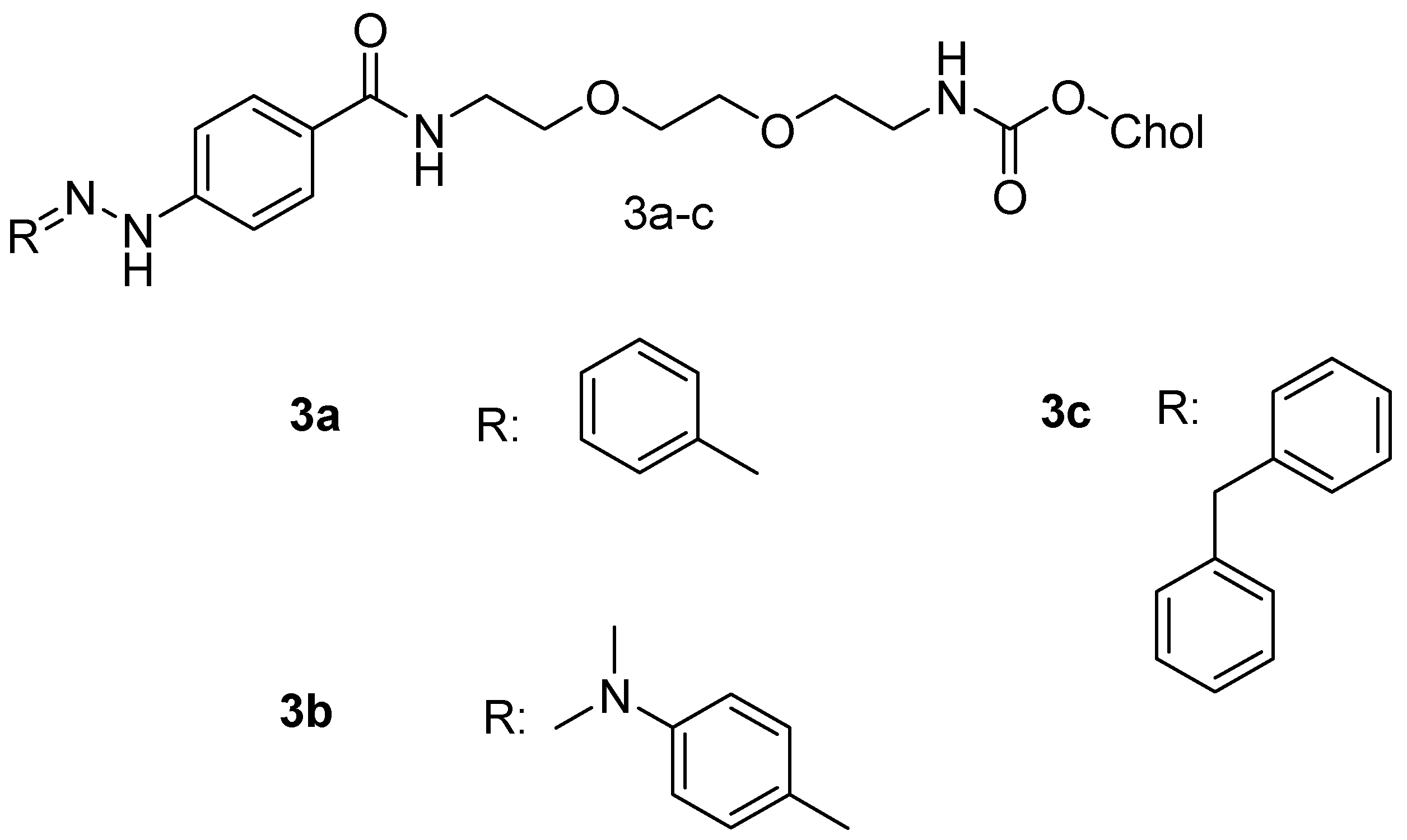
Sarkar et al. synthesized cholesterol-based hydrazone (CBH) tethered tiny amphiphiles with different carbonyl residues (
Figure 2) such as benzaldehyde (3a),
p-dimethylaminobenzaldehyde (3b) and benzophenone (3c). Compound
3a was the most stable of the three and its drug loading efficiency of doxorubicin (DOX) was 57%. Drug release studies of the DOX-loaded compound
3a showed that below the pH of 6.5, the drug-loaded vesicles released DOX better than at pH 7.0–8.0, possibly due to the hydrazone bond cleavage [
38]. The release pattern is important since most tumor cells have both their intracellular and extracellular pH below 6.5 [
17], meaning the anticancer drug is released right at the site of action enhancing its efficacy.
3.2. Nanoparticles
Liposomes have been used as a drug delivery system but suffer from drawbacks such as high leakage of the encapsulated drugs, high production cost, fast clearance from circulation and short stability [
39]. Heidari et al. prepared titania nanotubes (TNTs) in which 5-fluorouracil, a chemotherapeutic drug, was filled into the cylindrical empty space. Liposomes made of soy lecithin and cholesterol (
Scheme 2) were used to coat the drug-loaded TNTs capping the nanotube hole to prevent drug leakage. The drug loading was 100% in the nanotubes. The rate of release of 5-fluorouracil was influenced by the liposomal layers coating on the surface of the nanotubes. At TNT concentrations of <300 μg/mL and between 300 and 1500 μg/mL, about 90% and 80% of the HeLa cells were alive, respectively [
40]. The highest toxicity was noted above 3000 μg/mL [
41]. Cytotoxicity studies revealed that 5-fluorouracil loaded with compound
4 had a decreased IC
50 of 250 μg/mL when compared to about 470 μg/mL for the free 5-fluorouracil. Compound
4 displayed enhanced cellular internalization with effective delivery of 5-fluorouracil into the cells [
40].
DOX can be readily loaded into DNA nanoparticles for drug delivery [
42]. Choi et al. synthesized amphiphilic DNA-cholesterol/DNA-peptide hybrid duplex that assembled into DNA nanoparticles (c-DNA-p nanoparticles) in an aqueous solution. The binding efficiency studies were done by analyzing the fluorescent bands of free DOX and DOX/c-DNA-p complexes on 2% agarose gel electrophoresis. That 0.5 mM of the nanoparticles fully complexed with 3.5 mM DOX revealed the absence of free DOX, indicating a simple and efficient method of loading DOX into the nanoparticles. DOX was fully bound to c-DNA-p nanoparticles at neutral pH 7.4 but the drug began to dissociate from the DOX/c-DNA-p complex at acidic pH of 6.5. At pH 5.0, most of the DOX dissociated from the complex thus indicating the lowering of the binding affinity between DOX and the c-DNA-p nanoparticles which could facilitate cytosolic DOX release [
43].
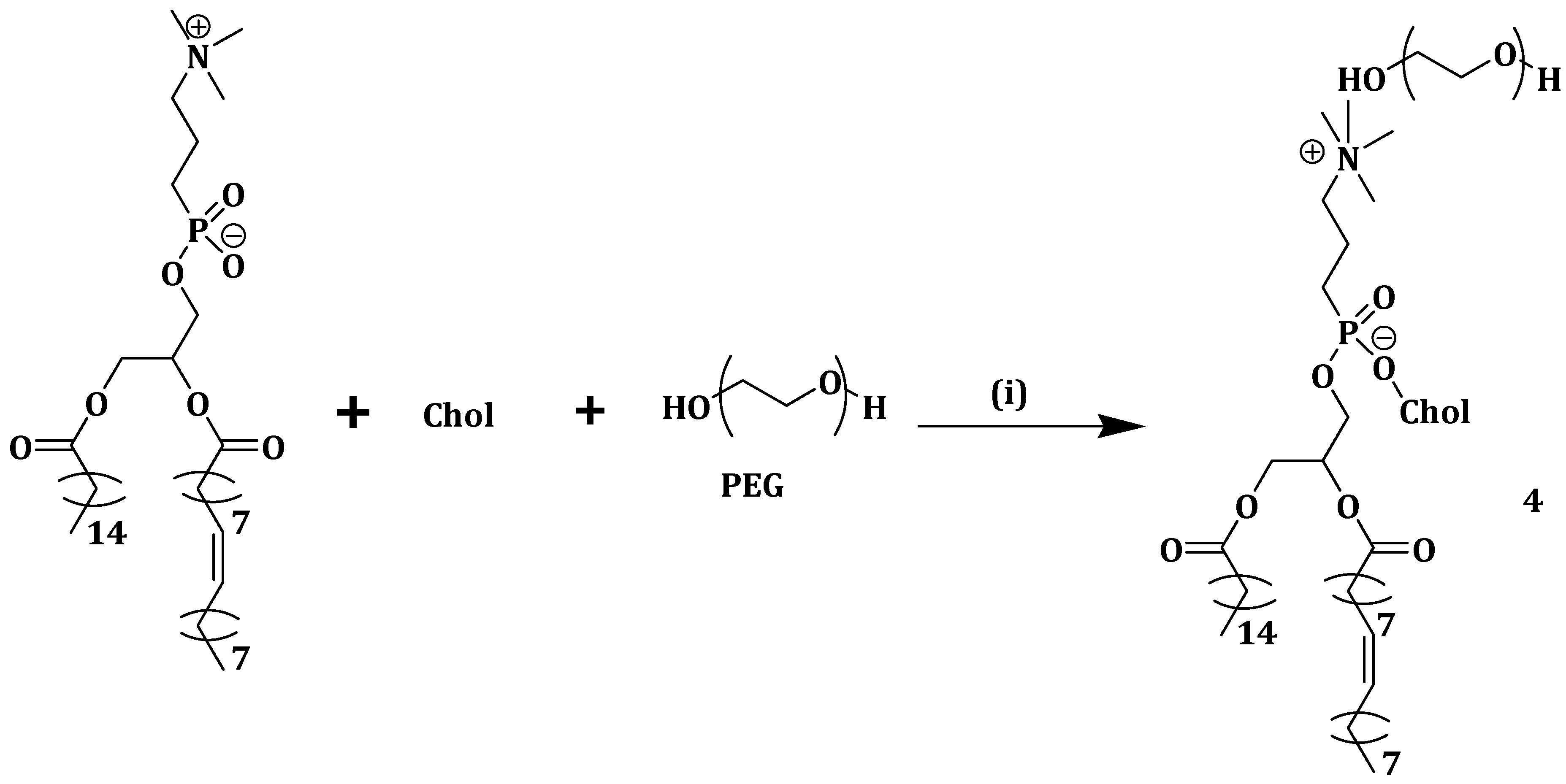
Nanoparticle instability and limited drug loading capacity in most cholesterol-containing polymeric nanoparticles lead to premature drug release into the plasma before reaching the tumor sites. Gonzalez-Fajardo et al. designed and synthesized a drug vehicle, P(NBCh9-bNBPEG), with several cholesteric and PEG side chains as hydrophobic and hydrophilic blocks, respectively. The brush-like architecture had the ability to form stable nanoparticles for the encapsulation of hydrophobic drugs [
44]. DOX was loaded into the P(NBCh9-b-NBPEG) nanoparticles (DOX-NPs) and in vivo studies were performed in a xenograft mouse tumor model. There was a significantly reduced tumor growth without side effects when the mice were treated with DOX-NPs. The low release of DOX from the nanoparticles in the blood as well as the reduced accumulation in the major organs like liver, spleen and heart revealed the improved safety profile of DOX-NPs when compared to the free drugs. The results revealed the potential of P(NBCh9-b-NBPEG) nanoparticles for the delivery of hydrophobic drugs with reduced toxicity [
44].
