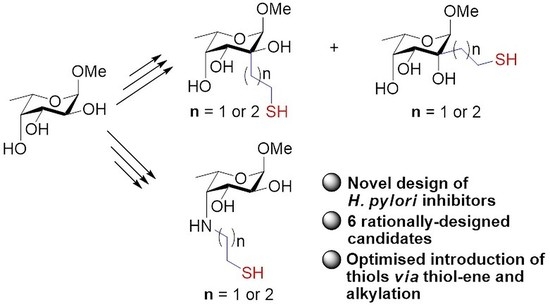
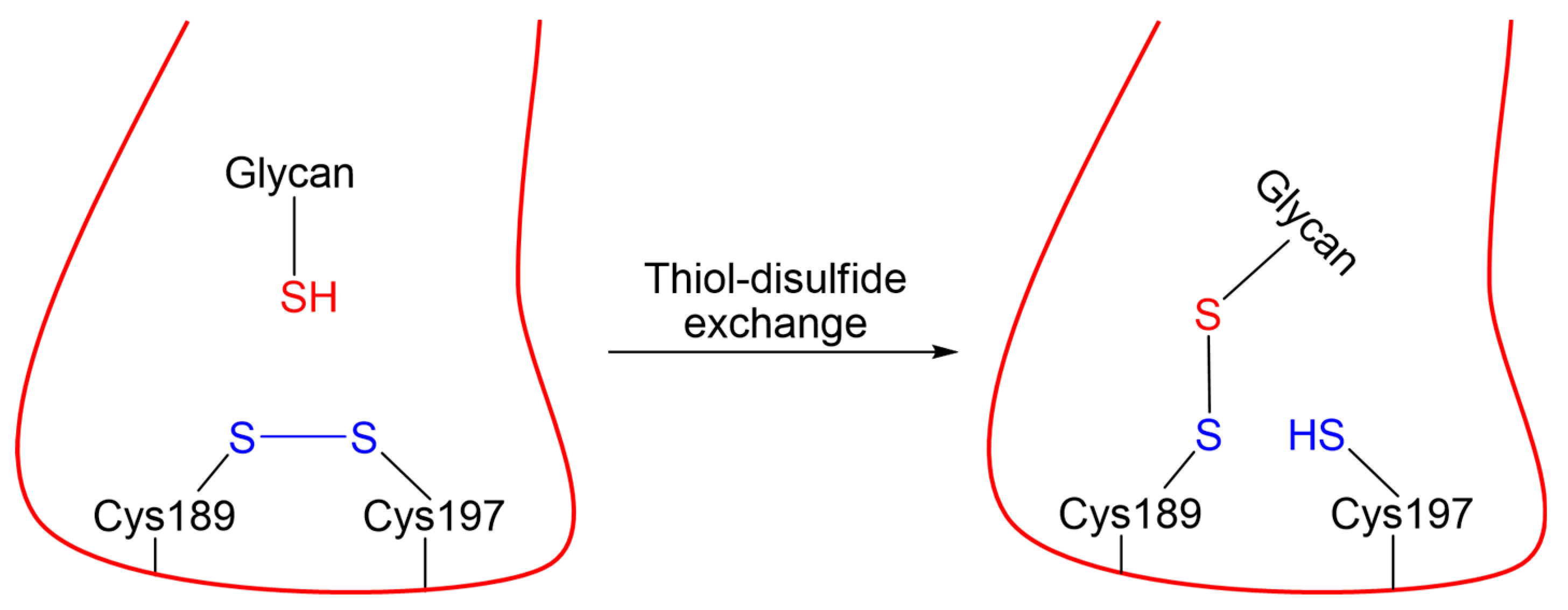
3. Materials and Methods
Reactions were monitored by thin-layer chromatography (TLC) on Merck DC-Alufolien plates precoated with silica gel 60 F254 in the eluents states in parentheses (
v/v). Visualisation was performed with UV-light (254 nm) and/or staining with 8% H2SO4/EtOH solution. All chemicals were purchased from commercial suppliers [Carbosynth Ltd. (Compton, Berkshire, UK), Fisher Scientific Ltd. (Blanchardstown, Co. Dublin, Ireland), Glycom A/S (Hørsholm, Denmark), Merck (Carrigtwohill, Co. Cork, Ireland), Sigma-Aldrich (Arklow, Co. Wicklow, Ireland), VWR (Blanchardstown, Co. Dublin, Ireland) and were used without purification. Dry solvents were obtained from a PureSolv-ENTM solvent purification system (Innovative Technology Inc., Hong Kong) or were used as purchased from Sigma-Aldrich (Arklow, Co. Wicklow, Ireland) in AcroSeal
® bottles. NMR spectra were recorded on Varian Inova spectrometers (Varian, Ltd., Palo Alto, CA, USA) at 25 °C. High-resolution mass spectrometry (HRMS) data were recorded on a Waters Micromass LCT LC-TOF instrument using electrospray ionisation (ESI) in positive mode. Specific rotations were recorded (Model 343) at the sodium D-line (589 nm) at 20 °C in a 1 dm cell at the stated concentration (
c 1.0 = 10 mg/mL) on a Perkin-Elmer polarimeter (PerkinElmer Ltd., Waltham, MA, USA), except for compound
17 which was recorded on a Schmidt-Haensch UniPol L 2000 polarimeter. Deprotected sugars were lyophilised using a Christ Alpha 1-2 LDplus (SciQuip Ltd., Shropshire, UK) freeze-dryer: pressure: 0.035 mbar; ice-condenser temperature: −55 °C. Each proton and carbon belonging to the monosaccharide ring systems was numbered according to conventional guidelines [
37].
3.1. General Procedure A (Preparation of Compounds 12–15)
Thioacetic acid (20 eq.) and azobisisobutyronitrile (1 eq.) were added to a solution of the alkene-bearing sugar (1 eq.) in dry, de-gassed 1,4-dioxane (0.5 mL) under N2. The reaction mixture was refluxed at 75 °C and after 3 h, the volatiles were removed in vacuo. Flash chromatography on silica gel yielded the desired product.
3.2. General Procedure B (Preparation of Compounds 1–4)
A freshly prepared solution of dry, de-gassed 0.4 M NaOMe/MeOH was added to a solution of the thioacetate-containing sugar in dry, de-gassed MeOH (1 mL) under N2 until pH 13 was reached. The mixture was stirred at room temperature for 5 min. The solution was then neutralised with Dowex® (H+) ion-exchange resin and the resin was filtered off and washed with MeOH. The filtrate was concentrated in vacuo to furnish the product.
3.3. Methyl 2-C-vinyl-α-l-fucopyranoside (8) and Methyl 6-deoxy-2-C-vinyl-α-l-talopyranoside (10)
Four Å molecular sieves (150 mg), 4-methylmorpholine N-oxide (180 mg, 1.53 mmol), and tetrapropylammonium perruthenate (27 mg, 77 µmol) were added sequentially to a solution of compound 7 (180 mg, 0.825 mmol) in dry CH2Cl2 (7.5 mL). The resulting mixture was stirred at room temperature. After 2 h, TLC analysis (CH2Cl2/acetone, 9:1) showed the disappearance of the starting material and the formation of a less polar spot. The mixture was filtered through a Celite®-silica-Celite® triple pad, and the filter was washed with CH2Cl2 (10 mL) and EtOAc (15 mL). The filtrate was concentrated in vacuo to afford a white solid which was used without further purification.
The crude was dissolved in dry toluene (10 mL) and the solution was cooled to 0 °C. 1 M vinylmagnesium bromide/THF (2.5 mL, 2.5 mmol) was then added dropwise. After 10 min, EtOH (1 mL) and sat. aq. NaHCO3 solution (10 mL) were added. The resulting mixture was extracted with EtOAc (2 × 20 mL), and the combined organic phase was dried over MgSO4. The solids were filtered off and the filtrate was concentrated in vacuo.
The crude residue was dissolved in MeOH (15 mL) and Dowex® (H+) acidic ion-exchange resin (400 mg) was added. The mixture was stirred at room temperature and after 16 h, the solids were filtered off and the filtrate was concentrated in vacuo. The residue was purified by flash column chromatography on silica gel (toluene/EtOAc, 3:7→EtOAc) to afford 8 (23 mg, 14%) and 10 (85 mg, 50%) over 3 steps as white amorphous solids.
