Design, Synthesis, and Structure–Activity Relationships of Thiazole Analogs as Anticholinesterase Agents for Alzheimer’s Disease

 ,
,  , and
, and
Abstract
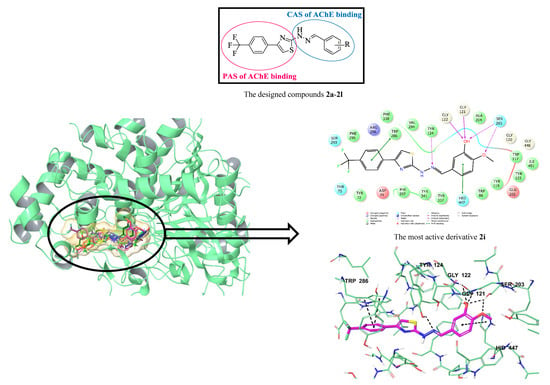
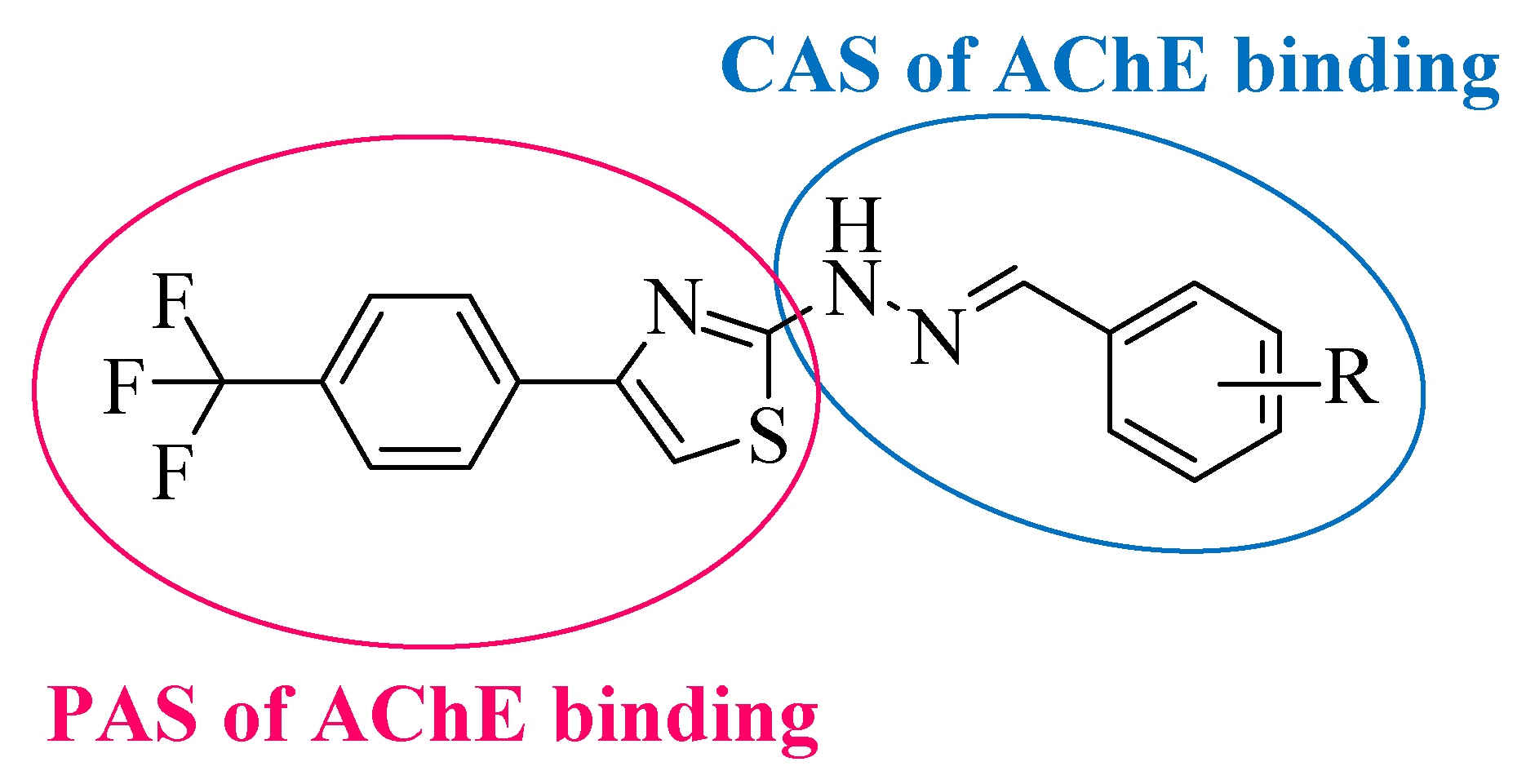
1. Introduction
2. Materials and Methods
2.1. Chemistry
2.1.1. General Procedure for the Synthesis of the Compounds
Synthesis of Substituted Thiosemicarbazones (1a–1l)
General Procedures of Target Compounds (2a–2l)
2.2. Cholinesterase Enzymes Inhibition Assay
2.3. Kinetic Studies of Enzyme Inhibition
2.4. Prediction of ADME Parameters and BBB Permeability
2.5. In Vitro BBB Permeability Assay
2.6. Molecular Docking
3. Results and Discussion
3.1. Chemistry
3.2. Anticholinesterase Enzymes Inhibition Assay
3.3. Kinetic Studies of Enzyme Inhibition
3.4. Prediction of ADME Parameters and BBB Permeability
3.5. In Vitro BBB Permeability Assay
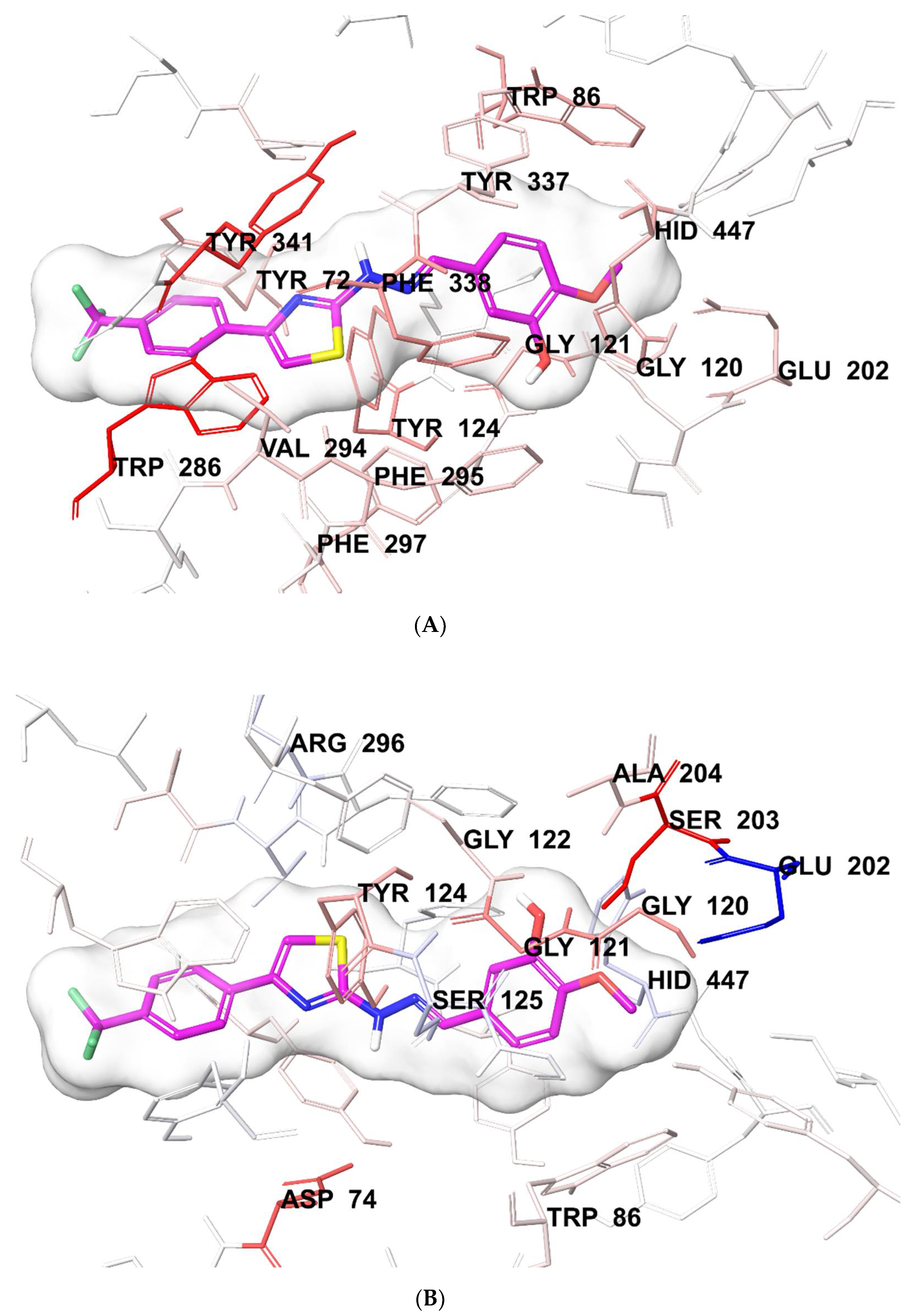
3.6. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kalaria, R.N.; Maestre, G.E.; Arizaga, R.; Friedland, R.P.; Galasko, D.; Hall, K.; Luchsinger, J.A.; Ogunniyi, A.; Perry, E.K.; Potocnik, F.; et al. Alzheimer’s disease and vascular dementia in developing countries: Prevalence, management, and risk factors. Lancet Neurol. 2008, 7, 812–826. [Google Scholar] [CrossRef]
- Rodgers, A.B. Alzheimer’s Disease: Unraveling the Mystery; Publication Number: 02-3782; National Institutes of Health Publications: Bethesda, MD, USA, 2002.
- Huber, A.; Stuchbury, G.; Bürkle, A.; Burnell, J.; Münch, G. Neuroprotective therapies for Alzheimer’s disease. Curr. Pharm. Des. 2006, 12, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.G.; Cappai, R.; Barnham, K.J. The redox chemistry of the Alzheimer’s disease amyloid β peptide. Biomembranes 2007, 768, 1976–1990. [Google Scholar] [CrossRef] [PubMed]
- Craig, L.A.; Hong, N.S.; McDonald, R.J. Revisiting the cholinergic hypothesis in the development of Alzheimer’s disease. Neurosci. Biobehav. Rev. 2011, 35, 1397–1409. [Google Scholar] [CrossRef]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef]