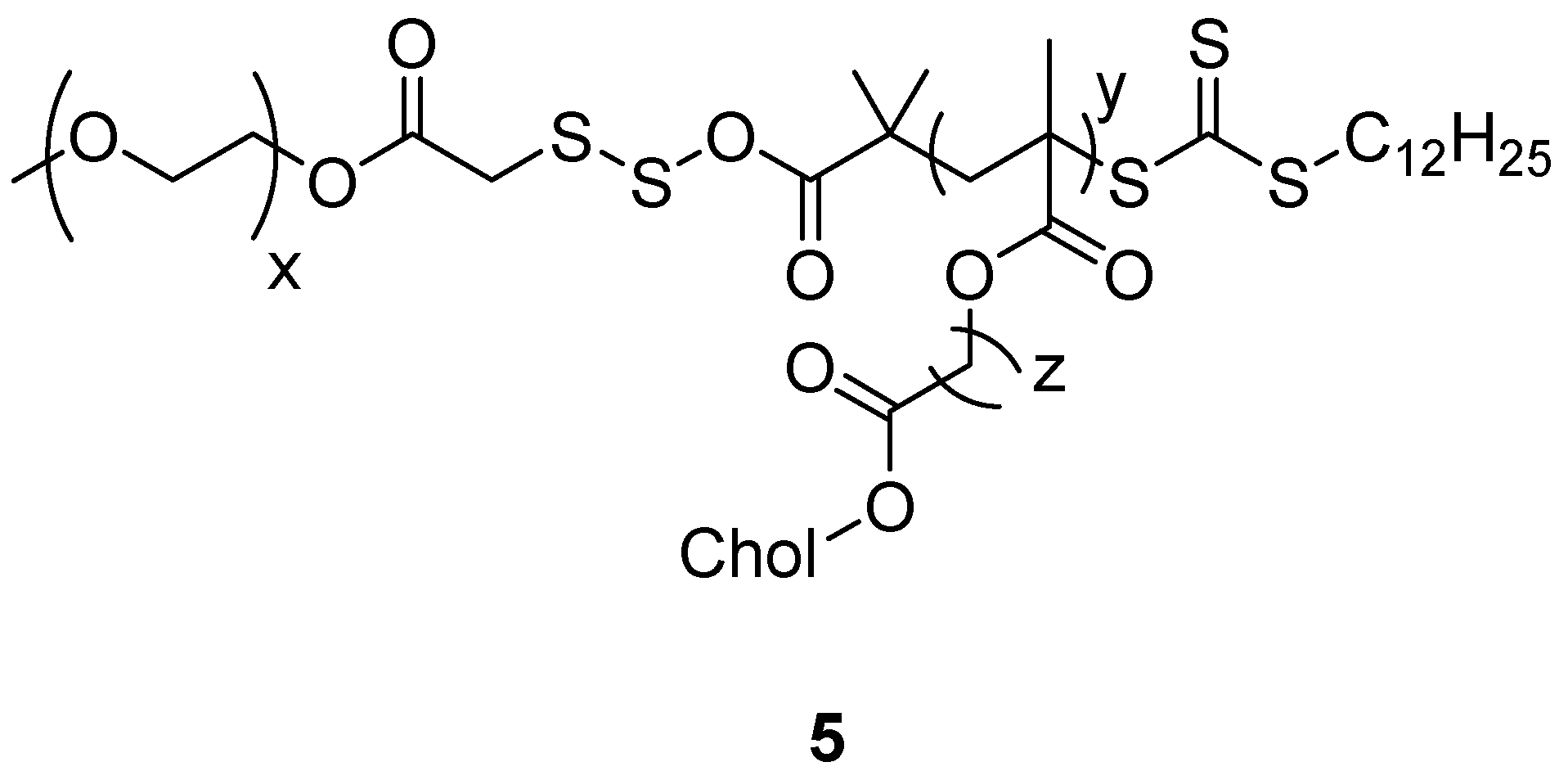
Nguyen et al. synthesized a novel amphiphilic cholesterol-based block polymer comprising of polymethacrylate with cholesterol and PEG having reducible disulfide bonds (PC5MA-SS-PEO) (
Figure 3) (
5) as a nanoparticulate redox-sensitive delivery system. DOX was encapsulated to the self-assembled nanoparticles and the drug loading was 18%
w/
w with 95% drug encapsulation efficiency. The DOX-loaded nanoparticles exhibited excellent stability and readily released DOX in dithiothreitol reductive conditions. Notably, the reducible nanoparticles
5 rapidly released DOX inside the tumor cells thereby inducing higher toxicity when compared to the non-reducible PC5MA-PEO-thioester nanoparticles. The in vivo images taken from A549 tumor-bearing severe combined immunodeficiency mice showed that the nanoparticles
5 accumulated preferably in the tumor tissues when compared to the healthy cells/tissues revealing that they are promising delivery systems for the delivery of anticancer drugs with reduced toxicity [
45].
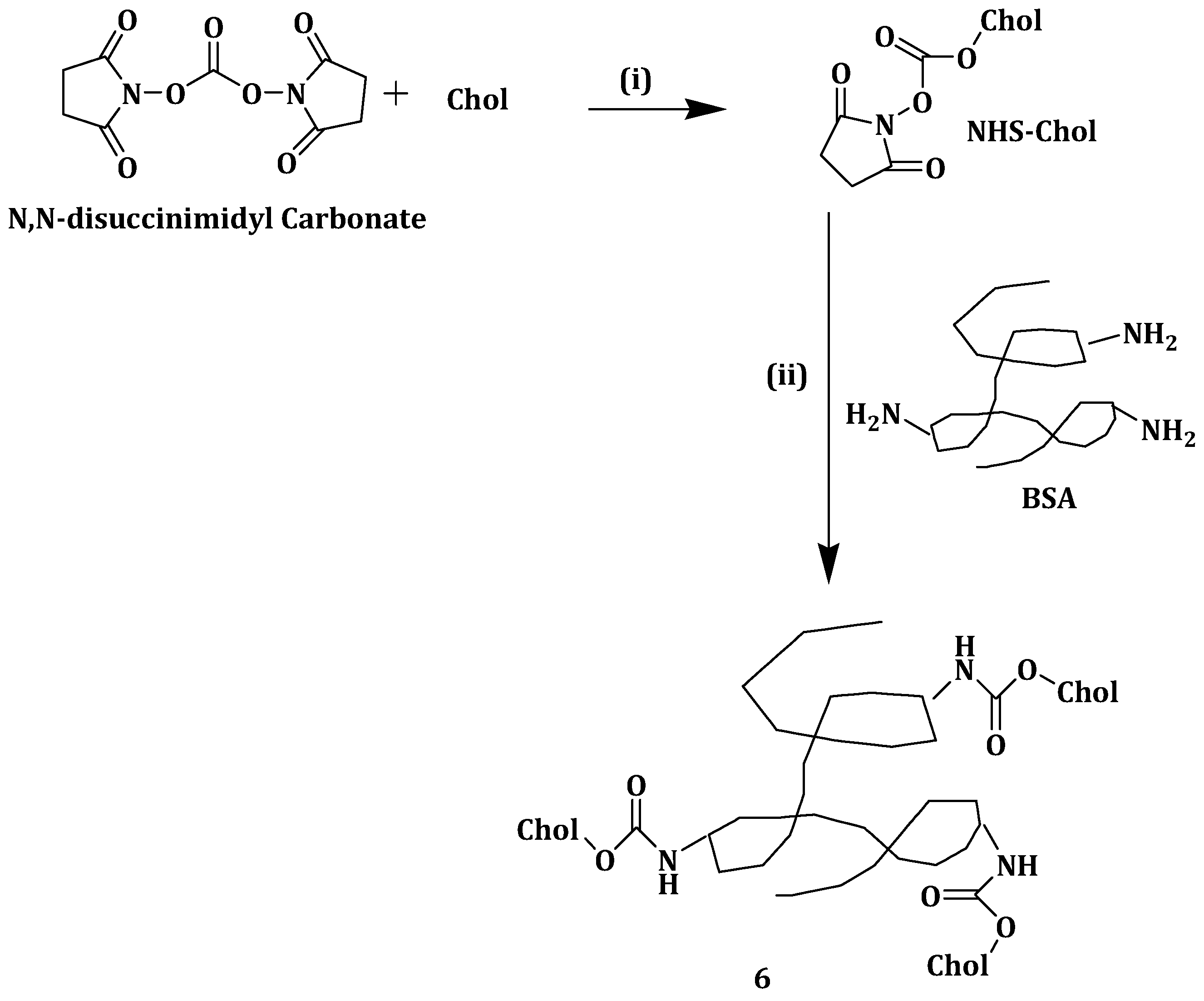
Abraxane is a nanoparticle composite of paclitaxel (PTX) and human serum albumin (HSA), approved for cancer [
46,
47]. The poor colloidal stability of abraxane in the blood has been found not to improve PTX serum half-life [
48,
49]. To circumvent the aforementioned problem, Battogtokh et al. synthesized cholesteryl bovine serum albumin (Chol-BSA) (
Scheme 3) nanoparticles (
6) as PTX carriers. When loaded with PTX, nanoparticles
6 showed a 94.8% drug loading efficiency and 37.9% drug loading capacity making the formulation a potential drug carrier than its closest competitors [
50]. The nanoparticles had an optimum size of 150 nm and a negative charge surface making them stable in aqueous medium [
51]. The release of PTX was two-fold slower from PTX-loaded nanoparticle
6 when compared to PTX-BSA over a period of 72h at 37 °C, showing the sustained drug release from nanoparticles
6. When incubated with B16F10 cells, there was 2- and 0.5-fold increase in PTX uptake from the PTX-loaded nanoparticles
6 when compared to PTX-Cre/EtOH and PTX–BSA, respectively. After incubating PTX-loaded nanoparticles
6 with MCF-7 cells for 1 h, the uptake of the nanoparticles was reported to be 3.5- and 1.4-fold higher when compared to PTX-Cre/EtOH and PTX–BSA, respectively. These findings revealed the capability of the PTX-loaded nanoparticles
6 in delivering more drugs to the cells [
50].