(8): Rf = 0.4 (EtOAc); [α]
−85 (c 0.5, CHCl3); 1H NMR (500 MHz, CDCl3) δ 6.29 (dd, J = 17.4, 11.0 Hz, 1H, CH2=CH), 5.53 (dd, J = 17.5, 1.9 Hz, 1H, CH2(A)=CH), 5.32 (dd, J = 11.1, 1.9 Hz, 1H, CH2(B)=CH), 4.48 (s, 1H, H-1), 3.99 (qd, J = 6.6, 1.8 Hz, 1H, H-5), 3.85 (br s, 1H, H-3 or H-4), 3.81 (br s, 1H, H-3 or H-4), 3.43 (s, 3H, OCH3), 2.65 (s, 1H, OH), 2.57 (d, J = 4.6 Hz, 1H, OH), 2.30 (d, J = 3.3 Hz, 1H, OH), 1.32 (d, J = 6.6 Hz, 3H, H-6); 13C NMR (126 MHz, CDCl3) δ 136.6 (CH2=CH), 116.8 (CH2=CH), 102.7 (C-1), 74.5 (C-2), 73.3 (C-3 or C-4), 71.4 (C-3 or C-4), 65.6 (C-5), 55.7 (OCH3), 16.1 (C-6); HRMS (ESI) m/z calculated for C9H16O5 [M + Na]+ 227.0895, found 227.0905.
(10): Rf = 0.5 (EtOAc); [α]−150 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.91 (dd, J = 17.4, 10.9 Hz, 1H, CH2=CH), 5.48 (dd, J = 17.4, 1.6 Hz, 1H, CH2(A)=CH), 5.31 (dd, J = 10.9, 1.6 Hz, 1H, CH2(B)=CH), 4.39 (s, 1H, H-1), 4.25 (s, 1H, OH), 3.97 (q, J = 6.6 Hz, 1H, H-5), 3.90 (d, J = 6.3 Hz, 1H, OH), 3.76-3.71 (m, 2H, H-3, H-4), 3.48 (m, 1H, OH), 3.34 (s, 3H, OCH3), 1.32 (d, J = 6.6 Hz, 3H, H-6); 13C NMR (126 MHz, CDCl3) δ 137.8 (CH2=CH), 116.7 (CH2=CH), 104.4 (C-1), 75.5 (C-2), 72.1, 68.8 (C-3, C-4), 66.0 (C-5), 55.4 (OCH3), 16.5 (C-6); HRMS (ESI) m/z calculated for C9H16O5 [M + Na]+ 227.0895, found 227.0894.
3.4. Methyl 2-C-allyl-α-l-fucopyranoside (9) and Methyl 2-C-allyl-6-deoxy-α-l-talopyranoside (11)
Four Å molecular sieves (150 mg), 4-methylmorpholine N-oxide (252 mg, 2.15 mmol), and tetrapropylammonium perruthenate (38 mg, 0.11 mmol) were added sequentially to a solution of 7 (252 mg, 1.15 mmol) in dry CH2Cl2 (10 mL). The resulting mixture was stirred at room temperature. After 1.5 h, TLC analysis (CH2Cl2/acetone, 9:1) showed the disappearance of the starting material and the formation of a less polar spot. The mixture was filtered through a Celite®-silica-Celite® triple pad, and the filter was washed with CH2Cl2 (10 mL) and EtOAc (15 mL). The filtrate was concentrated in vacuo to afford a white solid which was used without further purification.
The crude was dissolved in dry toluene (10 mL) and the solution was cooled to 0 °C before adding 1 M allylmagnesium bromide/Et2O (3.5 mL, 3.5 mmol) dropwise. After 10 min, EtOH (1 mL) and sat. aq. NaHCO3 solution (10 mL) were added. The resulting mixture was extracted with EtOAc (2 × 20 mL), and the organic phase was dried over MgSO4. The solids were filtered off and the filtrate was concentrated in vacuo.
The crude residue was dissolved in MeOH (15 mL) and Dowex® (H+) acidic ion-exchange resin (600 mg) was added. The mixture was stirred at room temperature and after 48 h, the solids were filtered off and the filtrate was concentrated in vacuo. The residue was purified by flash chromatography on silica gel (toluene/EtOAc, 1:1→EtOAc) to afford 9 (86 mg, 34%) and 11 (54 mg, 21%) over 3 steps as white amorphous solids.
(9): Rf = 0.3 (EtOAc); [α]−145 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.93 (m, 1H, CH2=CH), 5.25–5.02 (m, 2H, CH2=CH), 4.56 (s, 1H, H-1), 3.93 (q, J = 6.5 Hz, 1H, H-5), 3.88 (d, J = 3.5 Hz, 1H, H-3 or H-4), 3.81 (br s, 1H, H-3 or H-4), 3.61–3.47 (m, 1H, OH), 3.39 (s, 3H, OCH3), 2.86–2.58 (m, 4H, CH-CH2, 2 × OH), 1.30 (d, J = 6.5 Hz, 3H, H-6); 13C NMR (151 MHz, CDCl3) δ 133.9 (CH2=CH), 118.7 (CH2=CH), 101.1 (C-1), 73.45 (C-2), 73.41 (C-3 or C-4), 71.4 (C-3 or C-4), 65.6 (C-5), 55.6 (OCH3), 36.1 (CH-CH2), 16.0 (C-6); HRMS (ESI) m/z calculated for C10H18O5 [M + Na]+ 241.1052, found 241.1064.
(11): Rf = 0.4 (EtOAc); [α]−66 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.87 (m, 1H, CH2=CH), 5.18 (s, 1H, CH2(A)=CH), 5.15 (m, 1H, CH2(B)=CH), 4.45 (s, 1H, H-1), 3.92 (q, J = 6.6 Hz, 1H, H-5), 3.68–3.52 (m, 4H, H-3, H-4, 2 × OH), 3.35 (s, 3H, OCH3), 2.60 (m, 1H, CH-CH2(A)), 2.33 (m, 1H, CH-CH2(B)), 1.30 (d, J = 6.6 Hz, 1H, H-6); 13C NMR (151 MHz, CDCl3) δ 132.6 (CH2=CH), 119.4 (CH2=CH), 102.9 (C-1), 74.6 (C-2), 72.8 (C-3 or C-4), 69.7 (C-3 or C-4), 65.8 (C-5), 55.3 (OCH3), 38.8 (CH-CH2), 16.5 (C-6); HRMS (ESI) m/z calculated for C10H18O5 [M + Na]+ 241.1052, found 241.1054.
