
Novel Acetylcholinesterase Inhibitors Based on Uracil Moiety for Possible Treatment of Alzheimer Disease

 , , ,
, , ,
Abstract
1. Introduction
2. Results and Discussion
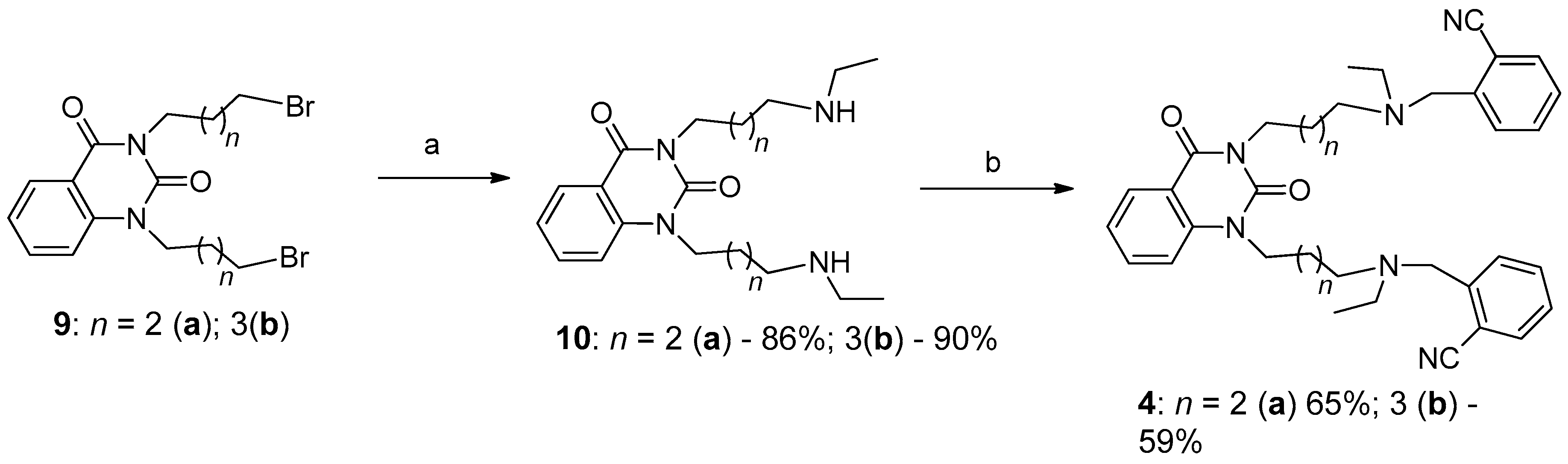
2.1. Preparation of 1,3-Bis[ω-(Substituted Benzylethylamino)Alkyl] Uracils
2.2. The Inhibitory Effects on Cholinesterases and Acute Toxicity of Uracils and Quinazoline-2,4-Diones
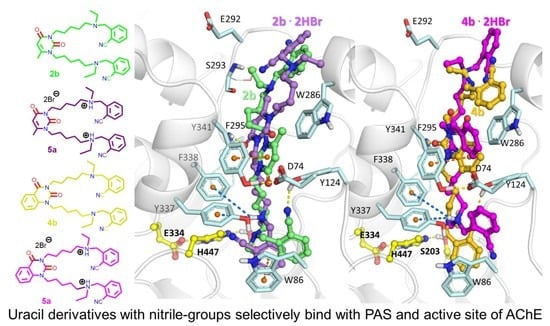
2.3. Molecular Docking Study of Title Compounds
2.4. Behavioral Tests
3. Experimental Section
3.1. Chemistry
3.1.1. General Methods
3.1.2. Initial Compounds for Preparation of Title Cholinesterase Inhibitors
3.1.3. Synthesis of Cholinesterase Inhibitors, 1,3-Bis[ω-(O-Nitrilebenzylethylamino)Alkyl]-6-Methyluracils and Quinazoline-2,4-Diones
3.2. Molecular Modeling
3.3. Biological Studies
3.3.1. In Vitro Cholinesterase Inhibition Assay
3.3.2. Acute Toxicity Evaluation and In Vivo BBB Permeation Assay
3.3.3. Memory Performance Study: Scopolamine Model
3.3.4. Behavioral Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- van Cauwenberghe, C.; van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet. Med. 2015, 18, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.; Santos, M.; Oliveira, C.; Shenk, J.; Nunomura, A.; Smith, M.; Zhu, X.; Perry, G. Alzheimer disease and the role of free radicals in the pathogenesis of the disease. CNS Neurol. Disord. Drug Targets 2008, 7, 3–10. [Google Scholar] [PubMed]
- Finder, V.H. Alzheimer’s Disease: A general introduction and pathomechanism. J. Alzheimer’s Dis. 2010, 22, S5–S19. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G. A century of Alzheimer’s disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Savelieff, M.G.; Lee, S.; Liu, Y.; Lim, M.H. Untangling amyloid-β, tau, and metals in Alzheimer’s disease. ACS Chem. Biol. 2013, 8, 856–865. [Google Scholar] [CrossRef]
- Trippier, P.C.; Labby, K.J.; Hawker, D.D.; Mataka, J.J.; Silverman, R.B. Target-and mechanism-based therapeutics for neurodegenerative diseases: Strength in numbers. J. Med. Chem. 2013, 56, 3121–3147. [Google Scholar] [CrossRef]
- Agis-Torres, A.; Sölhuber, M.; Fernandez, M.; Sanchez-Montero, J.M. Multi-target-directed ligands and other therapeutic strategies in the search of a real solution for Alzheimer’s disease. Curr. Neuropharmacol. 2014, 12, 2–36. [Google Scholar] [CrossRef]
- Petralia, M.C.; Battaglia, G.; Bruno, V.; Pennisi, M.; Mangano, K.; Lombardo, S.D.; Fagone, P.; Cavalli, E.; Saraceno, A.; Nicoletti, F.; et al. The role of macrophage migration inhibitory factor in Alzheimer’s disease: Conventionally pathogenetic or unconventionally protective? Molecules 2020, 25, 291. [Google Scholar] [CrossRef]