Cytotoxicity studies revealed that B16F10 and MCF-7 cells treated with 10 and 100 nM PTX-loaded nanoparticles
6 for 48 h exhibited lower cell viability when compared to the same cell lines that were treated with PTX-Cre/EtOH and PTX–BSA. No cytotoxic effect was displayed by nanoparticles
6. In vivo antitumor studies showed that the tumor volume in mouse model was smaller than that of mice administered with saline and other groups. Tumor volume of PTX-loaded nanoparticles
6 was reported to be 46.94% at day 8 after administration, compared to 78.23% and 77.35% of PTX-Cre/EtOH and PTX–BSA, respectively. The results indicated that PTX anti-tumor effect can be enhanced using nanoparticles
6 as a nanocarrier [
50].
Tamoxifen (TMX) is a drug used to treat estrogen receptor and progesterone receptor breast cancer [
52]. To improve its cell penetration, entrapment and pharmacokinetics, Mazumdar et al. developed mPEG-b-(CB-{g-chol}-co-LA), a self-assembling cholesterol grafted lipopolymer, synthesized from poly(ethyleneglycol)-block-2-methyl-2 carboxylpropylenecarboxylic acid-co-poly (L-lactide) [mPEG-b-(CB-{g-COOH}-co-LA)] copolymer followed by the incorporation of cholesterol through carbodiimide coupling. TMX release studies were done at pH 7.4 and 5.6, with the latter representing the acidic tumor medium. After 96 h at pH 7.4, 72% of TMX was released from [mPEG-b-(CB-{g-COOH}-co-LA)] while 55% was released from methoxy–poly (ethylene glycol)-poly(D,L-Lactide) (mPEG-PLA) (used for comparison). After 96 h of incubation at pH 5.6, about 97% of TMX was released from both nanoparticles [
53].
The TMX loaded mPEG-b-(CB-{g-chol}-coLA) lipopolymeric nanoparticles displayed higher cellular uptake efficiency and improved IC
50 values of 22.2 μM in the breast cancer cell lines in 4T1 and 18.8 μM in MCF-7 compared to IC
50 values of 27.6 μM and 23.5 μM, respectively, for the free TMX. The results also showed that at the above IC
50 values, TMX loaded lipopolymeric nanoparticles induced cell death and arrested cell cycle at G0/G1 phase at the same rate as the free TMX. The pharmacokinetics results showed approximately 2.5- and 2.7-fold increase in the half-life and mean residence time, respectively, of TMX after incorporating it into lipopolymeric nanoparticles. The findings indicate that mPEG-b-(CB-{gchol}-co-LA) lipopolymeric nanoparticles are useful for the internalization and enhanced residence time in breast cancer cell lines with reduced drug dose and side effects [
53].
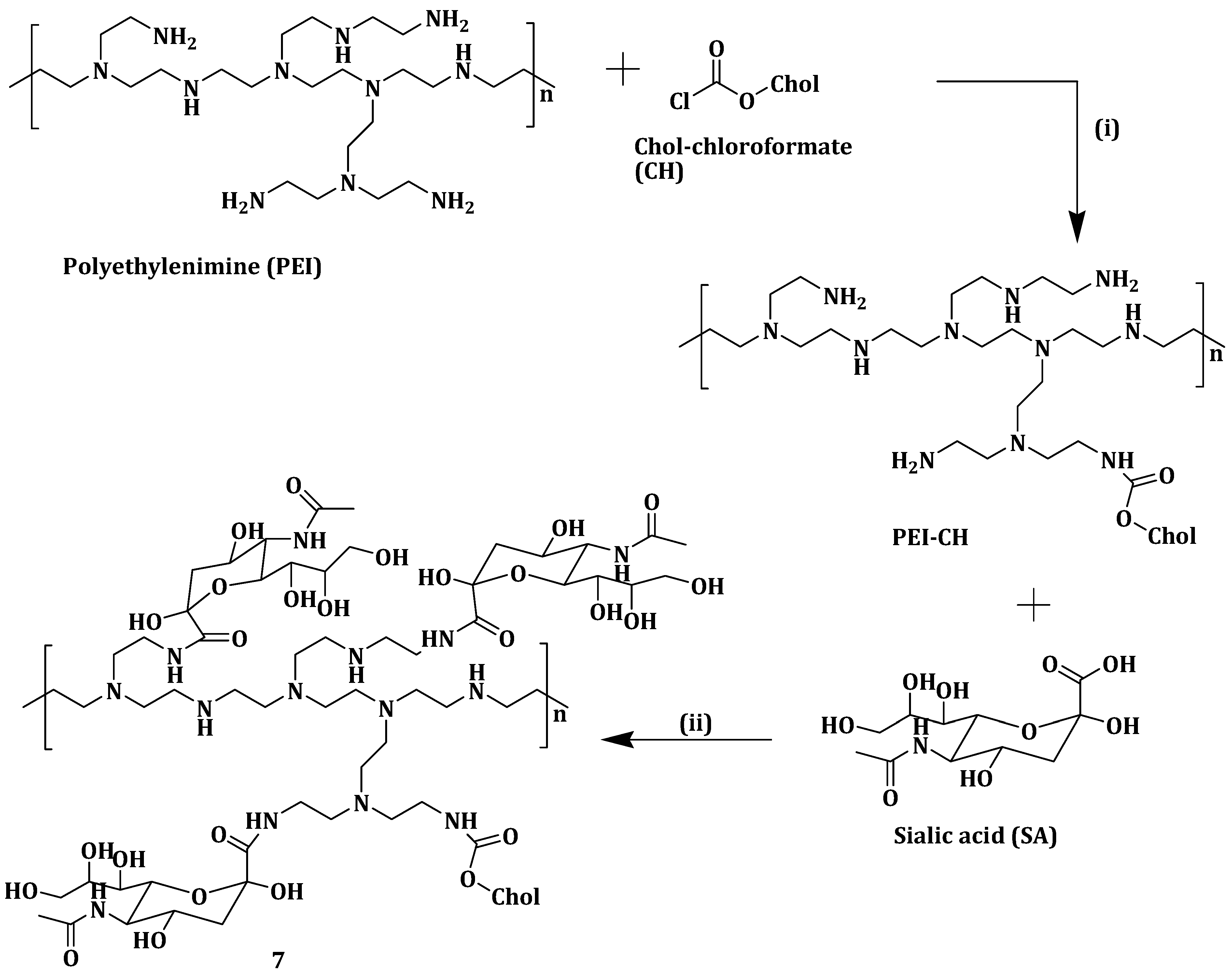
Zheng et al. prepared sialic acid-polyethyleneimine-cholesterol modified liposomes loaded with doxorubicin (DOX-SPCL) (
7) (
Scheme 4) to enhance sarcoma chemotherapy. DOX-loaded liposomes
7 selectively bind to tumor associated macrophages (TAMs) when compared to the conventional liposomal doxorubicin (DOX-CL) and PEGylated liposomal doxorubicin (DOX-PL). DOX-loaded liposomes
7 did not display precipitation or aggregation which is attributed to its high zeta potential (−4.5 mV). Cellular uptake and intracellular trafficking studies showed that DOX-loaded liposomes
7 being trapped in RAW264.7 cells lysosomes up to 1 h [
54].
Modification of the liposomes with sialic acid-polyethyleneimine-cholesterol enhanced their selective uptake through the binding of sialic acid and above all, the trafficking of DOX-loaded liposomes
7 into the cytosol is very critical for killing TAMs. The cytotoxicity of the liposomes was significant [
55]. However, it is important to indicate that polyethyleneimine display cellular toxicity due to its structure, charge, etc. Compared with other organs, DOX-loaded liposomes
7 accumulated in the tumors and promoted exhaustion of TAMs. DOX-loaded liposomes
7 reduced tumor volume by 92.7% in vivo [
54].
Cyclodextrins (CDs) are oligosaccharides whose free hydroxyl groups can be used for crosslinking either between CD molecules or with other molecules. CD nanosponges (NSPs) have been reported to improve absorption, solubility and control drug release [
56]. Drugs can be incorporated within the CD NSP structure, but their applications suffer from their inability to interact with cell membranes and also their non-binding nature. Cholesterol hydrogen succinate (CHS) is known for its protein interaction and cellular binding properties [
57]. Singh et al. functionalized β-cyclodextrin nanosponge (β-CD-NSP) surface with CHS in an attempt to enhance the cellular binding of CD-NSP. DOX was used as the model drug for cellular uptake and absorption capacity studies [
57].
β-CD-NSP-CHS exhibited a 5.8% higher drug adsorption capacity when compared to β-CD-NSP. The enhanced drug adsorption is attributed to the hydrophobic nature of the surface. The DOX release profiles at pH 6.8 and 1.2 of β-CD-NSP-DOX and β-CD-NSP-CHS-DOX were the same, suggesting that CHS grafting did not change the release pattern. The drug release from both nanosponges at pH 1.2 was fast with over 82% of the drug released within 15 min when compared to phosphate buffer at pH 6.8 in which less than 30% of the drug diffused from the NSP [
57].