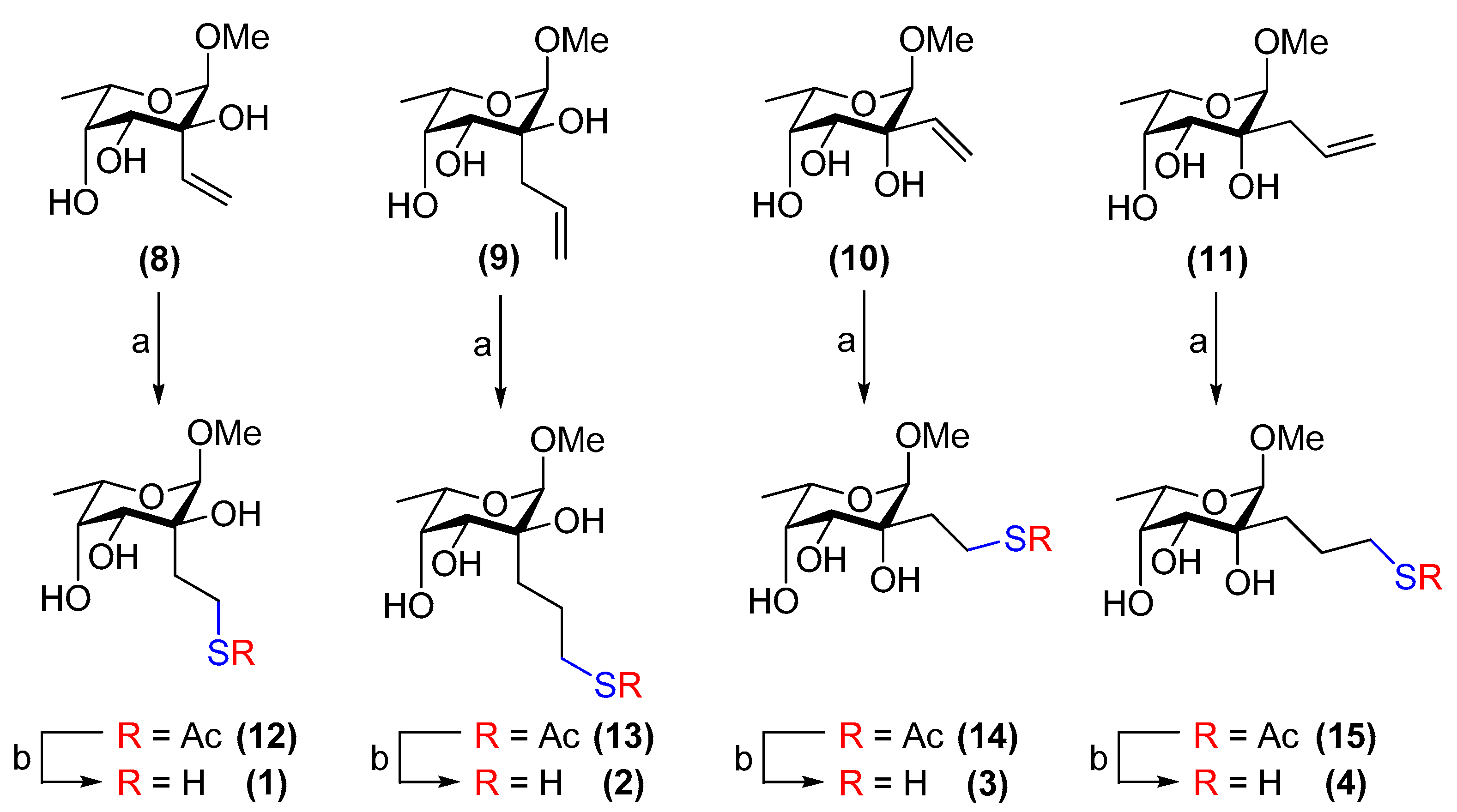
3.5. Methyl 2-C-[2-acetylthioethyl]-α-l-fucopyranoside (12)
Compound 8 (14 mg, 69 µmol) was subjected to General Procedure A and purified by flash chromatography on silica gel (toluene/EtOAc, 3:2→EtOAc) to afford 12 (14 mg, 73%) as a white amorphous solid. Rf = 0.6 (EtOAc); [α]−59 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 4.61 (s, 1H, H-1), 3.93 (qd, J = 6.6, 1.9 Hz, 1H, H-5), 3.88 (br s, 1H, H-3), 3.82 (br s, 1H, H-4), 3.42 (s, 3H, OCH3), 3.13 (ddd, J = 13.3, 11.1, 5.3 Hz, 1H, CH2(A)SAc), 3.02 (d, J = 3.8 Hz, 1H, OH), 2.90 (ddd, J = 13.3, 11.3, 5.4 Hz, 1H, CH2(B)SAc), 2.63 (d, J = 1.4 Hz, 1H, OH), 2.44 (m, 1H, OH), 2.32 (s, 3H, CH3(SAc)), 2.20 (ddd, J = 14.5, 11.4, 5.3 Hz, 1H, C(sugar)-CH2(A)), 2.06 (m, 1H, C(sugar)-CH2(B)), 1.30 (d, J = 6.7 Hz, 3H, H-6); 13C NMR (126 MHz, CDCl3) δ 196.7 (C=O), 101.2 (C-1), 73.6 (C-3), 73.4 (C-2), 71.3 (C-4), 65.8 (C-5), 55.8 (OCH3), 31.9 (CH2-SAc), 30.7 (CH3(SAc)), 23.9 (C(sugar)-CH2), 15.9 (C-6); HRMS (ESI) m/z calculated for C11H20OsS [M + Na]+ 303.0878, found 303.0878.
3.6. Methyl 2-C-[3-acetylthiopropyl]-α-l-fucopyranoside (13)
Compound 9 (67 mg, 0.31 mmol) was subjected to General Procedure A and purified by flash chromatography on silica gel (toluene/EtOAc, 1:1→EtOAc) to afford 13 (76 mg, 84%) as a white amorphous solid. Rf = 0.5 (EtOAc); [α]#x2212;74 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 4.55 (s, 1H, H-1), 3.93 (m, 1H, H-5), 3.85 (t, J = 3.9 Hz, 1H, H-3), 3.81 (br s, 1H, H-4), 3.56 (d, J = 4.1 Hz, 1H, OH), 3.40 (s, 3H, OCH3), 2.87 (m, 2H, CH2-SAc), 2.82 (m, 1H, OH), 2.72 (d, J = 1.3 Hz, 1H, OH), 2.32 (s, 3H, CH3(SAc)), 1.99–1.87 (m, 2H, -CH2-), 1.79 (m, 1H, C(sugar)-CH2(A)), 1.61 (m, 1H, C(sugar)-CH2(B)), 1.29 (d, J = 6.7 Hz, 3H, H-6); 13C NMR (126 MHz, CDCl3) δ 196.4 (C=O), 100.9 (C-1), 73.5 (C-2), 73.5 (C-3), 71.4 (C-4), 65.6 (C-5), 55.6 (OCH3), 30.7 (CH3(SAc)), 30.2 (-CH2-), 29.8 (CH2-SAc), 23.4 (C(sugar)-CH2), 15.9 (C-6); HRMS (ESI) m/z calculated for C12H22O6S [M + Na]+ 317.1035, found 317.1027.
3.7. Methyl 2-C-[2-acetylthioethyl]-6-deoxy-α-l-talopyranoside (14)
Compound 10 (50 mg, 0.25 mmol) was subjected to General Procedure A and purified by flash chromatography on silica gel (toluene/EtOAc, 3:2→EtOAc) to afford 14 (59 mg, 85%) as a white amorphous solid. Rf = 0.6 (EtOAc); [α]−72 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 4.57 (s, 1H, H-1), 4.14 (s, 1H, OH), 3.95–3.82 (m, 2H, H-5, OH), 3.74 (d, J = 9.2 Hz, 1H, OH), 3.67 (dd, J = 6.9, 3.0 Hz, 1H, H-4), 3.55 (dd, J = 9.3, 3.0 Hz, 1H, H-3), 3.35 (s, 3H, OCH3), 3.04 (ddd, J = 13.3, 11.4, 4.7 Hz, 1H, CH2(A)-SAc), 2.85 (ddd, J = 13.3, 11.2, 5.7 Hz, 1H, CH2(B)-SAc), 2.33 (s, 3H, CH3(SAc)), 2.05 (ddd, J = 14.1, 11.2, 4.7 Hz, 1H, C(sugar)-CH2(A)), 1.83 (ddd, J = 14.1, 11.2, 5.7 Hz, 1H, C(sugar)-CH2(B)), 1.30 (d, J = 6.6 Hz, 3H, H-6); 13C NMR (126 MHz, CDCl3) δ 196.9 (C=O), 102.6 (C-1), 74.6 (C-2), 72.7 (C-4), 69.7 (C-3), 65.7 (C-5), 55.3 (OCH3), 34.6 (CH2-SAc), 30.7 (CH3(SAc)), 23.0 (C(sugar)-CH2), 16.5 (C-6); HRMS (ESI) m/z calculated for C11H20OsS [M + Na]+ 303.0878, found 303.0872.
3.8. Methyl 2-C-[3-acetylthiopropyl]-6-deoxy-α-l-talopyranoside (15)
Compound 11 (38 mg, 0.17 mmol) was subjected to General Procedure A and purified by flash chromatography on silica gel (toluene/EtOAc, 3:2→EtOAc) to afford 15 (43 mg, 84%) as a white amorphous solid. Rf = 0.4 (toluene/EtOAc, 1:4); [α]−67 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 4.50 (s, 1H, H-1), 3.92–3.86 (m, 2H, H-5, OH), 3.79 (d, J = 7.1 Hz, 1H, OH), 3.65 (m, 1H, H-3 or H-4), 3.59 (d, J = 8.6 Hz, 1H, OH), 3.52 (br s, 1H, H-3 or H-4), 3.35 (s, 3H, OCH3), 2.90–2.82 (m, 2H, CH2-SAc), 2.33 (s, 3H, CH3(SAc)), 1.85 (ddd, J = 14.5, 7.0, 4.0 Hz, 1H, -CH2(A)-), 1.75 (m, 1H, C(sugar)-CH2(A)), 1.65–1.48 (m, 2H, -CH2(B)-, C(sugar)-CH2(B)), 1.29 (d, J = 6.6 Hz, 3H, H-6); 13C NMR (126 MHz, CDCl3) δ 196.4 (C=O), 102.5 (C-1), 74.5 (C-2), 72.7 (C-3 or C-4), 70.0 (C-3 or C-4), 65.6 (C-5), 55.3 (OCH3), 33.3 (-CH2-), 30.8 (CH3(SAc)), 29.7 (CH2-SAc), 22.4 (C(sugar)-CH2), 16.5 (C-6); HRMS (ESI) m/z calculated for C12H22O6S [M + Na]+ 317.1035, found 317.1024.