- Petralia, M.C.; Mazzon, E.; Fagone, P.; Basile, M.S.; Lenzo, V.; Quattropani, M.C.; Di Nuovo, S.; Bendtzen, K.; Nicoletti, F. The cytokine network in the pathogenesis of major depressive disorder. Close to translation? Autoimmun. Rev. 2020, 19, 102504. [Google Scholar] [CrossRef]
- Scarpini, E.; Schelterns, P.; Feldman, H. Treatment of Alzheimer’s disease: Current status and new perspectives. Lancet Neurol. 2003, 2, 539–547. [Google Scholar] [CrossRef]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Tönnies, E.; Trushina, E. Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef] [PubMed]
- Grossberg, G.T.; Tong, G.; Burke, A.D.; Tariot, P.N. Present algorithms and future treatments for Alzheimer’s disease. J. Alzheimers Dis. 2019, 67, 1157–1171. [Google Scholar] [CrossRef] [PubMed]
- Citron, M. Alzheimer’s disease: Strategies for disease modification. Nat. Rev. Drug Discov. 2010, 9, 387–398. [Google Scholar] [CrossRef]
- Panza, F.; Solfrizzi, V.; Frisardi, V.; Imbimbo, B.P.; Capurso, C.; D’Introno, A.; Colacicco, A.M.; Seripa, D.; Vendemiale, G.; Capurso, A.; et al. Beyond the neurotransmitter-focused approach in treating Alzheimer’s Disease: Drugs targeting β-amyloid and tau protein. Aging Clin. Exp. Res. 2009, 21, 386–406. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Dinamarca, M.C.; Alvarez, A. Amyloid-cholinesterase interactions. Implications for Alzheimer’s disease. FEBS J. 2008, 275, 625–632. [Google Scholar] [CrossRef]
- de Ferrari, G.V.; Canales, M.A.; Shin, I.; Weiner, L.M.; Silman, I.; Inestrosa, N.C. A structural motif of acetylcholinesterase that promotes amyloid β-peptide fibril formation. Biochemistry 2001, 40, 10447–10457. [Google Scholar] [CrossRef]
- Castro, A.; Martinez, A. Peripheral and Dual Binding Site Acetylcholinesterase inhibitors: Implications in treatment of Alzheimers disease. Mini Rev. Med. Chem. 2001, 1, 267–272. [Google Scholar] [CrossRef]
- Castro, A.; Martinez, A. Targeting beta-amyloid pathogenesis through acetylcholinesterase inhibitors. Curr. Pharm. Des. 2006, 12, 4377–4387. [Google Scholar] [CrossRef]
- Wang, C.; Wu, Z.; Cai, H.; Xu, S.; Liu, J.; Jiang, J.; Yao, H.; Wu, X.; Xu, J. Design, synthesis, biological evaluation and docking study of 4-isochromanone hybrids bearing N-benzyl pyridinium moiety as dual binding site acetylcholinesterase inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 5212–5216. [Google Scholar] [CrossRef]