Cytotoxicity studies revealed that β-CD-NSP-CHS exerted no cytotoxic effects in HeLa cells even at a high sample concentration of 100 µg/mL. The surface-modified β-CD-NSP was biocompatible and safe in drug delivery. Laser scanning confocal microscopic images showed high DOX accumulation in HeLa cells incubated with DOX-loaded β-CD-NSP-CHS samples when compared to HeLa cells treated with free DOX. This finding indicate that CHS played an important role in enhancing the drug penetration and cell internalization [
58]. The surface modified CD-NSP is useful in the delivering of small low water-soluble drugs thereby improving both their solubility and bioavailability [
57].
Mallick et al. prepared liposomes from cholesterol-phenylalanine-arginine-phenylalanine-lysine (Chol-FRFK) sequence and 1,2-dioleoyl-
sn-glycero-3-phoshphoethanolamine (DOPE) was used to formulate (Chol-FRFK/D liposomes) for delivery of an anticancer drug, Antimycin A (AMA), to the mitochondria. The Chol-FRFK/D liposomes had a higher cellular uptake and mitochondrial targeting compared to the two controls, DQAsomes and DOTAP/DOPE (D/D) liposomes. Treating A549 cells with Chol-FRFK/D liposomes showed an IC
50 of 10 µM compared to an IC
50 of 50 µM for free AMA [
15]. Drug encapsulation in the Chol-FRFK/D liposomes was 100% since AMA could be accommodated both in the lipid bilayer membrane and the inside of the core. The Chol-FRFK/D-AMA formulation was very cytotoxic that it caused mitochondria-mediated apoptosis in A549 cells. The Chol-FRFK/D liposomes are a promising anticancer delivery system for anticancer therapy [
15].
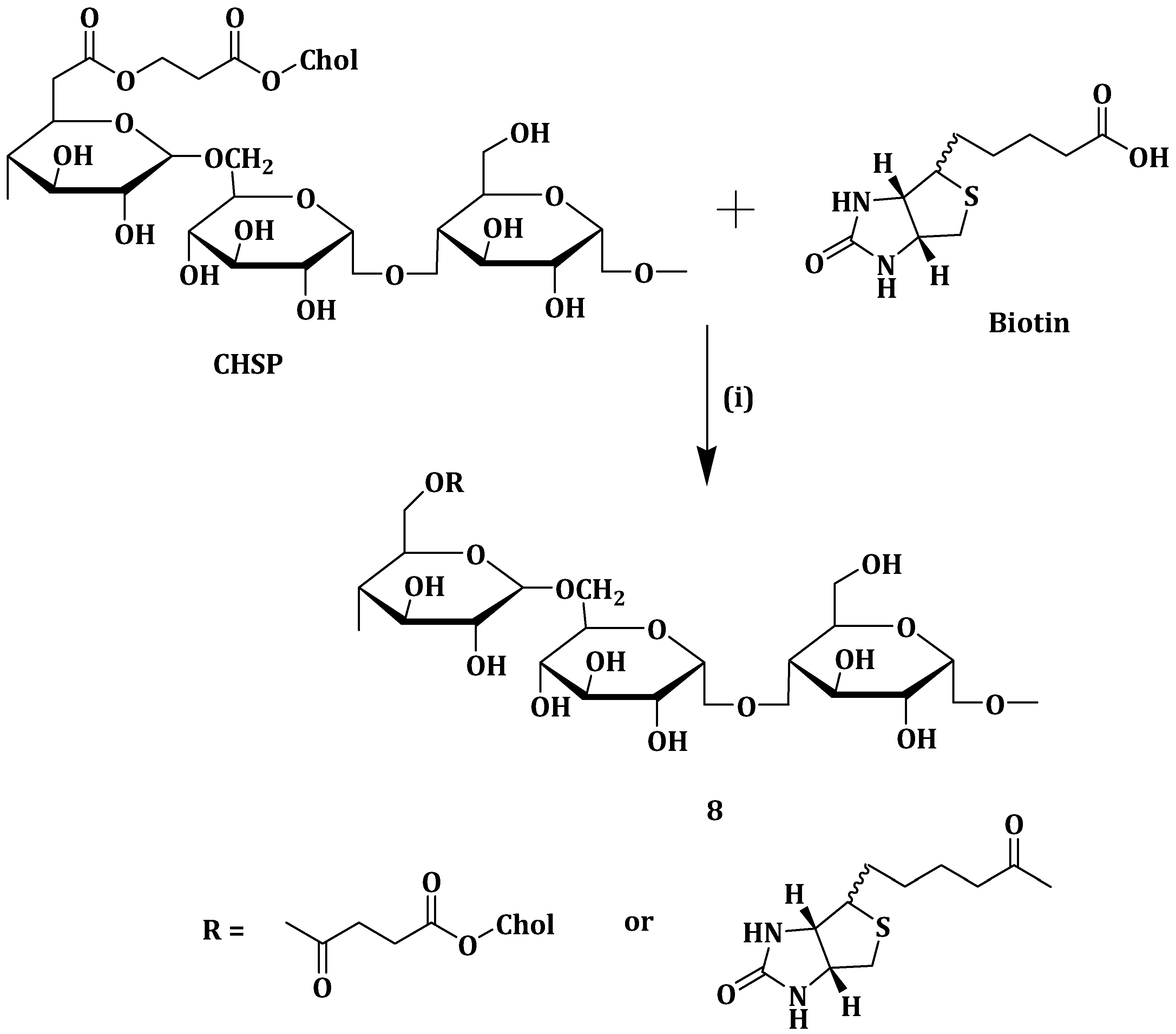
Cholesterol-modified pullulan nanoparticles (CHSP NPs) have been found to exhibit sustained drug release and excellent biocompatibility. Its major setback is that it does not actively target the tumor cells [
59]. To deal with the above set-back, Yang et al. synthesized biotin grafted CHSP copolymer (Bio-CHSP) (
Scheme 5) (
8) for targeted drug delivery. Mitoxantrone (MTO) is an anticancer agent use in treating various carcinomas and it has side effects such as vomiting, nausea, cardiotoxicity and anemia. The synthesized nanoparticles
8 was used as a carrier for MTO to decrease its toxic effects, enhance therapeutic efficacy and for sustained drug release [
60]. Nanoparticles
8 with different degree of substitution of biotin per hundred glucose units was synthesized (Bio-CHSP
-20.1, Bio-CHSP
-29.2 and Bio-CHSP
-38.9) and they had critical aggregation concentration values of 0.0055, 0.0017 and 0.00015 mg/mL, respectively. The nanoparticles
8 self-aggregated in aqueous media because of the hydrophobic cholesterol and biotin. MTO release at pH 7.4 and 3.5 from MTO-loaded nanoparticles
8 was slightly pH-dependent. MTO dissolved more in lower pH thus inducing drug release, thus MTO release was faster in pH of 3.5 when compared to pH 7.4 [
60].
Release studies showed that free MTO was released over 90% within 8 h. MTO was released rapidly in MTO-loaded nanoparticles
8 and then slowly after 12 h, suggesting sustained release ability of nanoparticles
8. In vivo toxicity of nanoparticles
8 was studied in mice. Judged from their eating habits, pathological changes and body weight data, the mice remained healthy throughout the experiment. Histopathological studies of major organs showed that there was no difference between the control and those administered with nanoparticles
8. This means that the nanoparticles did not induce acute toxicity after intravenous administration at a dose of 200 mg/kg. Together with the other in vitro results, nanoparticles
8 was biocompatible and safe for hydrophobic drug delivery [
60].
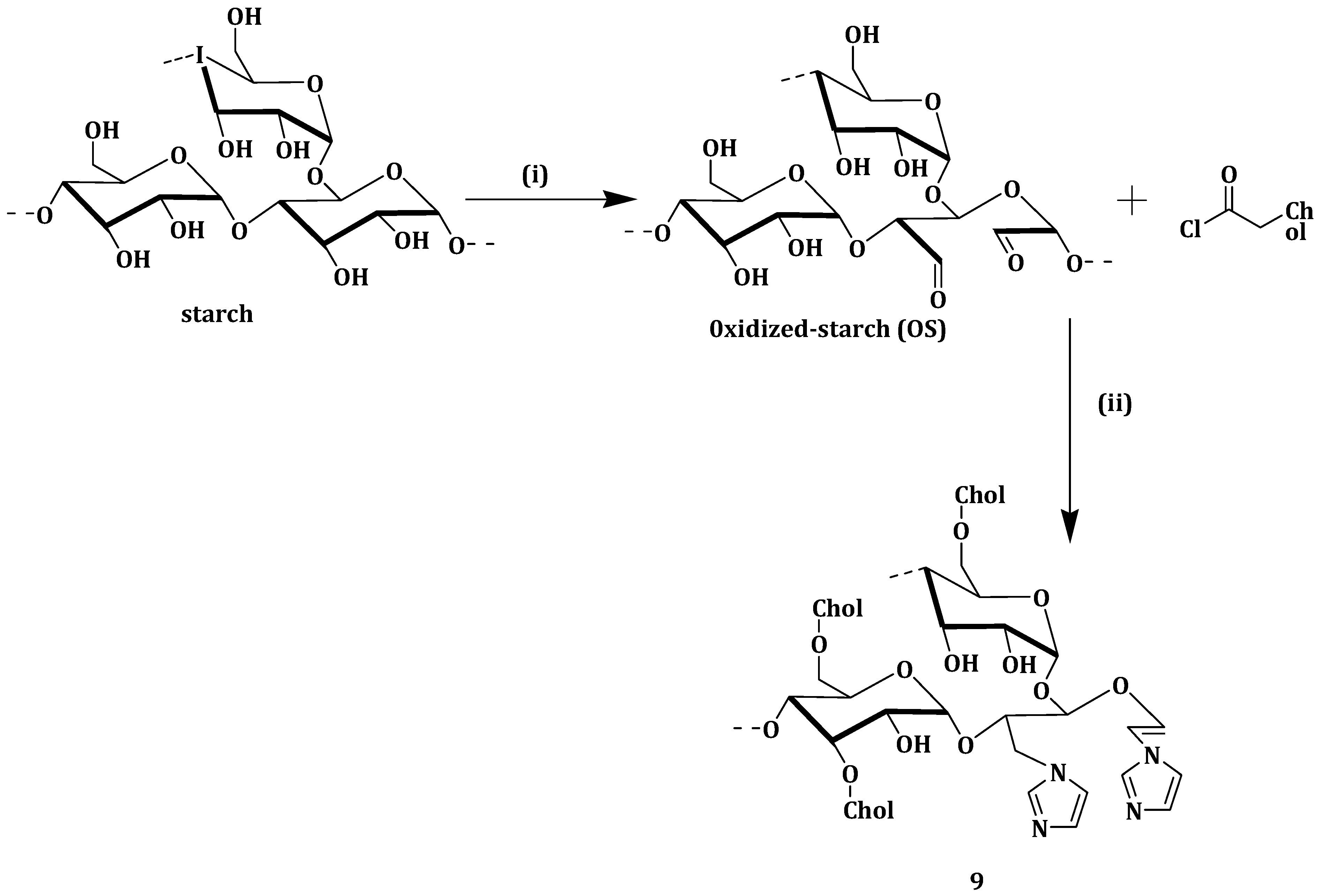
Xu et al. developed novel cholesterol/imidazole modified oxidized-starch (Cho-Imi-OS) (
Scheme 6) (
9) nanoparticles as a pH-sensitive and neutrally charged tumor-targeted drug delivery system, using curcumin as a model drug. The curcumin-loaded nanoparticles
9 drug loading and entrapment efficiencies were 4.16% and 17.84%, respectively. Curcumin was released more quickly at pH 5.5 (65% cumulative release) than at pH 7.4 (38% cumulative release) after 68 h. The faster cumulative release at pH 5.5 is very important since the pH in tumor cells endosomes is between 5.0 and 6.0, meaning most of the curcumin is released right at the tumor site for effective therapy [
61].