- Su, B.; Wang, X.; Nunomura, A.; Moreira, P.I.; Lee, H.G.; Perry, G.; Smith, M.A.; Zhu, X. Oxidative stress signaling in Alzheimer’s disease. Curr. Alzheimer Res. 2008, 5, 525–532. [Google Scholar] [CrossRef]
- Holmes, C. Systemic inflammation and a lzheimer’s disease. Neuropathol. Appl. Neurobiol. 2013, 39, 51–68. [Google Scholar] [CrossRef]
- Greenough, M.A.; Camakaris, J.; Bush, A.I. Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem. Int. 2013, 62, 540–555. [Google Scholar] [CrossRef]
- Bajda, M.; Więckowska, A.; Hebda, M.; Guzior, N.; Sotriffer, C.A.; Malawska, B. Structure-based search for new inhibitors of cholinesterases. Int. J. Mol. Sci. 2013, 14, 5608–5632. [Google Scholar] [CrossRef]
- Mason, J.M.; Kokkoni, N.; Stott, K.; Doig, A.J. Design strategies for anti-amyloid agents. Curr. Opin. Struct. Biol. 2013, 13, 526–532. [Google Scholar] [CrossRef]
- Shukla, V.; Skuntz, S.; Pant, H.C. Deregulated Cdk5 activity is involved in inducing Alzheimer’s disease. Arch. Med. Res. 2013, 43, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Atri, A.; Frölich, L.; Ballard, C.; Tariot, P.N.; Molinuevo, J.L.; Boneva, N.; Windfeld, K.; Raket, L.L.; Cummings, J.L. Effect of idalopirdine as adjunct to cholinesterase inhibitors on change in cognition in patients with Alzheimer disease: Three randomized clinical trials. JAMA 2018, 319, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, D.K.; Farlow, M.R.; Greig, N.H.; Sambamurti, K. Current drug targets for Alzheimer’s disease treatment. Drug Dev. Res. 2002, 56, 267–281. [Google Scholar] [CrossRef]
- Bajda, M.; Guzior, N.; Ignasik, M.; Malawska, B. Current drug targets for Alzheimer’s disease treatment. Curr. Med. Chem. 2011, 18, 4949–4975. [Google Scholar] [CrossRef]
- Alagille, D.; DaCosta, H.; Baldwin, R.M.; Tamagnan, G.D. 2-Arylimidazo [2, 1-b] benzothiazoles: A new family of amyloid binding agents with potential for PET and SPECT imaging of Alzheimer’s brain. Bioorg. Med. Chem. Lett. 2011, 21, 2966–2968. [Google Scholar] [CrossRef]