- Gupta, S.; Fallarero, A.; Järvinen, P.; Karlsson, D.; Johnson, M.S.; Vuorela, P.M.; Mohan, C.G. Discovery of dual binding site acetylcholinesterase inhibitors identified by pharmacophore modeling and sequential virtual screening techniques. Bioorg. Med. Chem. Lett. 2011, 21, 1105–1112. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Palomero, E.; Munoz, P.; Usan, P.; Garcia, P.; Delgado, E.; De Austria, C.; Valenzuela, R.; Rubio, L.; Medina, M.; Martinez, A. Potent beta-amyloid modulators. Neurodegener. Dis. 2008, 5, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Viayna, E.; Sola, I.; Bartolini, M.; De Simone, A.; Tapia-Rojas, C.; Serrano, F.G.; Sabaté, R.; Juárez-Jiménez, J.; Pérez, B.; Luque, F.J.; et al. Synthesis and multitarget biological profiling of a novel family of rhein derivatives as disease-modifying anti-Alzheimer agents. J. Med. Chem. 2014, 57, 2549–2567. [Google Scholar] [CrossRef] [PubMed]
- Semenov, V.E.; Zueva, I.V.; Mukhamedyarov, M.A.; Lushchekina, S.V.; Kharlamova, A.D.; Petukhova, E.O.; Mikhailov, A.S.; Podyachev, S.N.; Saifina, L.F.; Petrov, K.A.; et al. 6-Methyluracil derivatives as bifunctional acetylcholinesterase inhibitors for the treatment of Alzheimer’s disease. ChemMedChem. 2015, 10, 1863–1874. [Google Scholar] [CrossRef] [PubMed]
- Zueva, I.; Dias, J.; Lushchekina, S.; Semenov, V.; Mukhamedyarov, M.; Pashirova, T.; Babaev, V.; Nachon, F.; Petrova, N.; Nurullin, L.; et al. New evidence for dual binding site inhibitors of acetylcholinesterase as improved drugs for treatment of Alzheimer’s disease. Neuropharmacology 2019, 155, 131–141. [Google Scholar] [CrossRef]
- Fleming, F.F.; Yao, L.; Ravikumar, P.C.; Funk, L.; Shook, B.C. Nitrile-containing pharmaceuticals: Efficacious roles of the nitrile pharmacophore. J. Med. Chem. 2010, 53, 7902–7917. [Google Scholar] [CrossRef]
- Semenov, V.E.; Voloshina, A.D.; Toroptzova, E.M.; Kulik, N.V.; Zobov, V.V.; Giniyatullin, R.K.; Mikhailov, A.S.; Nikolaev, A.E.; Akamsin, V.D.; Reznik, V.S. Antibacterial and antifungal activity of acyclic and macrocyclic uracil derivatives with quaternized nitrogen atoms in spacers. Eur. J. Med. Chem. 2006, 41, 1093–1101. [Google Scholar] [CrossRef]
- Semenov, V.E.; Giniyatullin, R.K.; Mikhailov, A.S.; Nikolaev, A.E.; Kharlamov, S.V.; Latypov, S.K.; Reznik, V.S. Unusual reaction of macrocyclic uracils with paraformaldehyde. Eur. J. Org. Chem. 2011, 28, 5423–5426. [Google Scholar] [CrossRef]
- Semenov, V.E.; Nikolaev, A.E.; Krylova, E.S.; Sharafutdinova, D.R.; Reznik, V.S. 1,3-Dipolar cycloaddition reactions in the series of N-alkynyl-substituted uracils. Russ. J. Org. Chem. 2012, 48, 582–587. [Google Scholar] [CrossRef]
- Tian, W.Q.; Wang, Y.A. Mechanisms of Staudinger reactions within density functional theory. J. Org. Chem. 2004, 69, 4299–4308. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Feather-Stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Sussman, J.L.; Harel, M.; Frolow, F.; Oefner, C.; Goldman, A.; Toker, L.; Silman, I. Atomic structure of acetylcholinesterase from Torpedo Californica: A prototypic acetylcholine-binding protein. Science 1991, 253, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Inglis, F. The tolerability and safety of cholinesterase inhibitors in the treatment of dementia. Int. J. Clin. Pract. Suppl. 2002, 127, 45–63. [Google Scholar]