The nanoparticles without a drug and curcumin-loaded nanoparticles
9 showed hemolysis ratios of 1% and 2%, respectively, which are all within the standard level of 5%. A549 cells effective uptake of curcumin-loaded nanoparticles
9, when compared to free curcumin, was attributed to the nano-size of the curcumin-loaded nanoparticles
9. Curcumin-loaded nanoparticles
9 exhibited an IC
50 of 4.2 µg/mL, and the nanoparticles not loaded with drug maintained cell viability of more than 85% [
61].
Wu et al. prepared liposomes with hydrogenated soybean phospholipids (HSPC), 1,2-distearoyl-
sn-glycero-3-phosphoethanolamine-
N-[methoxy(polyethylene glycol)-2000] (DSPE-PEG
2000) and varying amounts of cholesterol (
Table 1) and loaded them with doxorubicin. The rate of drug release decreased as the cholesterol content in the liposomes increased. Liposomal cellular uptake in BxPC-3 and HPaSteC cells decreased with increase in the liposomal cholesterol content. The authors reported a decrease in the membrane rigidity of the liposomes as the cholesterol content increased. The aforementioned findings suggest that when the liposomal membranes are less rigid, there is an increase in surface contact with the cells resulting in a prolonged cellular uptake [
62].
Lip3 and Lip2 displayed moderate rigidity inhibiting tumor spheroid growth and exhibited high tumor penetration with excellent tumor growth inhibition in vivo. The results showed that moderate liposomal rigidity resulted in better diffusivity making cholesterol-tuned liposomes a promising system for delivery drugs to tumor cells [
62].
Qui et al. prepared and patented a method for preparing liposome comprising of a phospholipid and cholesterol-based compound to deliver chloroquine (CQ) and doxorubicin (DOX). Liposomes were prepared from 500 mg phosphatidylcholine and 100 mg cholesterol and had an average size of 129 nm. These liposomes were loaded with 20 mg of CQ and DOX (1:1
w/
w) and had a drug encapsulation of 96% and 97% and drug loading of 1.5% and 1.6% for CQ and DOX, respectively [
63].
Cytotoxicity tests were performed using free drugs (CQ and DOX) as controls and no significant difference between free doxorubicin and drug-loaded liposomes was observed. Both displayed an IC50 = 0.4 µg/mL (
Table 2) against human breast cancer cells (MCF-7). However, the drug-loaded liposomes were more potent on DOX-resistant MCF-7 cells (MCF-7/ADR) with IC
50 = 3.3 µg/mL when compared to free DOX with IC
50 = 13.7 µg/mL. Free CQ was less effective when compared to the drug-loaded liposomes on MCF-7 and MCF-7/ADR. The results followed a similar trend when the above formulations were used on human promyelocytic leukemia cells (HL60) and DOX-resistance human promyelocytic leukemia cell (HL60/ADR), as shown in
Table 2. The results showed the potential ability of the DOX and CQ-loaded liposomes in overcoming mediated multidrug resistance [
63].
3.3. Copolymers
Polymeric nanocarriers transport loaded drugs to the tumor cells [
64]. They suffer from several barriers during transportation such as inability to diffuse through the cell membrane [
65]. The biological pH stimulus can be used to facilitate drug release in acid-labile polymeric nanocarriers. Polymeric nanocarriers need to be constructed with strong bonds that can maintain an integrated structure [
66,
67].
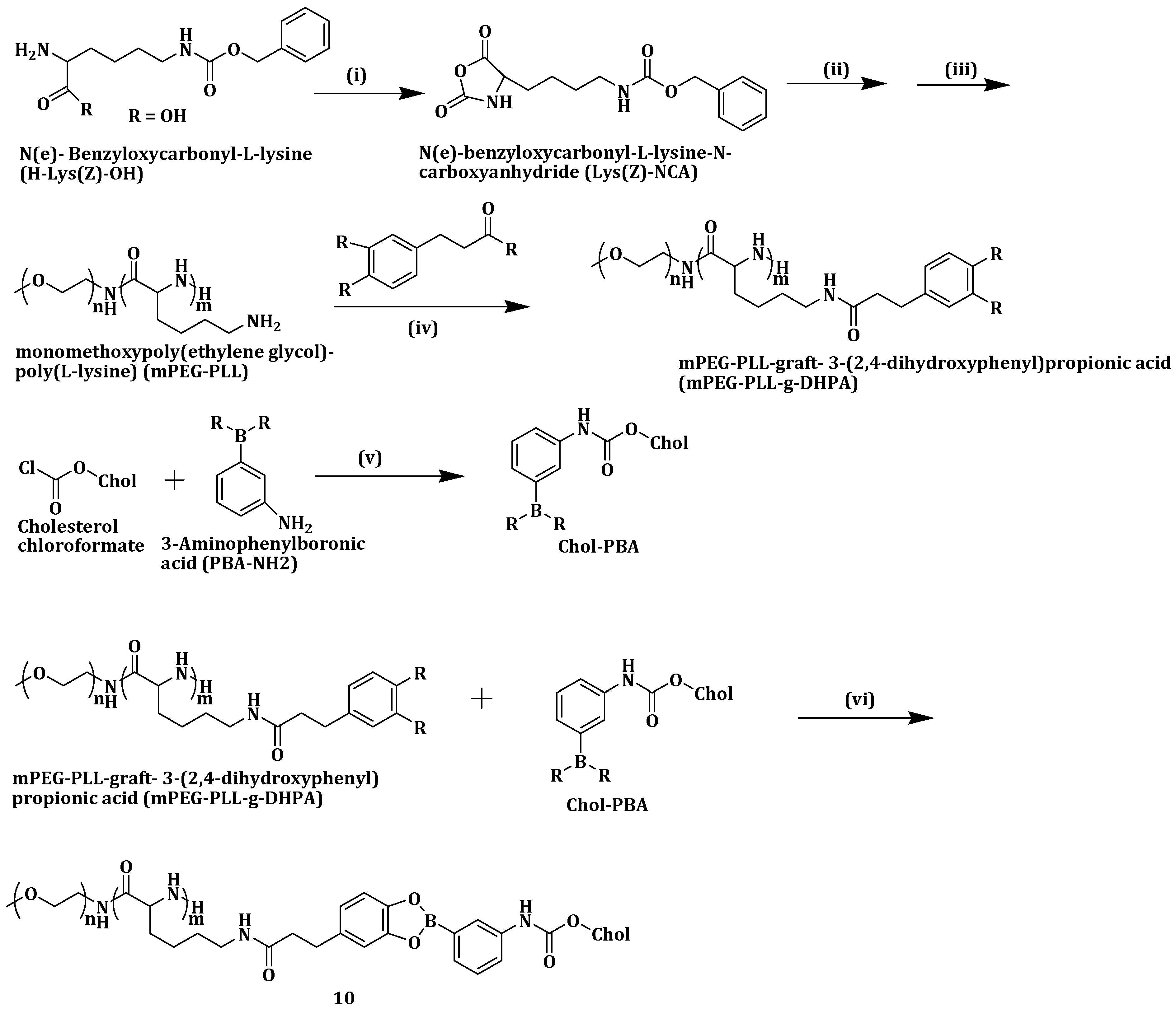
Yang et al. constructed a pH-responsive polymeric drug carrier by reversibly attaching phenylboronic acid-modified cholesterol (Chol-PBA) to catechol-pending methoxypoly (ethylene glycol)-block-poly(L-lysine) (
Scheme 7). The pH-dependent metastability of monomethoxypoly(ethylene glycol)-poly(L-lysine)-graft-3-(2,4-dihydroxyphenyl)propionic acid (mPEG-PLL-g-DHPA)/Chol-PBA (MPDP) (
10) nanoassemblies was evaluated using Nile Red Dye (NRD) at a pH of 7.4 and 5.0 over a period of 24 h. No color changes were detected in the solution at pH 7.4. However, at pH 5.0, red deposits were observed with the solution color fading and ultimately becoming pale yellow. The co-polymer,
10 did not change size at pH 7.4 but increased sharply at pH 5.0 from 196.2 to 954.8 nm, indicating its structural instability in the acidic environment. The boronate bond hydrolyzed in acidic medium precipitating cholesterol and loaded NRD [
68].
In vitro drug release studies were performed on
10 loaded with DOX. At a pH of 7.4, less than 40% of DOX was released within seven days, compared to 70% drug release at pH 5.0. Tumor HeLa cells viability in
10 remained very high above 90% at 500 µg/mL. Cellular uptake studies revealed that the internalization of the DOX-loaded
10 nanoassemblies into cells was via LDL receptor-mediated endocytosis [
68].