- Gan, C.; Zhou, L.; Zhao, Z.; Wang, H. Benzothiazole Schiff-bases as potential imaging agents for β-amyloid plaques in Alzheimer’s disease. Med. Chem. Res. 2013, 22, 4069–4074. [Google Scholar] [CrossRef]
- Xie, Y.; Deng, S.; Chen, Z.; Yan, S.; Landry, D.W. Identification of small-molecule inhibitors of the Aβ–ABAD interaction. Bioorg. Med. Chem. Lett. 2006, 16, 4657–4660. [Google Scholar] [CrossRef]
- Youssef, K.M.; Fawzy, I.M.; El-Subbagh, H.I. N-substituted-piperidines as Novel Anti-alzheimer Agents: Synthesis, antioxidant activity, and molecular docking study. Future J. Pharm. Sci. 2018, 4, 1–7. [Google Scholar] [CrossRef]
- Silva, D.; Chioua, M.; Samadi, A.; Agostinho, P.; Garcão, P.; Lajarín-Cuesta, R.; de los Ríos, C.; Iriepa, I.; Moraleda, I.; Gonzalez-Lafuente, L.; et al. Synthesis, pharmacological assessment, and molecular modeling of acetylcholinesterase/butyrylcholinesterase inhibitors: Effect against amyloid-β-induced neurotoxicity. ACS Chem. Neurosci. 2013, 4, 547–565. [Google Scholar] [CrossRef]
- Lan, J.S.; Zhang, T.; Liu, Y.; Yang, J.; Xie, S.S.; Liu, J.; Miao, Z.Y.; Ding, Y. Design, synthesis and biological activity of novel donepezil derivatives bearing N-benzyl pyridinium moiety as potent and dual binding site acetylcholinesterase inhibitors. Eur. J. Med. Chem. 2017, 133, 184–196. [Google Scholar] [CrossRef]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Matthew, C.F.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.S.; Tu, W.T.; Luo, M.; Shi, J.G. Molecular Docking and Design of Novel Heterodimers of Donepezil and Huperzine Fragments as Acetylcholinesterase Inhibitors. Chin. J. Struct. Chem. 2016, 35, 839–848. [Google Scholar]
- Vitorovic-Todorovic, M.D.; Koukoulitsa, C.; Juranic, I.O.; Mandic, L.M.; Drakulic, B.J. Structural Modifications of 4-Aryl-4-Oxo-2-Aminylbutanamides and Their Acetyl-and Butyrylcholinesterase Inhibitory Activity. Investigation of AChE-Ligand Interactions by Docking Calculations and Molecular Dynamics Simulations. Supplementary data for the article. Eur. J. Med. Chem. 2014, 81, 158–175. [Google Scholar] [CrossRef]