3.9. Methyl 2-C-[2-thioethyl]-α-l-fucopyranoside (1)
Compound 12 was subjected to General Procedure B (14 mg, 50 µmol) to afford 1 (11 mg, 92%) as a colourless, amorphous solid. Rf = 0.5 (CH2Cl2/MeOH, 19:1); [α]−78 (c 0.9, CH3OH); 1H NMR (400 MHz, CD3OD) δ 4.52 (s, 1H, H-1), 3.95 (m, 1H, H-5), 3.82 (d, J = 3.9 Hz, 1H, H-4), 3.70 (dd, J = 3.9, 1.6 Hz, 1H, H-3), 3.40 (s, 3H, OCH3), 2.73 (m, 1H, CH2(A)-SH), 2.59 (m, 1H, CH2(B)-SH), 2.25 (ddd, J = 14.5, 11.8, 5.3 Hz, 1H, C(sugar)-CH2(A)), 2.15 (ddd, J = 14.5, 11.8, 5.3 Hz, 1H, C(sugar)-CH2(B)), 1.25 (d, J = 6.6 Hz, 3H, H-6); 13C NMR (Taken from HSQC) δ 103.3 (C-1), 74.3 (C-4), 74.1 (C-3), 67.7 (C-5), 56.3 (OCH3), 39.9 (C(sugar)-CH2), 20.1 (CH2-SH), 16.8 (C-6); HRMS (ESI) m/z calculated for C9H18OsS [M + Na]+ 261.0773, found 261.0767.
3.10. Methyl 2-C-[3-thiopropyl]-α-l-fucopyranoside (2)
Compound 13 (33 mg, 0.11 mmol) was subjected to General Procedure B to afford 2 (26 mg, 92%) as a colourless, amorphous solid. Rf = 0.4 (CH2Cl2/MeOH, 19:1); [α]−78 (c 0.9, CH3OH); 1H NMR (400 MHz, CD3OD) δ 4.53 (s, 1H, H-1), 3.97 (m, 1H, H-5), 3.83 (d, J = 4.0 Hz, 1H, H-4), 3.72 (dd, J = 4.0, 1.9 Hz, 1H, H-3), 3.41 (s, 3H, OCH3), 2.52 (t, J = 7.3 Hz, 2H, CH2-SH), 2.00–1.90 (m, 2H, -CH2-), 1.83 (m, 1H, C(sugar)-CH2(A)), 1.65 (m, 1H, C(sugar)-CH2(B)), 1.25 (d, J = 6.7 Hz, 3H, H-6); 13C NMR (Taken from HSQC) δ 103.0 (C-1), 74.2 (C-4), 73.4 (C-3), 67.4 (C-5), 56.1 (OCH3), 32.1 (-CH2-), 29.3 (C(sugar)-CH2), 26.3 (CH2-SH), 16.6 (C-6); HRMS (ESI) m/z calculated for C10H20O5S [M + Na]+ 275.0929, found 275.0919.
3.11. Methyl 6-deoxy-2-C-[2-thioethyl]-α-l-talopyranoside (3)
Compound 14 (10 mg, 36 µmol) was subjected to General Procedure B to afford 3 (8 mg, 92%) as a colourless, amorphous solid. Rf = 0.5 (CH2Cl2/MeOH, 19:1); [α]−57 (c 0.9, CH3OH); 1H NMR (500 MHz, CD3OD) δ 4.48 (s, 1H, H-1), 3.89 (m, 1H, H-5), 3.61 (d, J = 2.5 Hz, 1H, H-4), 3.50 (d, J = 3.3 Hz, 1H, H-3), 3.37 (s, 3H, OCH3), 2.69 (td, J = 12.3, 4.4 Hz, 1H, CH2(A)-SH), 2.50 (td, J = 12.3, 5.3 Hz, 1H, CH2(B)-SH), 2.12 (ddd, J = 13.8, 12.3, 4.4 Hz, 1H, C(sugar)-CH2(A)), 1.82 (ddd, J = 13.8, 12.3, 5.2 Hz, 1H, C(sugar)-CH2(B)), 1.26 (d, J = 6.6 Hz, 3H, H-6); 13C NMR (Taken from HSQC) δ 105.1 (C-1), 74.8 (C-4), 71.0 (C-3), 68.0 (C-5), 55.9 (OCH3), 41.4 (C(sugar)-CH2), 19.3 (CH2-SH), 17.3 (C-6); HRMS (ESI) m/z calculated for C9H18OsS [M + Na]+ 261.0773, found 261.0773.
3.12. Methyl 6-deoxy-2-C-[3-thiopropyl]-α-l-talopyranoside (4)
Compound 15 (30 mg, 0.10 mmol) was subjected to General Procedure B to afford 4 (24 mg, 95%) as a colourless, amorphous solid. Rf = 0.4 (CH2Cl2/MeOH, 19:1); [α]−71 (c 0.8, CH3OH); 1H NMR (400 MHz, CD3OD) δ 4.48 (s, 1H, H-1), 3.90 (m, 1H, H-5), 3.62 (dd, J = 3.4, 1.0 Hz, 1H, H-4), 3.51 (d, J = 3.4 Hz, 1H, H-3), 3.38 (s, 3H, OCH3), 2.56–2.44 (m, 2H, CH2-SH), 1.97–1.73 (m, 2H, C(sugar)-CH2(A), -CH2-(A)), 1.67–1.53 (m, 2H, C(sugar)-CH2(B), -CH2-(B)), 1.27 (d, J = 6.6 Hz, 3H, H-6); 13C NMR (Taken from HSQC) δ 104.9 (C-1), 74.7 (C-4), 71.3 (C-3), 68.0 (C-5), 56.0 (OCH3), 34.8 (-CH2-), 29.0 (C(sugar)-CH2), 26.3 (CH2-SH), 17.3 (C-6); HRMS (ESI) m/z calculated for C10H20O5S [M + Na]+ 275.0929, found 275.0917.
3.13. Methyl 4-O-acetyl-2,3-O-(2′,3′-dimethoxybutane-2′,3′-diyl)-α-l-quinovopyranoside (17)
Compound 16 (6.09 g, 20.6 mmol) was placed under N2 and dissolved in dry CH2Cl2 (90 mL) and dry pyridine (14 mL). The solution was cooled to −40 °C and trifluoromethanesulfonic anhydride (4.2 mL, 25 mmol) was added. The reaction was stirred at −40 °C for 7 h, then allowed to reach room temperature. The organic phase was washed with sat. NaHCO3 solution (100 mL) and brine (100 mL), dried over MgSO4, filtered and concentrated in vacuo. The dark red/brown oil obtained was immediately used in the next step.
The crude and tetrabutylammonium acetate (7.55 g, 25.0 mmol) were placed under N2 and dry toluene (60 mL) was added. The reaction was stirred for 14 h at room temperature and the solvent was then removed under reduced pressure. The product was purified via flash chromatography on silica gel (cyclohexane/EtOAc, 4:1→1:1) to yield 17 a pale-yellow syrup (4.71 g, 68% over 2 steps). Rf = 0.7 (cyclohexane/EtOAc, 1:1); [α]+47 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3) δ 4.80 (t, J = 9.7 Hz, 1H, H-4), 4.69 (d, J = 3.6 Hz, 1H, H-1), 4.06 (t, J = 9.7 Hz, 1H, H-3), 3.84–3.72 (m, 2H, H-2, H-5), 3.41 (s, 3H, OCH3), 3.24 (s, 3H, OCH3), 3.22 (s, 3H, OCH3), 2.07 (s, 3H, CH3(OAc)), 1.32 (s, 3H, CH3(BDA)), 1.24 (s, 3H, CH3(BDA)), 1.17 (d, J = 6.3 Hz, 3H, H-6); 13C NMR (126 MHz, CDCl3) δ 169.9 (C=O), 100.0, 99.5 (2 × C(ketal)), 97.9 (C-1), 73.3 (C-4), 68.7 (C-2), 67.1 (C-3), 66.1 (C-5), 55.2, 48.0, 47.6 (3 × OCH3), 21.0 (CH3(OAc)), 17.9, 17.8 (2 × CH3(BDA)), 17.5 (C-6); HRMS (ESI) m/z calculated for C15H26O8 [M + Na]+ 357.1525, found 357.1523.