Synthetic and natural polymers have been used for the intravenous delivery of anticancer agents to the tumor sites [
69]. Poly(lactide-co-glycolide) (PLGA) is a biodegradable and biocompatible polymer for tumor-targeted drug delivery. Lee et al. developed poly(lactide-co-glycolide)-cholesterol based nanoparticles (PLGA-C NPs) (
11) (
Figure 4) for intravenous delivery of curcumin to the tumor and cell imaging. Curcumin-loaded nanoparticles
11 size was maintained in both PBS of pH 7.4 and serum (50%
v/
v), which is suitable for efficient drug delivery [
70]. Curcumin release was performed at pH 5.5 (for endocytic compartments), 6.8 (for tumor environment) and 7.4 (for normal physiological conditions). The release of curcumin was in a pH-dependent manner with the amount of drug release after 144 h being 60.7%, 43.7% and 11.8% at pH 5.5, 6.8 and 7.4, respectively. The sustained and pH-dependent drug release mechanism of the drug-loaded nanoparticles influenced its drug tumor-targeting capability [
70].
The cytotoxicity of the nanoparticles was evaluated in Hep-2 cells derived from human laryngeal carcinoma. Both PLGA NPs and the nanoparticles
11 showed no cytotoxic effects making them safe formulation for intravenous drug delivery. In vitro cellular uptake was assessed in Hep-2 cells by encapsulating Dil, a hydrophobic fluorescent dye, into the NPs. The nanoparticles
11 cellular accumulation efficiency was 1.6-fold higher when compared to the nanoparticles when incubated for 4 h. After 24 h incubation, the cellular distribution of the nanoparticles done by CLSM showed stronger fluorescence intensity in
11 when compared to PLGA NPs. In vivo tumor targeting of
11 was shown by Near-infrared fluorescence (NIRF) images of Hep-2 tumor-xenografted mouse model. The results revealed the usefulness of
11 in targeting tumor cells [
70].
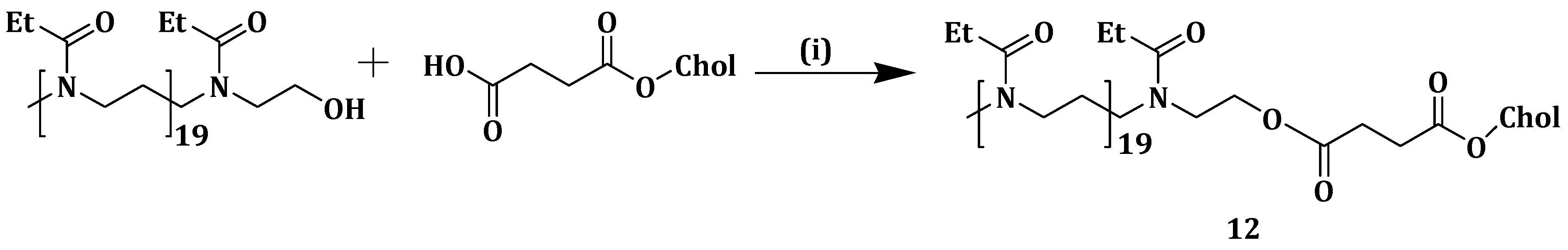
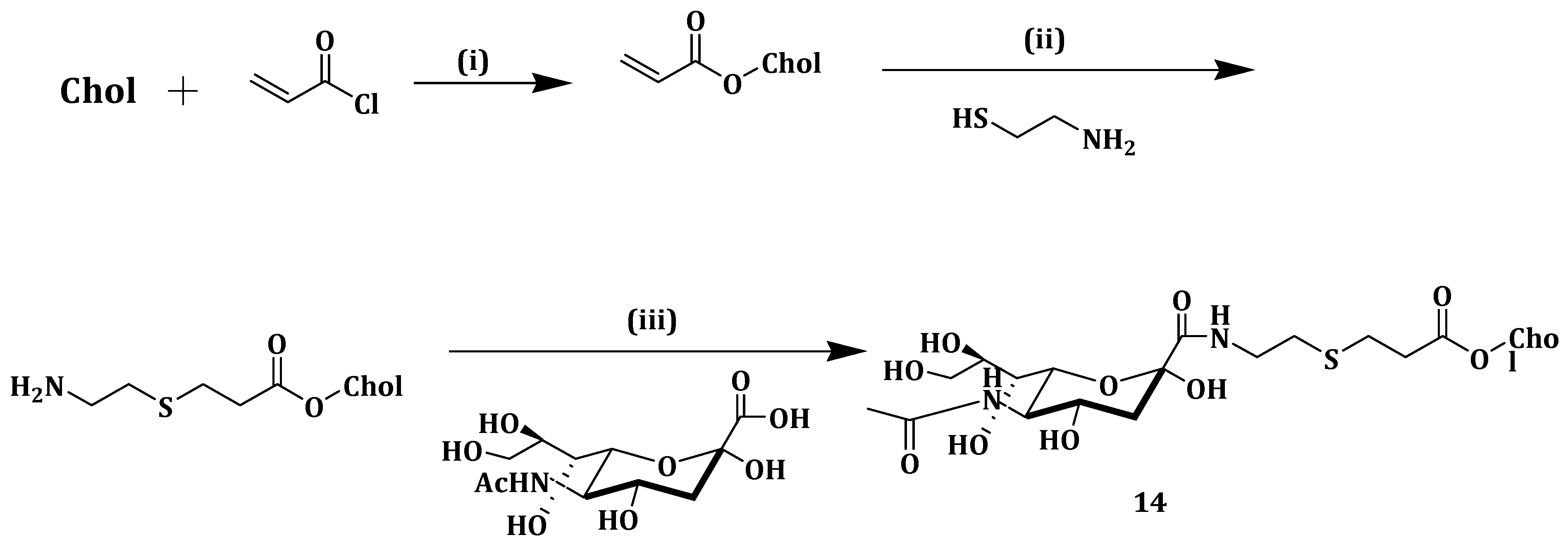
Incorporation of PEG into liposome surfaces is reported to increase drug bioavailability and the circulation time [
71]. PEGylation of liposomes suffers from drawbacks such as loss of response in low pH environment and liposomal cellular uptake hindrance [
72,
73]. Poly(2-ethyl-2-oxazoline) (PEtOz), a long-chain macromolecule polymer, is biocompatible, flexible and low cost and has been reported as a suitable pH-sensitive drug delivery vehicle [
74,
75]. Similar to PEG, PEtOz increases liposome stability in vitro and in vivo, thus it can be used as a PEG substitute [
76]. Xu et al. constructed pH-sensitive liposomes from poly (2-ethyl-2-oxazoline) cholesterol hemisuccinate (PEtOz-CHEMS) (
Scheme 8) (
12) and encapsulated DOX, an anticancer drug. DOX-loaded liposomes
12 displayed slow DOX release when compared to the conventional DOX liposomes (CL-DOX), CHEMS modified DOX liposomes (CH-DOX) at pH 7.4. DOX release increased rapidly as the pH decreased and more than 90% was released within 4 h at pH 5.4. This revealed the significant change in release kinetics at low pH and the stability of the formulation at normal physiological pH, after PEtOzylation of liposomes [
77].
Cellular uptake studies after incubating A375 cells with the different liposomes showed that DOX-loaded compound
12 liposomes exhibited higher fluorescence intensity at pH 6.5 when compared to pH 7.4. PEtOzylation increased the cellular uptake of the liposomes with a maximum DOX accumulation, and, at pH 7.4, DOX-loaded liposomes
12 exhibited cellular uptake higher than that of CH-DOX, CL-DOX and PEG-DOX liposomes. CLSM analysis of A375 cells incubated with the different liposomes showed that PEtOz-DOX liposomes exhibited enhanced cellular uptake, and DOX release into the cytoplasm for PEtOzylated liposomes. A375 cells inhibition increased when incubated by the various liposomes at 37 °C under different pH. However, DOX-loaded liposomes
12 had higher cytotoxic effect at pH 6.4 when compared to pH 7.4 while CH-DOX, CL-DOX and PEG-DOX liposomes showed no difference in the inhibitions even at the different pH mediums. The results revealed the potential of PEtOz in modifying liposomes for drug delivery [
77].
The pH around and inside tumor cells is different from that of normal ones. The extracellular pH around cancer cells is reported to be weakly acidic around 6.5–7.0, while, inside tumor cells, the pH is between 5.0 and 6.5 [
78]. Researchers have focused on developing pH-triggered drug delivery vehicles that are stable at normal physiological pH of 7.4 and can release drugs rapidly in the weakly acidic environment [
79,
80]. In their quest for a novel hydrophobic pH-sensitive drug carrier with lower toxicity and higher efficiency, Yang et al. designed and synthesized three triblock copolymers poly(ethylene glycol) methyl ether-b-peptide-g-cholesterol (mPEG-b-P-g-Chol) (
Scheme 9) (
13a–
c), which self-assembled into micelles in an aqueous medium. The peptides functioned as the pH-sensitive part and the internal core comprising of cholesterol offered space for drug loading. DOX was loaded as the model drug [
81]. The three polymers (mPEG-P1-Chol
13a, mPEG-P2-Chol
13b and mPEG-P3 Chol
13c, which differed only in shape, being linear, y-shaped and fork-shaped, respectively) showed very low CMC values revealing that the micelles could remain intact even in extremely dilute volume of the body’s systemic circulation. The three DOX-loaded polymers exhibited larger particle sizes at a lower pH of 5.0 when compared to pH 7.4. The DOX loading in the three copolymers
13a–
c micelles was about 15.7%, 20.2% and 23.1% in weight, respectively, thus drug loading content and entrapment efficiency increased with polymer complexity [
81].