- Neochoritis, C.; Tsoleridis, C.A.; Stephanidou-Stephanatou, J. 1-Arylaminoimidazole-2-thiones as intermediates in the synthesis of imidazo [2,1-b][1,3,4] thiadiazines. Tetrahedron 2008, 64, 3527–3533. [Google Scholar] [CrossRef]
- Ibrar, A.; Khan, A.; Ali, M.; Sarwar, R.; Mehsud, S.; Farooq, U.; Halimi, M.A.S.; Khan, I.; Al-Harrasi, A. Combined in vitro and in silico studies for the anticholinesterase activity and pharmacokinetics of coumarinyl thiazoles and oxadiazoles. Front. Chem. 2018, 6, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, R.K.P.; Sasi, M.V.; Gupta, S.K.; Krishnamurthy, S.; Ayyannan, S.R. Design, synthesis, and pharmacological evaluation of 2-amino-5-nitrothiazole derived semicarbazones as dual inhibitors of monoamine oxidase and cholinesterase: Effect of the size of aryl binding site. J. Enzyme Inhib. Med. Chem. 2018, 33, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.Y.; Yang, Q.; Zhang, Y.G.; Li, J.J.; Yang, G.L. Design, synthesis and evaluation of N-acyl-4-phenylthiazole-2-amines as acetylcholinesterase inhibitors. Acta Pharm. Sin. 2014, 49, 813–818. [Google Scholar]
- Tian, Y.F.; Chen, J.T.; Li, J.J.; Zhang, Y.C.; Cao, T.T.; Ma, Z.Y. Design, synthesis and evaluation of new L-proline derivatives as acetylcholinesterase inhibitors. Acta Pharm. Sin. 2015, 50, 719–724. [Google Scholar]
- Zhang, X.Z.; Xu, Y.; Jian, M.M.; Yang, K.; Ma, Z.Y. Synthesis, in vitro assays, molecular docking, theoretical ADMET prediction, and evaluation of 4-methoxy-phenylthiazole-2-amine derivatives as acetylcholinesterase inhibitors. Med. Chem. Res. 2019, 28, 1683–1693. [Google Scholar] [CrossRef]
- Xu, Y.; Jian, M.M.; Han, C.; Yang, K.; Bai, L.; Cao, F.; Ma, Z.Y. Design, synthesis and evaluation of new 4-arylthiazole-2-amine derivatives as acetylcholinesterase inhibitors. Bioorg. Med. Chem. Lett. 2020, 30, 126985. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Feather-Stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Saglık, B.N.; Ilgın, S.; Ozkay, Y. Synthesis of new donepezil analogues and investigation of their effects on cholinesterase enzymes. Eur. J. Med. Chem. 2016, 124, 1026–1040. [Google Scholar] [CrossRef] [PubMed]