3.14. Methyl 2,3-O-(2′,3′-dimethoxybutane-2′,3′-diyl)-α-l-quinovopyranoside (18)
Compound 17 (3.33 g, 9.96 mmol) was dissolved in MeOH (100 mL) and NaOMe (293 mg, 5.06 mmol) was added. The reaction was stirred for 19 h at room temperature and then Amberlite® IR120 resin (H+ form) was added to neutralise the solution. The mixture was filtered, and the filtrate was concentrated under reduced pressure. The resulting yellow syrup was purified via flash chromatography on silica gel (toluene→toluene/EtOAc, 1:1) to yield 18 as a white foam (2.74 g, 94%). Rf = 0.4 (cyclohexane/EtOAc, 1:1); [α]+59 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3) δ 4.68 (d, J = 3.5 Hz, 1H, H-1), 3.95 (dd, J = 10.4, 9.2 Hz, 1H, H-3), 3.74–3.63 (m, 2H, H-2, H-5), 3.40 (s, 3H, OCH3), 3.35 (m, 1H, H-4), 3.27 (s, 3H, OCH3), 3.24 (s, 3H, OCH3), 2.23 (d, J = 2.8 Hz, 1H, OH), 1.33 (s, 3H, CH3(BDA)), 1.30 (s, 3H, CH3(BDA)), 1.28 (d, J = 6.3 Hz, 3H, H-6); 13C NMR (126 MHz, CDCl3) δ 100.0, 99.6 (2 × C(ketal)), 98.1 (C-1), 73.8 (C-4), 69.5 (C-3), 68.6 (C-2), 67.9 (C-5), 55.1, 48.1, 48.0 (3 × OCH3), 18.0, 17.8 (2 × CH3(BDA)), 17.7 (C-6); HRMS (ESI) m/z calculated for C13H24O7 [M + Na]+ 315.1420, found 315.1410.
3.15. Methyl 4-azido-4-deoxy-2,3-O-(2′,3′-dimethoxybutane-2′,3′-diyl)-α-l-fucopyranoside (19)
Compound 18 (3.96 g, 13.6 mmol) was placed under an N2 atmosphere and dissolved in dry CH2Cl2 (37 mL) and dry pyridine (8 mL). The solution was cooled to -20 °C and trifluoromethanesulfonic anhydride (2.6 mL, 16 mmol) was added slowly. The reaction was stirred at -20 °C for 4 h, then allowed to reach room temperature. The solution was then diluted with CH2Cl2 (35 mL) and washed with sat. aq. NaHCO3 (70 mL) and brine (70 mL). The combined aqueous phase was extracted with CH2Cl2 (70 mL) and the collective organic layer was dried over MgSO4, filtered and concentrated. The crude was used immediately in the next step.
The crude triflate and sodium azide (5.31 g, 81.7 mmol) were placed under N2 and dry DMF (110 mL) was added. The resulting suspension was stirred at 60 °C for 17 h and the solvent was then removed in vacuo. EtOAc (100 mL) and water (100 mL) were added to the resulting residue. The organic layer was separated and then washed with brine (100 mL). The collective aqueous phase was extracted with EtOAc (2 × 50 mL) and the combined organic layer was dried over MgSO4, filtered and concentrated under reduced pressure. The product was purified by flash chromatography on silica gel (cyclohexane/EtOAc, 3:1) to yield 19 as a pale-yellow syrup (3.00 g, 70% over 2 steps). Rf = 0.4 (cyclohexane/EtOAc, 3:1); [α]+86 (c 1.0, CHCl3); 1H NMR (300 MHz, CDCl3) δ 4.71 (d, J = 3.6 Hz, 1H, H-1), 4.21 (dd, J = 10.5, 3.5 Hz, 1H, H-3), 4.11 (dd, J = 10.5, 3.6 Hz, 1H, H-2), 3.99 (m, 1H, H-5), 3.64 (dd, J = 3.5, 3.2 Hz, 1H, H-4), 3.37 (s, 3H, OCH3), 3.27 (s, 3H, OCH3), 3.26 (s, 3H, OCH3), 1.31 (s, 3H, CH3(BDA)), 1.29 (s, 3H, CH3(BDA)), 1.26 (d, J = 6.5 Hz, 3H, H-6); 13C NMR (101 MHz, CDCl3) δ 100.3, 100.2 (2 × C(ketal)), 98.4 (C-1), 67.0 (C-3), 65.50 (C-5), 65.49 (C-2), 63.8 (C-4), 55.4, 48.14, 48.11 (2 × OCH3), 17.9, 17.8 (2 × CH3(BDA)), 17.4 (C-6); HRMS (ESI) m/z calculated for C13H23N3O6 [M + Na]+ 340.1485, found 340.1469.
3.16. Methyl 4-amino-4-deoxy-2,3-O-(2′,3′-dimethoxybutane-2′,3′-diyl)-α-l-fucopyranoside (20)
Compound 19 (2.74 g, 8.63 mmol) was dissolved in EtOH (15 mL). 10% Pd/C (0.54 g, 0.51 mmol) suspended in EtOH (5 mL) was added and the mixture was stirred under H2 (5 bar) at room temperature for 22 h. The suspension was then filtered through Celite® and the filtrate was concentrated. Flash chromatography on silica gel (CH2Cl2/MeOH, 49:1) yielded 20 as a white solid (2.28 g, 91%). Rf = 0.7 (CH2Cl2/MeOH, 19:1); mp = 87–89 °C; [α]+41 (c 1.0, MeOH); 1H NMR (300 MHz, CD3OD) δ 4.66 (d, J = 2.2 Hz, 1H, H-1), 4.07–3.97 (m, 3H, H-2, H-3, H-5), 3.38 (s, 3H, OCH3), 3.25 (s, 3H, OCH3), 3.24 (s, 3H, OCH3), 2.98 (t, J = 1.7 Hz, 1H, H-4), 1.28–1.27 (m, 6H, 2 × CH3(BDA)), 1.21 (d, J = 6.6 Hz, 3H, H-6); 13C NMR (126 MHz, CD3OD) δ 101.5, 101.3 (2 × C(ketal)), 99.7 (C-1), 67.8 (C-2 or C-3), 67.4 (C-5), 66.1 (C-2 or C-3), 55.4 (OCH3), 54.1 (C-4), 48.22, 48.15 (2 × OCH3), 18.1, 18.0 (2 × CH3(BDA)), 16.9 (C-6); HRMS (ESI) m/z calculated for C13H25NO6 [M + H]+ 292.1760, found 292.1761.
3.17. 2-Bromoethyl Thioacetate (21)
Dry THF (40 mL) was added to potassium thioacetate (2.00 g, 17.5 mmol) under N
2. 1,2-Dibromoethane (3.1 mL, 36 mmol) was added and the mixture was refluxed at 90 °C for 22 h. The suspension was then filtered through Celite
®, and the filtrate was concentrated under reduced pressure. The resulting yellow residue was purified via flash chromatography on silica gel (cyclohexane/EtOAc, 19:1), yielding compound
21 as a colourless oil (1.18 g, 37%). R
f = 0.5 (cyclohexane/EtOAc, 10:1);
1H NMR (400 MHz, CDCl
3) δ 3.44 (m, 2H, CH
2-Br), 3.30 (m, 2H, CH
2-S), 2.35 (s, 3H, CH
3(SAc));
13C NMR (101 MHz, CDCl
3) δ 194.7 (C=O
(SAc)), 31.4 (CH
2), 30.7 (CH
3(SAc)), 30.1 (CH
2). While the chemical shifts of the
1H NMR data match those reported in literature, we observed the CH
2 signals as multiplets rather than triplets [
28]. No
13C NMR data have been reported previously.