In vitro drug release of the three polymers in solutions of pH 7.4 and 5.0 showed that DOX release was pH dependent. After 24 h, only about 35%, 30% and 26% of DOX was released from micelles
13a–
c, respectively. The three polymers exhibited DOX release of about 90%, 84% and 80%, respectively, in 148 h. The enhanced positive charge on micelles surface at pH 5.0 caused the histidine moieties to experience electrostatic repulsion increasing the swelling and drug release. At 400 mg/L polymer concentration, cell viability against NIH 3T3 cells was 97.7%, 95.5% and 95.4%, and 96.8%, 95.9% and 95.0% against HepG2 cells, for
13a–
c, respectively [
81]. The DOX-loaded micelles exhibited lower cytotoxicity which increased slowly in lower concentrations suggesting a slow and time-controlled DOX release from the micelles. Although the free DOX-HCl showed the lowest cell viability, it was toxic to the tumor and the normal cells. The slight slowing down of the inhibition effect of DOX-loaded micelles
13 revealed their capability to protect the drug. The results showed the potential of
13 in delivering anticancer drugs [
81].
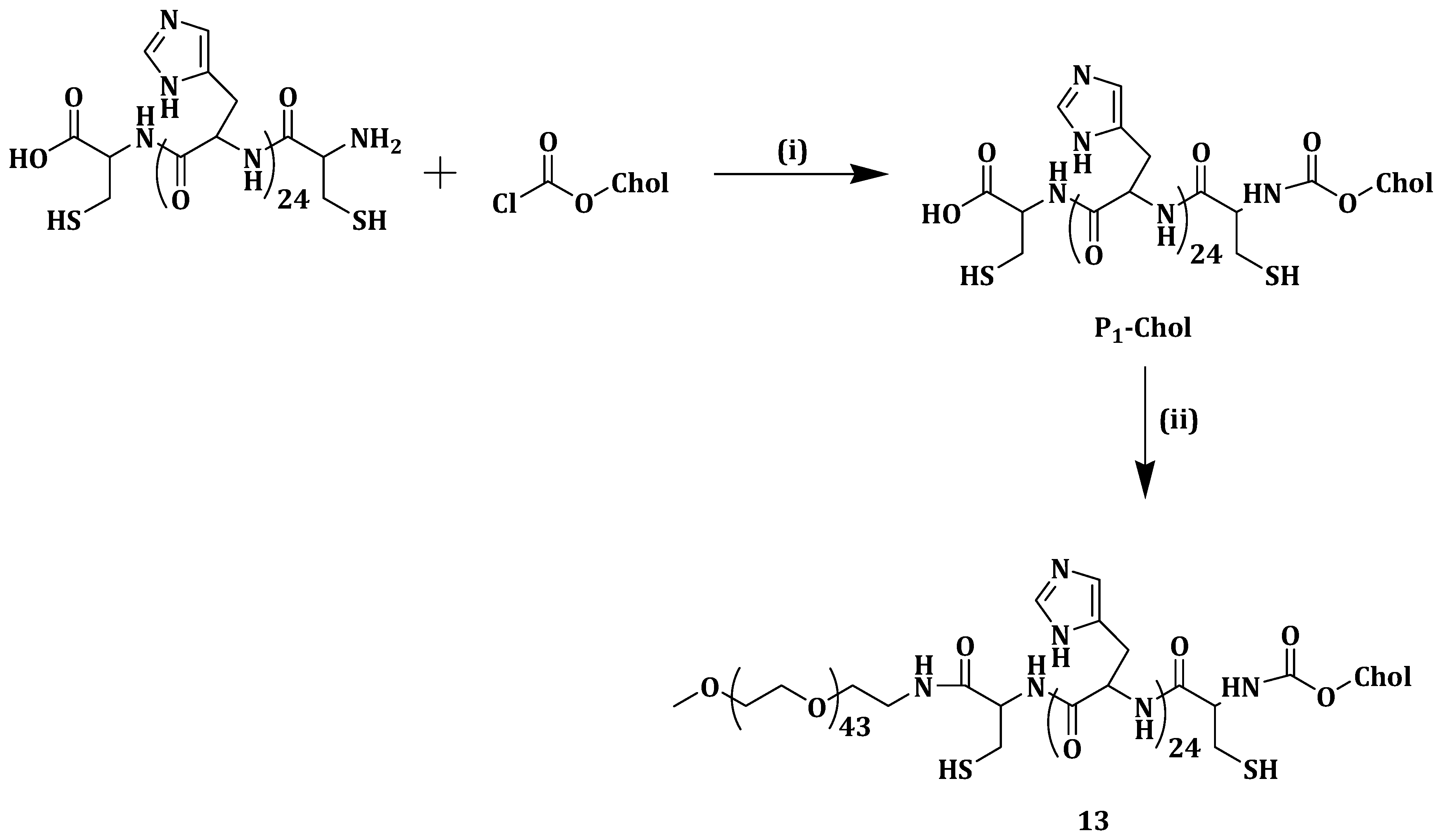
Tumor cells are surrounded by different kinds of cells such as fibroblasts and inflammatory cells, mainly lymphocytes and macrophages [
82]. The mass population around the tumor cells is termed tumor-associated macrophages (TAMs) and tends to promote tumor growth [
83]. Siglec-1 is a member of the Sialic acid (SA) binding family that is overexpressed on TAMs. SA can be used as a ligand for the enhanced distribution of anticancer drugs to TAMs since studies have revealed that Siglecs function as endocytic receptors [
84]. In trying to target TAMs and improve the tumor selectivity and specific toxicity of epirubicin (EPI), Zhou et al. synthesized a sialic acid-cholesterol conjugate (SA-Chol) (
Scheme 10) (
14) decorated onto EPI-loaded liposomes surface. Efficacy of
14 was studied in comparison with the sialic acid-octadecylamine conjugate (SA-ODA) [
85].
When injected with 0.125 mmol/kg SA-ODA, mice death occurred within 1 min but no death was observed when the same dosage of
14 was administered to the mice used in the study. The aforementioned findings revealed the toxicity of SA-ODA when compared to
14, making it a more favorable drug delivery system. In vitro release assay was done for the EPI-loaded
14 liposomes (EPI-SAL), EPI solution (EPI-S), conventional liposomal EPI (EPI-CL) and PEGylated liposomal EPI (EPI-PL). No significant EPI leaked from any of the liposomes and this suggested that incorporating
14 in the liposomes delayed EPI release to some extent. All the formulations proved to be stable for three months between 2 and 6 °C with no change in size and entrapment efficiency [
85].
In vitro study on S180 and RAW264.7 cells using EPI-loaded liposomes
14 showed IC
50 values of 1.41 and 0.47 µM (
Table 3). However, EPI-loaded liposomes
14 had a better inhibitory ratio than EPI-CL especially with regard to RAW264.7 cells suggesting that SA moiety modification increased EPI cytotoxicity. In vitro cellular uptake studies revealed that
14 modification increased EPI cellular uptake in RAW264.7 cells when compared to EPI-CL. In RAW264.7 cells, SA solution inhibited EPI-loaded liposomes
14 cellular uptake due to the unavailability of SA receptor on the cells surface. Siglec-1 is overexpressed on the macrophage surface and it interacts with SA on the EPI-loaded compound
14 liposomes surface [
85].
In vivo circulation studies showed that modifying the liposomes with
14 did not change its pharmacokinetic behavior. In vivo uptake of EPI formulations by TAMs showed that delivery of EPI to TAMs was greatly improved by EPI-loaded liposomes
14. Compared with the other formulations, tumor-bearing mice injected with EPI-loaded liposomes
14 lived longer up to 60 days, denoting lower toxicity of EPI-loaded the liposomes. TAMs were identified by CD68 cells and fewer of them were noted when a group of mice was treated with
14, indicating that it delivered EPI into TAMs thus destroying the macrophages. The results revealed that
14 has the potential to effectively deliver f EPI to TAMs [
85].
The potential of RNAi gene silencing technology in biomedical applications has been employed for a wide variety of diseases, especially genetic disorders and cancer [
86]. Small interfering RNAs (siRNAs) can be chemically modified as a way of improving their therapeutic properties [
8]. This makes them not to be easily degraded in the serum, and, thus, they are delivered to the target cells and tissues. The bioavailability and biodistribution of siRNA were discovered to be altered by substances such as antibodies α-tocopherol and cholesterol [
87]. Chernikov et al. investigated gene-silencing activity and carrier-free biodistribution of cholesterol-conjugated nuclease-resistant anti-MDR1 siRNA (Chol-siRNA) (
Figure 5) (
15) in healthy and tumor-bearing mice. Intravenous administration of fluorescent
15 showed that it rapidly spread in the mouse bloodstream within 5 min. Other administration routes such as intramuscular, intrapertoneal and subcutaneous showed slow spreading of fluorescent
15 [
88].
Attaching cholesterol to siRNA significantly reduced its accumulation in the kidneys but at the same time enhanced its accumulation in the liver. Reduced accumulation and slower excretion in the kidneys indicate the ability of siRNA to accumulate in the tumor. It was confirmed by CLSM that the tumor and liver cells had
15 in their cytoplasm [
34]. P-glycoprotein levels were reduced by 60% in human KB-8-5 xenograft that overexpressed the MDR1 gene after five days of injection. The Pglycoprotein level in tumors was successfully reduced by cholesterol-siMDR regardless of how the
15 was administered. These results showed great promise in using compound
15 as a therapeutic agent for reversing drug resistance in tumors [
88].