- Demir Özkay, Ü.; Can, Ö.D.; Sağlık, B.N.; Acar Çevik, U.; Levent, S.; Özkay, Y.; Ilgın, S.; Atlı, Ö. Design, synthesis, and AChE inhibitory activity of new benzothiazole-piperazines. Bioorg. Med. Chem. Lett. 2016, 26, 5387–5394. [Google Scholar] [CrossRef] [PubMed]
- Acar Çevik, U.; Levent, S.; Sağlık, B.N.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis of Novel 4-(Dimethylaminoalkyl) piperazine-1-carbodithioa te Derivatives as Cholinesterase Inhibitors. Lett. Drug Des. Discov. 2017, 14, 528–539. [Google Scholar] [CrossRef]
- Levent, S.; Acar Çevik, U.; Sağlık, B.N.; Özkay, Y.; Can, Ö.D.; Demir Özkay, Ü.; Uçucu, Ü. Anticholinesterase activity screening of some novel dithiocarbamate derivatives including piperidine and piperazine moieties. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 469–474. [Google Scholar] [CrossRef]
- Hussein, W.; Sağlık, B.N.; Levent, S.; Korkut, B.; Ilgın, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis and biological evaluation of new cholinesterase inhibitors for Alzheimer’s disease. Molecules 2018, 23, 2033. [Google Scholar] [CrossRef]
- Tok, F.; Koçyiğit Kaymakçıoğlu, B.; Sağlık, B.N.; Levent, S.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis and biological evaluation of new pyrazolone Schiff bases as monoamine oxidase and cholinesterase inhibitors. Bioorg. Chem. 2019, 84, 41–50. [Google Scholar] [CrossRef]
- Acar Çevik, U.; Sağlık, B.N.; Levent, S.; Osmaniye, D.; Kaya Çavuşoğlu, B.; Özkay, Y.; Kaplancıklı, Z.A. Synthesis and AChE-inhibitory activity of new benzimidazole derivatives. Molecules 2019, 24, 861. [Google Scholar] [CrossRef]
- Osmaniye, D.; Sağlık, B.N.; Acar Çevik, U.; Levent, S.; Kaya Çavuşoğlu, B.; Özkay, Y.; Kaplancıklı, Z.A.; Turan-Zitouni, G. Synthesis and AChE inhibitory activity of novel thiazolylhydrazone derivatives. Molecules 2019, 24, 2392. [Google Scholar] [CrossRef]
- QikProp, version 4.8; Schrödinger, LLC.: New York, NY, USA, 2016.