3.18. Methyl 4-[(2-acetylthioethyl)amino]-4-deoxy-2,3-O-(2′,3′-dimethoxybutane-2′,3′-diyl)-α-l-fucopyranoside (22)
Compound 20 (0.50 g, 1.7 mmol) and K2CO3 (1.19 g, 8.57 mmol) were placed under N2 and dry DMF (5 mL) was added. Alkyl bromide 21 (610 mg, 3.33 mmol) in dry DMF (5 mL) was added followed by KI (286 mg, 1.72 mmol) and the resulting suspension was stirred at room temperature for 4 days. The solvent was removed in vacuo and water (25 mL) was added. The aqueous phase was extracted with EtOAc (3 × 25 mL) and the combined organic layer was dried over Na2SO4, filtered and concentrated. The product was purified by flash chromatography on silica gel (toluene/EtOAc, 7:3), yielding 22 as a pale-yellow syrup (271 mg, 40%). Rf = 0.4 (cyclohexane/EtOAc, 1:1); [α]−8.2 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 4.71 (d, J = 3.8 Hz, 1H, H-1), 4.12 (dd, J = 10.9, 4.4 Hz, 1H, H-3), 3.98–3.90 (m, 2H, H-2, H-5), 3.37 (s, 3H, OCH3), 3.24 (s, 3H, OCH3), 3.22 (s, 3H, OCH3), 3.07 (m, 1H, NH-CH2(A)), 2.98 (m, 1H, NH-CH2(B)), 2.93–2.84 (m, 2H, CH2-SAc), 2.73 (dd, J = 4.5, 4.4 Hz, 1H, H-4), 2.32 (s, 3H, CH3(SAc)), 1.31 (s, 3H, CH3(BDA)), 1.25 (s, 3H, CH3(BDA)), 1.24 (d, J = 6.7 Hz, 3H, H-6); 13C NMR (101 MHz, CDCl3) δ 196.1 (C=O(SAc)), 100.1, 99.8 (2 × C(ketal)), 98.3 (C-1), 67.1 (C-5), 66.5 (C-3), 65.6 (C-2), 60.1 (C-4), 55.1 (OCH3), 50.3 (CH2-SAc), 48.01, 47.99 (2 × OCH3), 30.8 (CH3(SAc)), 29.7 (NH-CH2), 17.91, 17.89 (2 × CH3(BDA)), 17.5 (C-6); HRMS (ESI) m/z calculated for C17H31NO7S [M + Na]+ 416.1719, found 416.1721.
3.19. Methyl 4-[(2-acetylthioethyl)amino]-4-deoxy-α-l-fucopyranoside (23)
Compound 22 (20 mg, 52 µmol) was stirred in TFA/H2O (1.7 mL, 9:1, v/v) at room temperature for 2.5 h. The solvents were then removed through co-evaporation with toluene under reduced pressure. The resulting residue was dissolved in CH2Cl2 (1.8 mL) and stirred with Amberlyst® A21 resin (197 mg) at room temperature for 18 h. The resin was then filtered off and washed with MeOH. The filtrate was concentrated in vacuo and the product was purified by flash chromatography on silica gel (EtOAc→EtOAc/MeOH, 19:1), furnishing 23 as a colourless syrup (11 mg, 77%). Rf = 0.5 (EtOAc/MeOH, 9:1); 1H NMR (500 MHz, CDCl3) δ 4.70 (d, J = 4.0 Hz, 1H, H-1), 4.06 (m, 1H, H-5), 3.63 (dd, J = 9.8, 4.6 Hz, 1H, H-3), 3.40 (s, 3H, OCH3), 3.38 (dd, J = 9.8, 4.0 Hz, 1H, H-2), 3.23 (m, 1H, CH2(A)-SAc), 3.04–2.95 (m, 2H, NH-CH2), 2.73–2.64 (m, 2H, H-4, CH2(B)-SAc), 2.36 (s, 3H, CH3(SAc)), 1.26 (d, J = 6.7 Hz, 3H, H-6); 13C NMR (126 MHz, CDCl3) δ 195.8 (C=O(SAc)), 99.6 (C-1), 71.3 (C-2), 70.7 (C-3), 66.3 (C-5), 62.6 (C-4), 55.6 (OCH3), 50.7 (CH2-SAc), 30.83 (CH3(SAc)), 30.82 (NH-CH2), 17.6 (C-6); HRMS (ESI) m/z calculated for C11H21NO5S [M + H]+ 280.1219, found 280.1208.
3.20. 2-Bromoethyl thiobenzoate (24)
Potassium thiobenzoate (0.51 g, 2.9 mmol) was placed under N2 and dry THF (28 mL) was added followed by 1,2-dibromoethane (2.4 mL, 28 mmol). The reaction was refluxed at 90 °C for 3 h, cooled to room temperature and the solids were removed by filtration through Celite®. The filtrate was concentrated in vacuo and purified by flash chromatography on silica gel (cyclohexane/toluene, 4:1). Compound 24 was obtained as a colourless oil (603 mg, 86%). Rf = 0.4 (cyclohexane/toluene, 7:3); 1H NMR (300 MHz, CDCl3) δ 7.99–7.92 (m, 2H, Ar-H), 7.58 (m, 1H, Ar-H), 7.52–7.39 (m, 2H, Ar-H), 3.67–3.40 (m, 4H, CH2-Br, CH2-S); 13C NMR (101 MHz, CDCl3) δ 190.7 (C=O(SBz)), 136.6 (Ar-C(quat)), 133.9, 128.9, 127.5 (3 × Ar-CH), 31.2 (CH2-S), 30.2 (CH2-Br).
3.21. Methyl 4-[(2-benzoylthioethyl)amino]-4-deoxy-2,3-O-(2′,3′-dimethoxybutane-2′,3′-diyl)-α-l-fucopyranoside (25)
Compound 20 (351 mg, 1.20 mmol) was placed under N2 and dissolved in dry DMF (6 mL). K2CO3 (183 mg, 1.33 mmol) was added followed by a solution of alkyl bromide 24 (326 mg, 1.33 mmol) in dry DMF (6 mL). KI (97 mg, 0.58 mmol) was then added and the reaction was stirred at 80 °C for 20 h. The solvent was then removed in vacuo and water (30 mL) was added to the resulting residue. The product was extracted with EtOAc (3 × 30 mL) and the combined organic layer was dried over MgSO4, filtered and concentrated under reduced pressure. Flash chromatography on silica gel (toluene/acetone, 19:1) yielded compound 25 as a colourless syrup (267 mg, 49%). Rf = 0.5 (toluene/EtOAc, 1:1); [α]+14 (c 1.0, CHCl3); 1H NMR (400 MHz, CDCl3) δ 8.00–7.93 (m, 2H, Ar-H), 7.56 (m, 1H, Ar-H), 7.49–7.38 (m, 2H, Ar-H), 4.73 (d, J = 3.8 Hz, 1H, H-1), 4.14 (dd, J = 10.8, 4.4 Hz, 1H, H-3), 3.99 (dd, J = 10.8, 3.8 Hz, 1H, H-2), 3.94 (m, 1H, H-5), 3.38 (s, 3H, OCH3), 3.32–3.14 (m, 8H, 2 × OCH3, NH-CH2(A), NH-CH2(B)), 3.03–2.96 (m, 2H, CH2-SBz), 2.79 (dd, J = 4.4, 1.6 Hz, 1H, H-4), 1.31 (s, 3H, CH3(BDA)), 1.26 (d, J = 6.7 Hz, 3H, H-6), 1.24 (s, 3H, CH3(BDA)); 13C NMR (101 MHz, CDCl3) δ 192.1 (C=O(SBz)), 137.3 (Ar-C(quat)), 133.4, 128.7, 127.3 (3 × Ar-CH), 100.1, 99.8 (2 × C(ketal)), 98.3 (C-1), 67.2 (C-5), 66.6 (C-3), 65.6 (C-2), 60.1 (C-4), 55.1 (OCH3), 50.4 (CH2-SBz), 48.0 (2 × OCH3), 29.6 (NH-CH2), 17.90, 17.88 (2 × CH3(BDA)), 17.5 (C-6). HRMS (ESI) m/z calculated for C22H33NO7S [M + H]+ 456.2056, found 456.2068.
3.22. Methyl 4-[(2-benzoylthioethyl)amino]-4-deoxy-α-l-fucopyranoside (26)
Compound 25 (52 mg, 0.11 mmol) was stirred in TFA/H2O (3.7 mL, 9:1, v/v) at room temperature for 3 h. The solvents were then co-evaporated with toluene under reduced pressure. The resulting residue was re-dissolved in CH2Cl2 (5 mL) and stirred at room temperature with Amberlyst® A21 resin (507 mg) for 20 h. The resin was then filtered off and washed with CH2Cl2/MeOH (1:1, v/v). The filtrate was concentrated in vacuo and purified via flash chromatography on silica gel (toluene/EtOAc, 1:4→EtOAc), yielding 26 as a white amorphous solid (30 mg, 76%). Rf = 0.3 (EtOAc); [α]−120 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.98–7.93 (m, 2H, Ar-H), 7.57 (m, 1H, Ar-H), 7.48–7.41 (m, 2H, Ar-H), 4.69 (d, J = 4.0 Hz, 1H, H-1), 4.06 (m, 1H, H-5), 3.63 (dd, J = 9.8, 4.7 Hz, 1H, H-3), 3.42–3.37 (m, 4H, H-2, OCH3), 3.31 (m, 1H, CH2(A)-SBz), 3.22–3.19 (m, 2H, NH-CH2), 2.81–2.71 (m, 2H, H-4, CH2(B)-SBz), 1.26 (d, J = 6.7 Hz, 3H, H-6); 13C NMR (126 MHz, CDCl3) δ 191.8 (C=O(SBz)), 137.0 (Ar-C(quat)), 133.7 (Ar-CH), 128.8 (Ar-CH), 127.4 (Ar-CH), 99.6 (C-1), 71.3 (C-2), 70.7 (C-3), 66.3 (C-5), 62.6 (C-4), 55.6 (OCH3), 50.8 (CH2-SBz), 30.7 (NH-CH2), 17.6 (C-6). HRMS (ESI) m/z calculated for C16H23NO5S [M + H]+ 342.1375, found 342.1366.