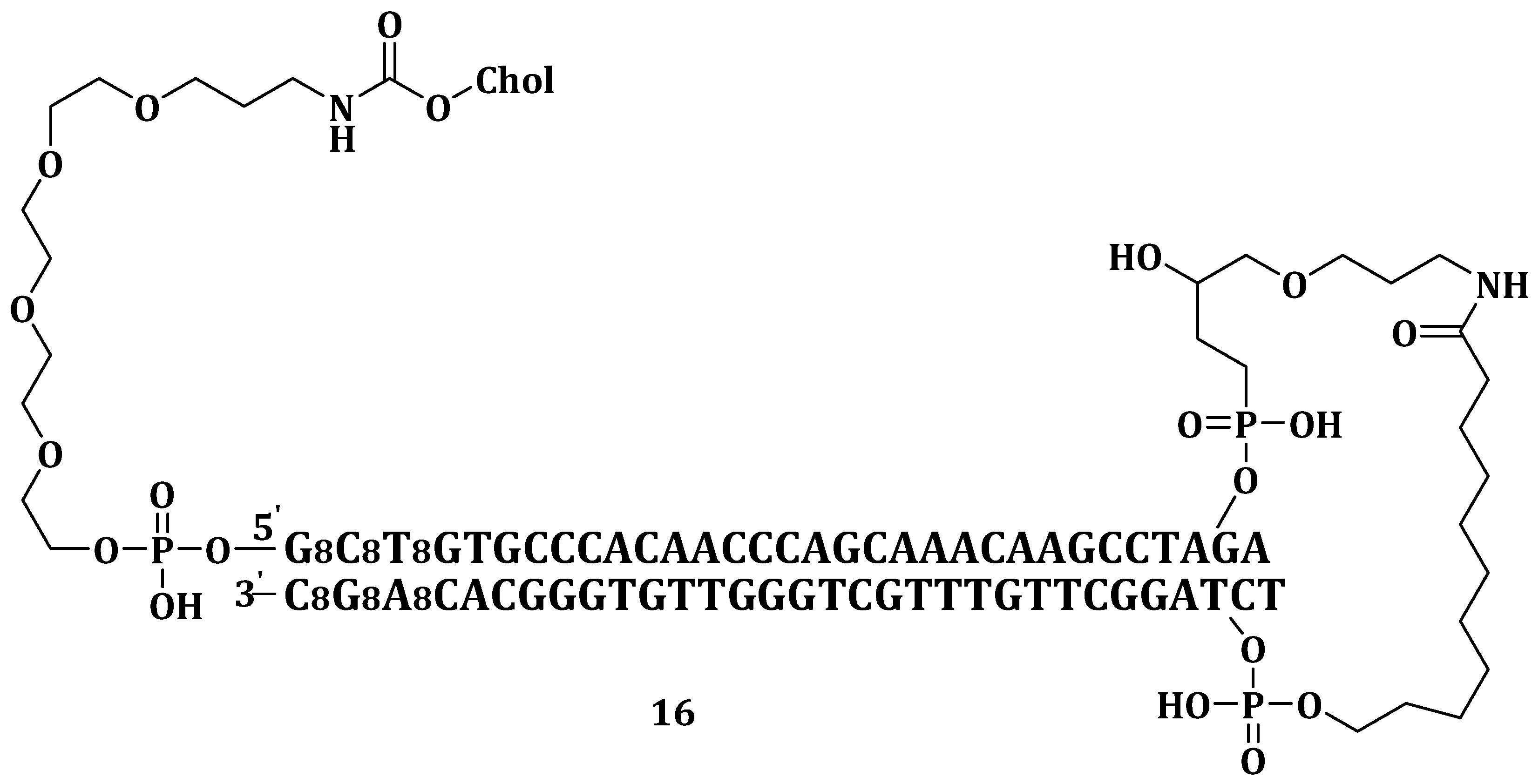
Sun et al. invented and patented an in vivo delivery system containing cholesterol and small DNA molecules (Dbait) (Chol-Dbait) (
Figure 6) (
16) and endosomolytic agents for nucleic acid. The components of the delivery system contribute to its capability to escape the endosome into the cytosol, impair the repair of DNA of damaged chromosomes thereby enhancing cancer treatment. The delivery system can be administered through a variety of routes but most preferably by intratumoral or subcutaneous injection. It required the assistance of a chloroquine as a fusogenic agent (which is administered separately) for its efficient release from the endosomes [
89].
Irrespective of the route of administration,
16, with or without chloroquine, was not toxic to the mice even up to doses of 800 mg/kg/injection. Lowest tumor growth was observed when SK28 xenografted human melanoma was first pre-treated with chloroquine followed by co-treatment with
16 and irradiated chloroquine. The formulation gave the best relative ratio of efficiency dose/toxicity dose of <0.037 when compared to 0.5 for the Dbait/PEI11K formulation. These results revealed
16, a therapeutic agent suitable for clinical trials [
89].
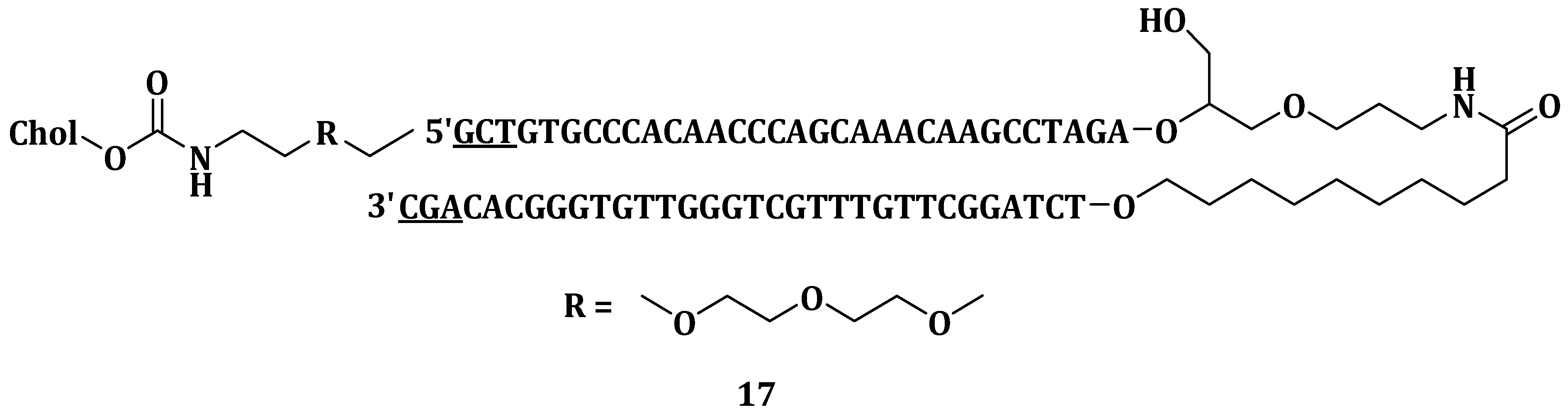
Dutreix et al. invented therapeutic whereby hyperthemic treatment was combined with nucleic acid molecule mimicking double strand breaks (Dbait molecules
17) for the treatment of solid tumors (
Figure 7). The component contained Dbait, cholesterol (to facilitate endocytosis), chloroquine (as the endosomolytic agent) and an antitumoral agent for DNA damaging, most preferably selected from topoisomerases I or II inhibitors. Mice treated with the combination of
17 and hyperthermia exhibited an 80% survival chance compared to 30% survival of those treated which either of the treatment. The combination treatment also decreased tumor size since it caused the highest tumor destruction at degenerative and necrosis stages when compared to treatment with either hyperthermia or
17 alone [
86].




























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
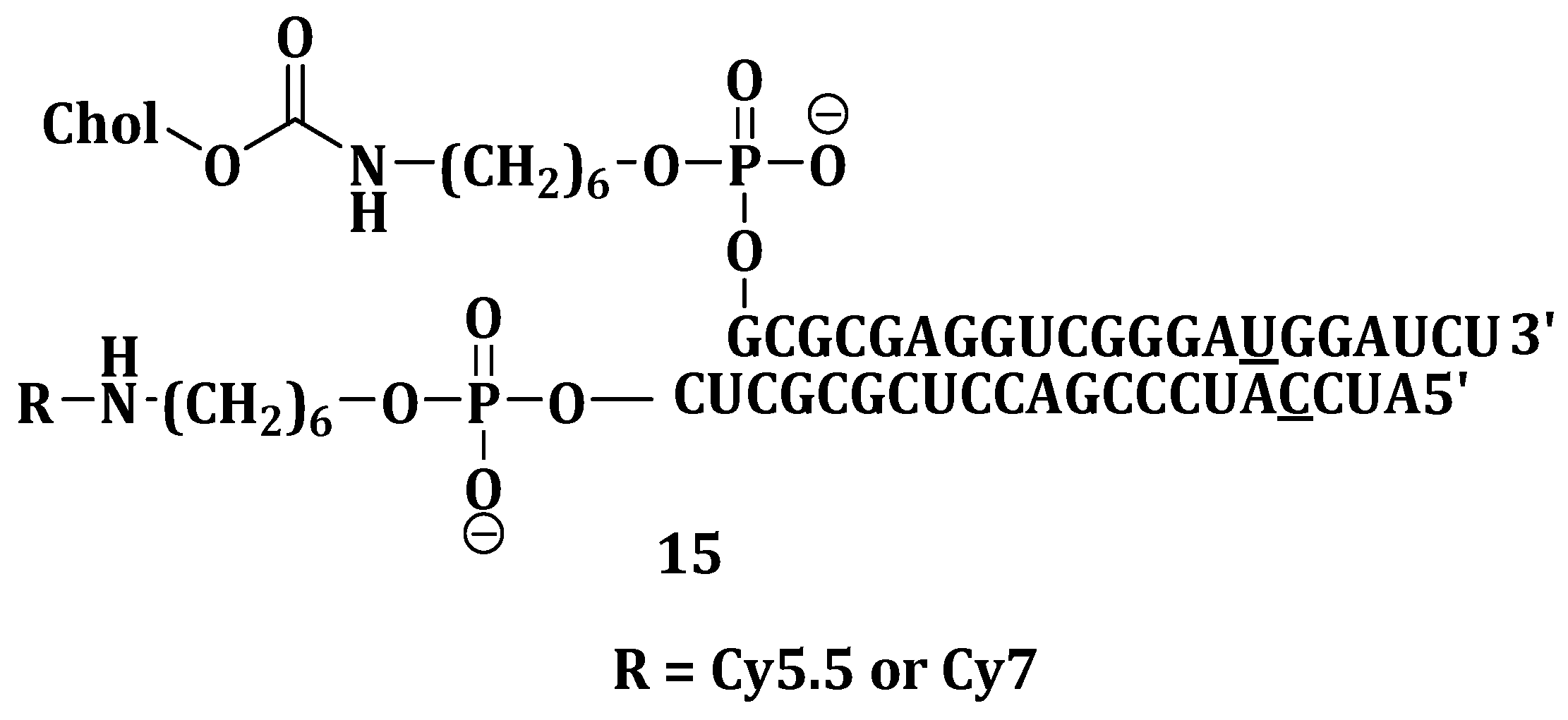{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}