- Yan, G.; Hao, L.; Niu, Y.; Huang, W.; Wang, W.; Xu, F.; Liang, L.; Wang, C.; Jin, H.; Xu, P. 2-Substituted-thio-N-(4-substituted-thiazol/1H-imidazol-2-yl) acetamides as BACE1 inhibitors: Synthesis, biological evaluation and docking studies. Eur. J. Med. Chem. 2017, 137, 462–475. [Google Scholar] [CrossRef]
- Caliandro, R.; Pesaresi, A.; Cariati, L.; Procopio, A.; Oliverio, M.; Lamba, D. Kinetic and structural studies on the interactions of Torpedo californica acetylcholinesterase with two donepezil-like rigid analogues. J. Enzyme Inhib. Med. Chem. 2018, 33, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Henchoz, Y.; Bard, B.; Guillarme, D.; Carrup, P.A.; Veuthey, J.L.; Martel, S. Analytical tools for the physicochemical profiling of drug candidates to predict absorption/distribution. Anal. Bioanal. Chem. 2009, 394, 707–729. [Google Scholar] [CrossRef] [PubMed]
- Kerns, E.H. High throughput physicochemical profiling for drug discovery. J. Pharm. Sci. 2001, 90, 1838–1858. [Google Scholar] [CrossRef] [PubMed]
- Kerns, E.H.; Di, L. Physicochemical profiling: Overview of the screens. Drug Discov. Today Technol. 2004, 1, 343–348. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Franco, L.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from structure. Adv. Drug Deliv. Rev. 2002, 54, 355–366. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Taylor, R. Organic fluorine hardly ever accepts hydrogen bonds. Chem-A Eur. J. Med. Chem. 1997, 3, 89–98. [Google Scholar] [CrossRef]
- Dvira, H.; Silman, I.; Harel, M.; Rosenberry, T.L.; Sussman, J.L. Acetylcholinesterase: From 3D structure to function. Chem. Biol. Interact. 2010, 187, 10–22. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Fan, K.; McConnell, O.J.; Carter, C.T. High throughput artificial membrane permeability assay for blood–brain barrier. Eur. J. Med. Chem. 2003, 38, 223–232. [Google Scholar] [CrossRef]
- Ning, X.; Guo, Y.; Wang, X.; Ma, X.; Tian, C.; Shi, X.; Zhu, R.; Cheng, C.; Du, Y.; Ma, Z.; et al. Design, synthesis, and biological evaluation of (e)-3, 4-dihydroxystyryl aralkyl sulfones and sulfoxides as novel multifunctional neuroprotective agents. J. Med. Chem. 2014, 57, 4302–4312. [Google Scholar] [CrossRef] [PubMed]
- Colovic, M.B.; Krstic, D.; Lazarevic-Pactil, T.; Bondzic, A.M.; Vasic, V. Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr. Neuropharmacol. 2013, 11, 315–335. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, M.; Stavrakov, G.; Philipova, L.; Zheleva, D.; Yordanov, N.; Doytchinova, I. Galantamine derivatives with indole moiety: Docking, design, synthesis and acetylcholinesterase inhibitory activity. Bioorg. Med. Chem. 2015, 23, 5382–5389. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Y.; Esteban, G.; Brogi, S.; Shionoya, M.; Wang, I.; Campiani, G.; Unzeta, M.; Inokuchi, T.; Butini, S.; Marco-Contelles, J. Donepezil-like multifunctional agents: Design, synthesis, molecular modeling and biological evaluation. Eur. J. Med. Chem. 2015, 121, 864–879. [Google Scholar] [CrossRef]