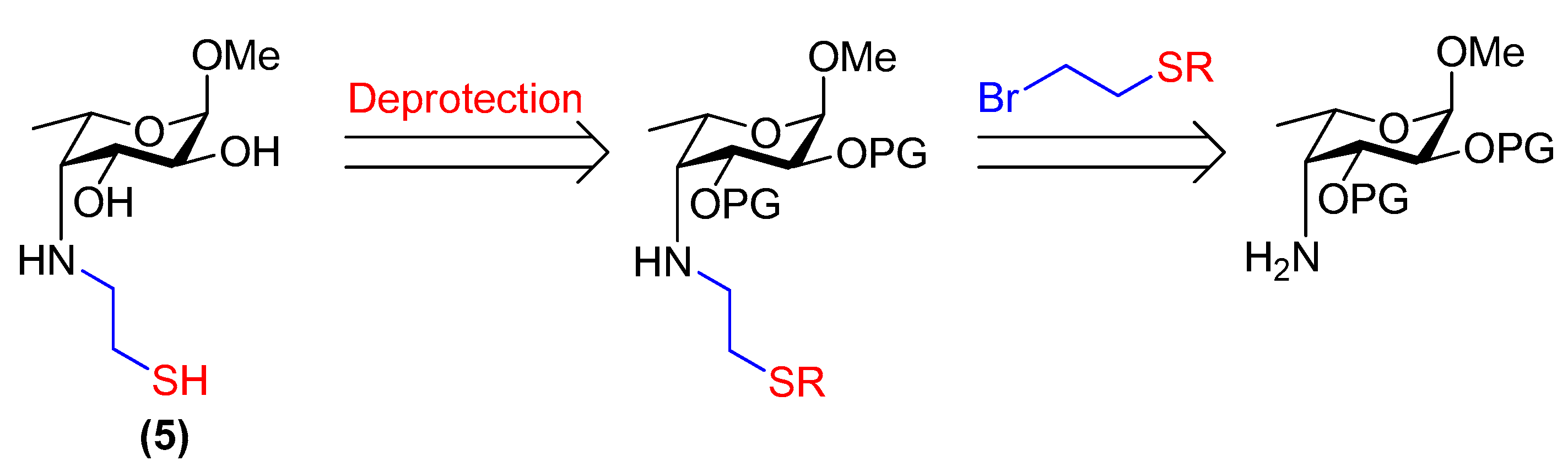
3.23. Methyl 4-deoxy-4-(2-thioethyl)amino-α-l-fucopyranoside (5)
Compound 26 (20 mg, 57 µmol) was placed under N2. Dry, de-gassed MeOH (0.58 mL) was added followed by freshly prepared, de-gassed 1 M NaOMe/MeOH (0.15 mL, 0.15 mmol). The reaction was stirred at room temperature for 15 min and then neutralised with Amberlite® IR120 resin (H+ form). The resin was filtered off and the filtrate was concentrated. The product was purified by flash chromatography through a short column of silica gel (CH2Cl2/MeOH, 19:1→9:1) under an N2 atmosphere and using de-gassed solvents. Compound 5 was obtained as a colourless residue (8 mg, 58% by mass, 95% thiol, 5% disulfide). Rf = 0.2 (EtOAc/MeOH, 19:1); [α]+2.7 (c 0.75, H2O); 1H NMR (500 MHz, D2O) δ 4.79 (d, 1H, H-1, underneath HDO peak), 4.14 (m, 1H, H-5), 3.97 (dd, J = 10.4, 4.3 Hz, 1H, H-3), 3.68 (dd, J = 10.4, 4.0 Hz, 1H, H-2), 3.40 (s, 3H, OCH3), 3.09 (m, 1H, CH2(A)-SH), 2.99 (d, J = 4.3 Hz, 1H, H-4), 2.85 (m, 1H, CH2(B)-SH), 2.77–2.67 (m, 2H, NH-CH2), 1.31 (d, J = 6.7 Hz, 3H, H-6); 13C NMR (126 MHz, D2O) δ 99.2 (C-1, JC-1,H-1 = 170.9 Hz), 68.8 (C-3), 68.0 (C-2), 65.9 (C-5), 61.8 (C-4), 55.0 (OCH3), 53.4 (CH2-SH), 23.3 (NH-CH2), 16.5 (C-6); HRMS (ESI) m/z calculated for C9H19NO4S [M + H]+ 238.1113, found 238.1119.
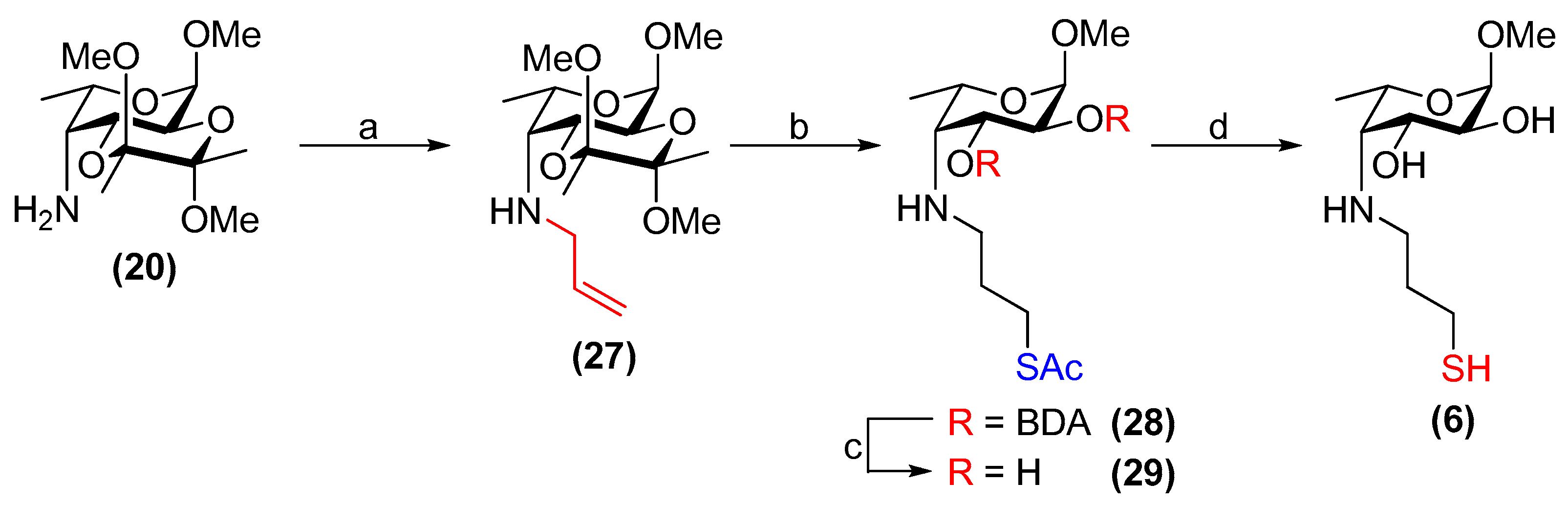
3.24. Methyl 4-allylamino-4-deoxy-2,3-O-(2′,3′-dimethoxybutane-2′,3′-diyl)-α-l-fucopyranoside (27)
Compound 20 (374 mg, 1.28 mmol) and K2CO3 (196 mg, 1.42 mmol) were subjected to an N2 atmosphere. Dry DMF (7.5 mL) was added followed by allyl bromide (0.13 mL, 1.5 mmol). The resulting suspension was stirred at room temperature for 21 h and the solvent was then removed under reduced pressure. Water (25 mL) was added and aqueous phase was extracted with EtOAc (3 × 25 mL). The combined organic layer was dried over Na2SO4, filtered and concentrated in vacuo. The crude was purified by flash chromatography on silica gel (toluene/EtOAc, 4:1→7:3) to yield 27 as a pale-yellow, waxy solid (290 mg, 68%). Rf = 0.3 (cyclohexane/EtOAc, 1:1); [α]+28 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 5.89 (m, 1H, CH2=CH), 5.19 (dq, J = 17.2, 1.4 Hz, 1H, CH2(A)=CH), 5.06 (dq, J = 10.2, 1.4 Hz, 1H, CH2(B)=CH), 4.72 (d, J = 3.8 Hz, 1H, H-1), 4.13 (dd, J = 10.8, 4.5 Hz, 1H, H-3), 3.99 (dd, J = 10.8, 3.8 Hz, 1H, H-2), 3.91 (m, 1H, H-5), 3.40 (ddt, J = 14.3, 5.4, 1.6 Hz, 1H, NH-CH2(A)), 3.37 (s, 3H, OCH3), 3.27 (m, 1H, NH-CH2(B)), 3.24 (s, 3H, OCH3), 3.21 (s, 3H, OCH3), 2.75 (dd, J = 4.5, 4.3 Hz, 1H, H-4), 1.30 (s, 3H, CH3(BDA)), 1.25–1.24 (m, 6H, CH3(BDA), H-6); 13C NMR (126 MHz, CDCl3) δ 137.5 (CH2=CH), 115.7 (CH2=CH), 100.0, 99.8 (2 × C(ketal)), 98.3 (C-1), 67.3 (C-5), 66.5 (C-3), 65.6 (C-2), 59.5 (C-4), 55.1 (OCH3), 53.8 (NH-CH2), 47.98, 47.94 (2 × OCH3), 17.90, 17.88 (2 × CH3(BDA)), 17.80 (C-6); HRMS (ESI) m/z calculated for C16H29NO6 [M + H]+ 332.2073, found 332.2079.