- Semenov, V.E.; Galiullina, L.F.; Lodochnikova, O.A.; Kataeva, O.N.; Gubaidullin, A.T.; Chernova, A.V.; Efremov, Y.Y.; Latypov, S.K.; Reznik, V.S. Triuracils-1,3-bis[ω-(N-methyluracil-1-yl)alkyl]thymines and their 5,5’-cyclic counterparts. Eur. J. Org. Chem. 2007, 2007, 4578–4593. [Google Scholar] [CrossRef]
- Nikolaev, A.E.; Semenov, V.E.; Lodochnikova, O.A.; Latypov, S.K.; Reznik, V.S. Synthesis and structure of the pyrimidinophanes with a sulfur atom in the spacer. Mendeleev Commun. 2010, 20, 4–6. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.J.; et al. General atomic and molecular electronic-structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Lushchekina, S.V.; Makhaeva, G.F.; Novichkova, D.A.; Zueva, I.V.; Kovaleva, N.V.; Richardson, R.J. Supercomputer modeling of dual-site acetylcholinesterase (AChE) inhibition. Supercomput. Front. Innov. 2018, 5, 89–97. [Google Scholar]
- Wang, J.-C.; Lin, J.-H.; Chen, C.-M.; Perryman, A.L.; Olso, A.J. Robust scoring functions for protein-ligand interactions with quantum chemical charge models. J. Chem. Inf. Model. 2011, 51, 2528–2537. [Google Scholar] [CrossRef]
- Park, P.; Sung, N.K.; Cho, A.E. Importance of accurate charges in binding affinity calculations: A case of neuraminidase series. Bull. Korean Chem. Soc. 2013, 34, 545–548. [Google Scholar] [CrossRef][Green Version]
- Füsti-Molnár, L.; Merz, K.M., Jr. An efficient and accurate molecular alignment and docking technique using ab initio quality scoring. J. Chem. Phys. 2008, 129, 025102. [Google Scholar] [CrossRef] [PubMed]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef] [PubMed]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [PubMed]
- Masson, P.; Lushchekina, S.; Schopfer, L.M.; Lockridge, O. Effects of viscosity and osmotic stress on the reaction of human butyrylcholinesterase with cresyl saligenin phosphate, a toxicant related to aerotoxic syndrome: Kinetic and molecular dynamics studies. Biochem. J. 2013, 454, 387–399. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Weiss, E.S. An abridged table of pobits for use in the graphic solution of the dosage-effect curve. Am. J. Public Health Nations Health 1948, 38, 22–24. [Google Scholar] [CrossRef]
- Smith, G. Animal models of Alzheimer’s disease: Experimental cholinergic denervation. Brain Res. 1988, 472, 103–118. [Google Scholar] [CrossRef]