- Genest, D.; Rochais, C.; Lecoutey, C.; Jana Sopkova-de Oliveira, S.; Ballandonne, C.; Butt-Gueulle, S.; Legay, R.; Since, M.; Dallemagne, P. Design, synthesis and biological evaluation of novel indano-and thiaindano-pyrazoles with potential interest for Alzheimer’s disease. Med. Chem. Commun. 2013, 4, 925–931. [Google Scholar] [CrossRef]
- Al-Rashid, Z.F.; Hsung, R.P. A computational view on the significance of E-ring in binding of (+)-arisugacin A to acetylcholinesterase. Bioorg. Med. Chem. 2015, 25, 4848–4853. [Google Scholar] [CrossRef]
- Alipour, M.; Khoobi, M.; Foroumadi, A.; Nadri, H.; Moradi, A.; Sakhteman, A.; Ghandi, M.; Shafiee, A. Novel coumarin derivatives bearing N-benzyl pyridinium moiety: Potent and dual binding site acetylcholinesterase inhibitors. Bioorg. Med. Chem. 2012, 20, 7214–7222. [Google Scholar] [CrossRef]
- Cheung, J.; Gary, E.N. Structures of human acetylcholinesterase bound to dihydrotanshinone I and territrem B show peripheral site flexibility. ACS Med. Chem. Lett. 2013, 4, 1091–1096. [Google Scholar] [CrossRef]
- Chiou, S.Y.; Huang, C.F.; Hwang, M.T.; Lin, G. Comparison of Active Sites of Butyrylcholinesterase and Acetylcholinesterase Based on Inhibition by Geometric Isomers of Benzene-di-N-Substituted Carbamates. J. Biochem. Molecular Toxicology 2009, 23, 303–308. [Google Scholar] [CrossRef]
- Glide; Version 7.1; Schrödinger, LLC.: New York, NY, USA, 2016.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | AChE % Inhibition | AChE IC50 (µM) | BChE % Inhibition | BChE IC50 (µM) | ||
|---|---|---|---|---|---|---|
| 10−3 M | 10−4 M | 10−3 M | 10−4 M | |||
| 2a | 93.425 ± 1.652 | 90.465 ± 1.322 | 0.063 ± 0.003 | 52.380 ± 0.935 | 42.466 ± 0.978 | >100 |
| 2b | 90.285 ± 1.451 | 82.151 ± 1.478 | 0.056 ± 0.002 | 55.488 ± 0.875 | 40.541 ± 0.710 | >100 |
| 2c | 78.215 ± 1.025 | 45.108 ± 0.971 | >100 | 43.612 ± 0.955 | 32.466 ± 0.721 | >1000 |
| 2d | 89.462 ± 2.041 | 82.445 ± 1.695 | 0.147 ± 0.006 | 51.950 ± 0.994 | 43.795 ± 0.895 | >100 |
| 2e | 90.611 ± 1.815 | 85.387 ± 1.730 | 0.040 ± 0.001 | 60.150 ± 1.039 | 41.575 ± 0.907 | >100 |
| 2f | 71.658 ± 1.395 | 40.487 ± 0.890 | >100 | 40.858 ± 0.895 | 31.495 ± 0.820 | >1000 |
| 2g | 93.461 ± 1.632 | 88.347 ± 1.604 | 0.031 ± 0.001 | 63.515 ± 1.155 | 40.388 ± 0.845 | >100 |
| 2h | 77.561 ± 1.758 | 41.947 ± 0.976 | >100 | 45.795 ± 0.926 | 34.306 ± 0.738 | >1000 |
| 2i | 95.207 ± 1.502 | 92.130 ± 1.798 | 0.028 ± 0.001 | 65.015 ± 1.470 | 41.518 ± 0.870 | >100 |
| 2j | 91.326 ± 2.107 | 84.945 ± 1.369 | 0.138 ± 0.005 | 50.811 ± 0.988 | 39.628 ± 0.865 | >100 |
| 2k | 65.298 ± 1.045 | 40.825 ± 0.940 | >100 | 40.171 ± 0.902 | 25.631 ± 0.633 | >1000 |
| 2l | 62.797 ± 1.277 | 35.369 ± 0.895 | >100 | 38.654 ± 0.815 | 23.208 ± 0.759 | >1000 |
| Donepezil | 99.254 ± 2.104 | 97.426 ± 1.890 | 0.021 ± 0.001 | - | - | - |
| Tacrine | - | - | - | 98.255 ± 1.895 | 95.465 ± 1.344 | 0.006 ± 0.0002 |
| Comp. | MW | RB | DM | MV | DHB | AHB | PSA | logP | logS | PCaco | logBB | PMDCK | CNS | PM | %HOA | VRF | VRT |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2a | 363.357 | 5 | 6.352 | 1073.112 | 2 | 4.750 | 57.634 | 4.295 | −6.085 | 1363.075 | −0.292 | 5986.232 | 0 | 2 | 100 | 0 | 1 |