3.25. Methyl 4-[(3-acetylthiopropyl)amino]-4-deoxy-2,3-O-(2′,3′-dimethoxybutane-2′,3′-diyl)-α-l-fucopyran -oside (28)
Compound 27 (281 mg, 0.849 mmol) and 1,2-dihydroxybenzene (116 mg, 1.05 mmol) were placed under N2 and dissolved in dry CH2Cl2 (1.1 mL). Thioacetic acid (0.12 mL, 1.7 mmol) was added followed by 1 M Et3B/hexanes (1.3 mL, 1.3 mmol). The reaction was stirred for 2.5 h at room temperature and then the volatiles were then removed under reduced pressure. The product was purified by flash chromatography on silica gel (toluene/EtOAc, 9:1→3:2), yielding 28 as a pale-yellow syrup (287 mg, 83%). Rf = 0.2 (cyclohexane/EtOAc, 3:2); [α]+8.4 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 4.71 (d, J = 3.8 Hz, 1H, H-1), 4.11 (dd, J = 10.8, 4.4 Hz, 1H, H-3), 3.97 (dd, J = 10.8, 3.8 Hz, 1H, H-2), 3.92 (m, 1H, H-5), 3.36 (s, 3H, OCH3), 3.24 (s, 3H, OCH3), 3.21 (s, 3H, OCH3), 3.01–2.91 (m, 2H, CH2-SAc), 2.81–2.68 (m, 2H, NH-CH2), 2.66 (dd, J = 4.5, 4.4 Hz, 1H, H-4), 2.31 (s, 3H, CH3(SAc)), 1.83–1.68 (m, 2H, -CH2-), 1.30 (s, 3H, CH3(BDA)), 1.25 (s, 3H, CH3(BDA)), 1.23 (d, J = 6.7 Hz, 3H, H-6); 13C NMR (126 MHz, CDCl3) δ 196.1 (C=O(SAc)), 100.0, 99.8 (2 × C(ketal)), 98.3 (C-1), 67.2 (C-5), 66.6 (C-3), 65.6 (C-2), 60.8 (C-4), 55.1 (OCH3), 50.3 (CH2-SAc), 47.98, 47.97 (2 × OCH3), 30.7 (CH3(SAc)), 30.2 (-CH2-), 26.9 (NH-CH2), 17.92, 17.88 (2 × CH3(BDA)), 17.6 (C-6); HRMS (ESI) m/z calculated for C18H33NO7S [M + H]+ 408.2056, found 408.2076.
3.26. Methyl 4-[(3-acetylthiopropyl)amino]-4-deoxy-α-l-fucopyranoside (29)
Compound 28 (272 mg, 0.667 mmol) was stirred in TFA/H2O (22 mL, 9:1, v/v) at room temperature for 3 h. The solvents were then co-evaporated with toluene in vacuo. The resulting residue re-dissolved in CH2Cl2 (25 mL) and stirred at room temperature with Amberlyst® A21 resin (2.42 g) for 16 h. The resin was then filtered off and washed with CH2Cl2/MeOH (1:1, v/v). The filtrate was concentrated in vacuo and purified via flash chromatography on silica gel (EtOAc→EtOAc/MeOH, 4:1) to yield compound 29 as a yellow syrup (133 mg, 68%). Rf = 0.3 (EtOAc/MeOH, 9:1); [α]−136 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 4.69 (d, J = 4.0 Hz, 1H, H-1), 4.05 (m, 1H, H-5), 3.63 (dd, J = 9.8, 4.7 Hz, 1H, H-3), 3.39 (m, 4H, H-2, OCH3), 3.04–2.89 (m, 3H, NH-CH2, CH2(A)-SAc), 2.69 (dd, J = 4.8, 4.7 Hz, 1H, H-4), 2.58 (m, 1H, CH2(B)-SAc), 2.33 (s, 3H, CH3(SAc)), 1.82–1.73 (m, 2H, -CH2-), 1.26 (d, J = 6.7 Hz, 3H, H-6); 13C NMR (126 MHz, CDCl3) δ 196.1 (C=O(SAc)), 99.6 (C-1), 71.1 (C-2), 70.4 (C-3), 66.2 (C-5), 62.6 (C-4), 55.6 (OCH3), 50.2 (CH2-SAc), 30.9 (-CH2-), 30.8 (CH3(SAc)), 26.7 (NH-CH2), 17.6 (C-6); HRMS (ESI) m/z calculated for C12H23NO5S [M + H]+ 294.1375, found 294.1364.
3.27. Methyl 4-deoxy-4-[(3-thiopropyl)amino]-α-l-fucopyranoside (6)
Compound 29 (27 mg, 91 µmol) was placed under N2 and dissolved in dry, de-gassed MeOH (0.90 mL). Freshly prepared, de-gassed 1 M NaOMe/MeOH (0.19 mL, 0.19 mmol) was then added and the reaction was stirred at room temperature for 30 min. The solution was then diluted with dry, de-gassed MeOH (1.0 mL), neutralised with activated Amberlite® IR120 resin (H+ form), filtered and concentrated in vacuo. Compound 6 was obtained as a yellow/orange residue (20 mg, 88% by mass, 88% thiol, 12% disulfide). Rf = 0.4 (CH2Cl2/MeOH, 9:1); [α]−180 (c 1.0, H2O); 1H NMR (400 MHz, D2O) δ 4.79 (d, 1H, H-1, underneath HDO peak), 4.12 (m, 1H, H-5), 3.97 (dd, J = 10.5, 4.6 Hz, 1H, H-3), 3.66 (dd, J = 10.5, 4.0 Hz, 1H, H-2), 3.40 (s, 3H, OCH3), 3.03–2.88 (m, 2H, H-4, CH2(A)-SH), 2.79 (m, 1H, CH2(B)-SH), 2.61 (t, J = 7.0 Hz, 2H, NH-CH2), 2.01–1.79 (m, 2H, -CH2-), 1.31 (d, J = 6.8 Hz, 3H, H-6); 13C NMR (126 MHz, D2O) δ 99.2 (C-1, JC-1,H-1 = 170.8 Hz), 68.7 (C-3), 68.0 (C-2), 66.1 (C-5), 62.2 (C-4), 55.0 (OCH3), 49.8 (CH2-SH), 32.5 (-CH2-), 21.4 (NH-CH2), 16.5 (C-6); HRMS (ESI) m/z calculated for C10H21NO4S [M + H]+ 252.1270, found 252.1261.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