- Corcoran, K.A.; Lu, Y.; Turner, R.S.; Maren, S. Overexpression of hAPPswe impairs rewarded alternation and contextual fear conditioning in a transgenic mouse model of Alzheimer’s disease. Learn. Mem. 2002, 9, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Deacon, R.M.J.; Rawlins, J.N.P. T-maze alternation in the rodent. Nat. Protoc. 2006, 1, 7–12. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compound are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | IC50 [nM] | AChE Selectivity b | LD50, mg/kg c | |
|---|---|---|---|---|
| AChE | BChE | |||
| 1ad | 47 ± 3 | 50,000 ± 3000 | 1064 | 281 |
| 1bd | 3.5 ± 0.3 | 35,000 ± 500 | 10,000 | 51 |
| 1cd | 83 ± 10 | 90,000 ± 3000 | 1084 | 93 |
| 1dd | 67 ± 8 | 20,000 ± 150 | 298 | 59 |
| 1ed | 5.6 ± 0.7 | 20,000 ± 2000 | 35,714 | 49 |
| 1fd | 1400 ± 90 | 200,000 ± 2000 | 143 | 58 |
| 2a | 7 ± 0.5 | 65,000 ± 500 | 9285 | 284 |
| 2b | 7.3 ± 0.6 | 100,000 ± 12,000 | 14,286 | 138 |
| 2c | 29 ± 0.3 | 10,000 ± 150 | 345 | 282 |
| 3 | 480 ± 30 | 1800 ± 170 | 3.8 | 146 |
| 13 | 2320 ± 170 | 2160 ± 150 | 0.9 | 10 |
| 4a | 5520 ± 400 | 2620 ± 200 | 0.5 | 107 |
| 4b | 38 ± 3 | 30,000 ± 500 | 789 | 1.1 |
| 2b·2HBr | 1.7 ± 2 | 942 ± 12 | 5541 | 13.1 |
| 2c·2HBr | 24 ± 2 | 10,000 ± 700 | 417 | 11.3 |
| 4b·2HBr | 46 ± 5 | 3920 ± 200 | 85 | 265 |
| Donepezil | 48 ± 3 | 7900 ± 600 | 165 | 5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Semenov, V.E.; Zueva, I.V.; Mukhamedyarov, M.A.; Lushchekina, S.V.; Petukhova, E.O.; Gubaidullina, L.M.; Krylova, E.S.; Saifina, L.F.; Lenina, O.A.; Petrov, K.A. Novel Acetylcholinesterase Inhibitors Based on Uracil Moiety for Possible Treatment of Alzheimer Disease. Molecules 2020, 25, 4191. https://doi.org/10.3390/molecules25184191
Semenov VE, Zueva IV, Mukhamedyarov MA, Lushchekina SV, Petukhova EO, Gubaidullina LM, Krylova ES, Saifina LF, Lenina OA, Petrov KA. Novel Acetylcholinesterase Inhibitors Based on Uracil Moiety for Possible Treatment of Alzheimer Disease. Molecules. 2020; 25(18):4191. https://doi.org/10.3390/molecules25184191
Chicago/Turabian StyleSemenov, Vyacheslav E., Irina V. Zueva, Marat A. Mukhamedyarov, Sofya V. Lushchekina, Elena O. Petukhova, Lilya M. Gubaidullina, Evgeniya S. Krylova, Lilya F. Saifina, Oksana A. Lenina, and Konstantin A. Petrov. 2020. "Novel Acetylcholinesterase Inhibitors Based on Uracil Moiety for Possible Treatment of Alzheimer Disease" Molecules 25, no. 18: 4191. https://doi.org/10.3390/molecules25184191
APA StyleSemenov, V. E., Zueva, I. V., Mukhamedyarov, M. A., Lushchekina, S. V., Petukhova, E. O., Gubaidullina, L. M., Krylova, E. S., Saifina, L. F., Lenina, O. A., & Petrov, K. A. (2020). Novel Acetylcholinesterase Inhibitors Based on Uracil Moiety for Possible Treatment of Alzheimer Disease. Molecules, 25(18), 4191. https://doi.org/10.3390/molecules25184191