| 2b | 363.357 | 5 | 8.087 | 1073.112 | 2 | 4.750 | 57.634 | 4.295 | −6.085 | 1363.095 | −0.292 | 5986.324 | 0 | 2 | 100 | 0 | 1 |
| 2c | 379.356 | 6 | 6.600 | 1090.479 | 3 | 5.500 | 77.329 | 3.641 | −5.694 | 602.692 | −0.756 | 2477.781 | 1 | 3 | 100 | 0 | 0 |
| 2d | 377.383 | 5 | 7.236 | 1122.809 | 1 | 4.750 | 42.093 | 5.142 | −6.596 | 4525.117 | 0.293 | 10,000 | 1 | 2 | 100 | 1 | 1 |
| 2e | 377.383 | 5 | 6.313 | 1113.128 | 1 | 4.750 | 43.433 | 5.066 | −6.219 | 4525.117 | 0.313 | 10,000 | 1 | 2 | 100 | 1 | 1 |
| 2f | 407.410 | 6 | 5.347 | 1189.498 | 1 | 5.500 | 50.440 | 5.192 | −6.655 | 4525.108 | 0.231 | 10,000 | 1 | 3 | 100 | 1 | 1 |
| 2g | 407.410 | 6 | 7.535 | 1179.309 | 1 | 5.500 | 51.624 | 5.098 | −6.351 | 4525.108 | 0.245 | 10,000 | 1 | 3 | 100 | 1 | 1 |
| 2h | 391.367 | 4 | 7.047 | 1084.671 | 1 | 5.500 | 53.698 | 4.576 | −5.904 | 4526.666 | 0.374 | 10,000 | 1 | 1 | 100 | 0 | 1 |
| 2i | 393.383 | 6 | 7.551 | 1142.737 | 2 | 5.500 | 65.447 | 4.388 | −6.249 | 1454.418 | −0.334 | 6420.985 | 0 | 3 | 100 | 0 | 1 |
| 2j | 393.383 | 6 | 6.825 | 1132.218 | 2 | 5.500 | 64.300 | 4.374 | −5.890 | 1674.494 | −0.220 | 7477.311 | 0 | 3 | 100 | 0 | 1 |
| 2k | 437.436 | 7 | 7.596 | 1265.955 | 1 | 6.250 | 59.089 | 5.273 | −6.619 | 4525.108 | 0.177 | 10,000 | 1 | 4 | 100 | 1 | 1 |
| 2l | 423.409 | 7 | 5.877 | 1207.466 | 2 | 6.250 | 73.261 | 4.414 | −6.177 | 1428.142 | −0.386 | 6295.69 | 0 | 4 | 100 | 0 | 1 |
| Classification | Type of BBB Permeation | Compound | Type of BBB Permeation |
|---|---|---|---|
| CNS+ | High BBB permeation Pe (10−6 cm s−1) > 4.0 | 2i | CNS+ High BBB permeation |
| CNS>− | Low BBB permeation Pe (10−6 cm s−1) < 2.0 | ||
| CNS± | BBB permeation uncertain 2.0 < Pe (10−6 cm s−1) < 4.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sağlık, B.N.; Osmaniye, D.; Acar Çevik, U.; Levent, S.; Kaya Çavuşoğlu, B.; Özkay, Y.; Kaplancıklı, Z.A. Design, Synthesis, and Structure–Activity Relationships of Thiazole Analogs as Anticholinesterase Agents for Alzheimer’s Disease. Molecules 2020, 25, 4312. https://doi.org/10.3390/molecules25184312
Sağlık BN, Osmaniye D, Acar Çevik U, Levent S, Kaya Çavuşoğlu B, Özkay Y, Kaplancıklı ZA. Design, Synthesis, and Structure–Activity Relationships of Thiazole Analogs as Anticholinesterase Agents for Alzheimer’s Disease. Molecules. 2020; 25(18):4312. https://doi.org/10.3390/molecules25184312
Chicago/Turabian StyleSağlık, Begüm Nurpelin, Derya Osmaniye, Ulviye Acar Çevik, Serkan Levent, Betül Kaya Çavuşoğlu, Yusuf Özkay, and Zafer Asım Kaplancıklı. 2020. "Design, Synthesis, and Structure–Activity Relationships of Thiazole Analogs as Anticholinesterase Agents for Alzheimer’s Disease" Molecules 25, no. 18: 4312. https://doi.org/10.3390/molecules25184312
APA StyleSağlık, B. N., Osmaniye, D., Acar Çevik, U., Levent, S., Kaya Çavuşoğlu, B., Özkay, Y., & Kaplancıklı, Z. A. (2020). Design, Synthesis, and Structure–Activity Relationships of Thiazole Analogs as Anticholinesterase Agents for Alzheimer’s Disease. Molecules, 25(18), 4312. https://doi.org/10.3390/molecules25184312