Semi-Continuous Flow Biocatalysis with Affinity Co-Immobilized Ketoreductase and Glucose Dehydrogenase


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. Ketoreductase and Glucose Dehydrogenase Expression
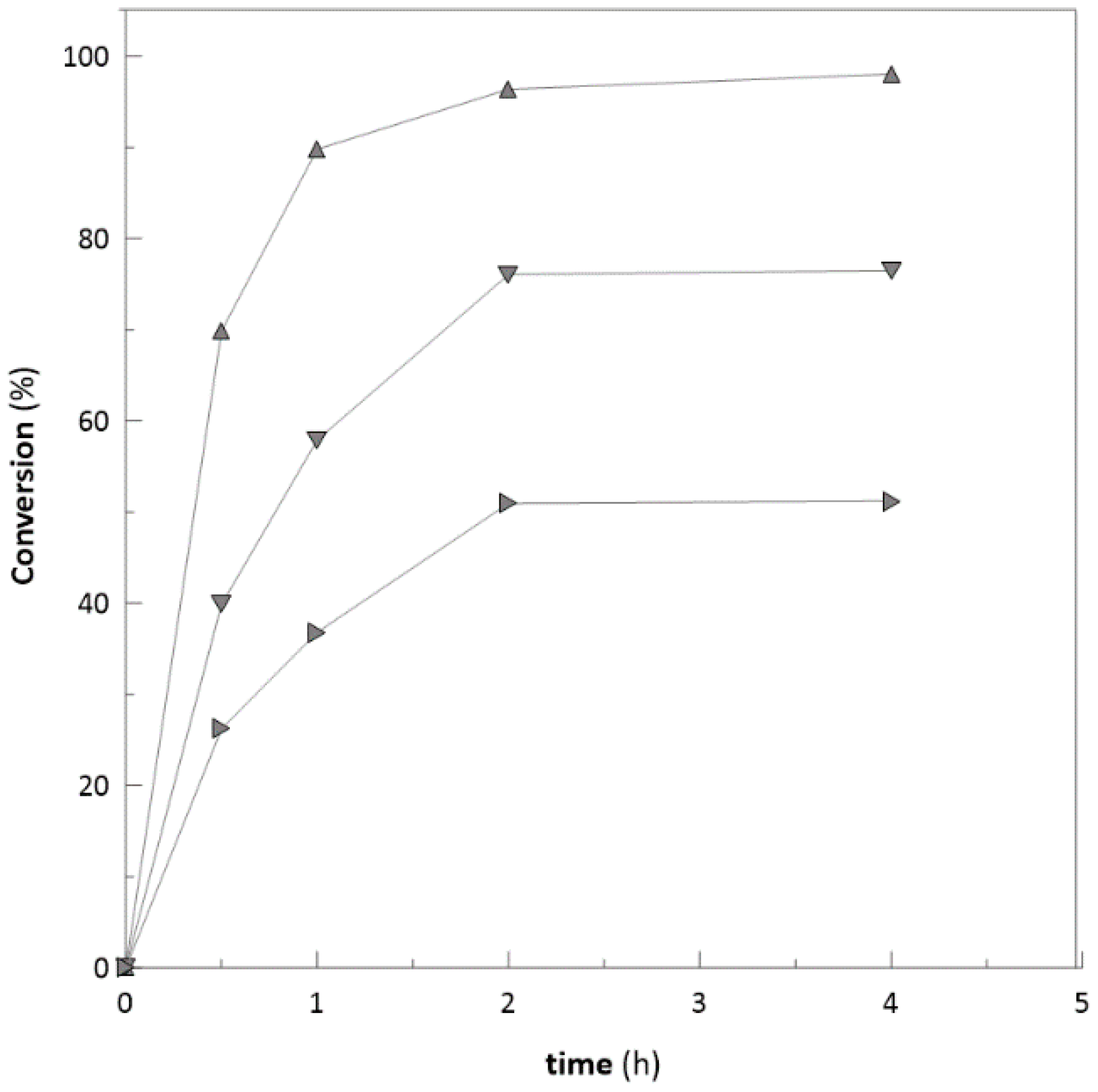
2.2. Biotransformation with Free Enzymes
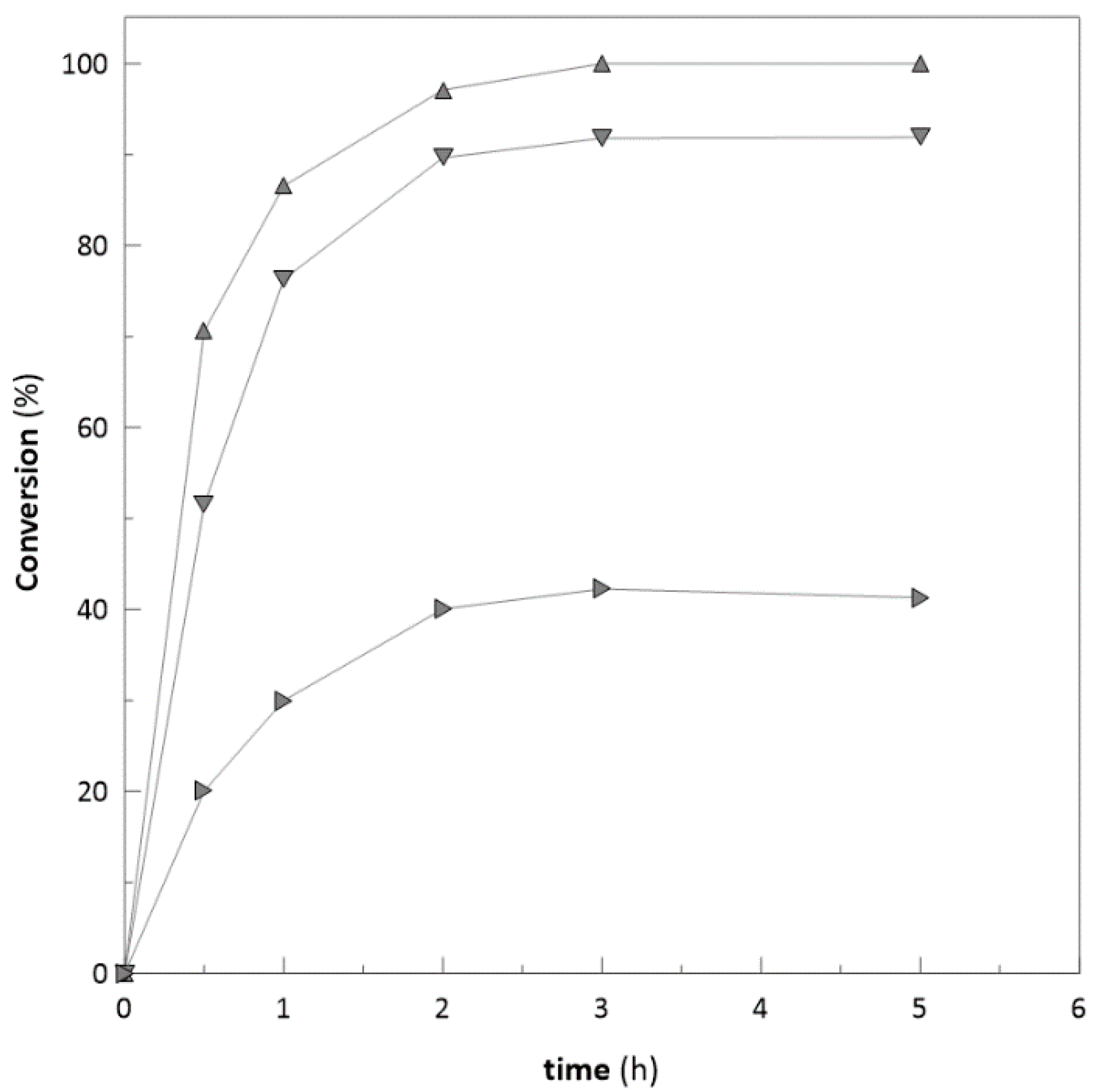
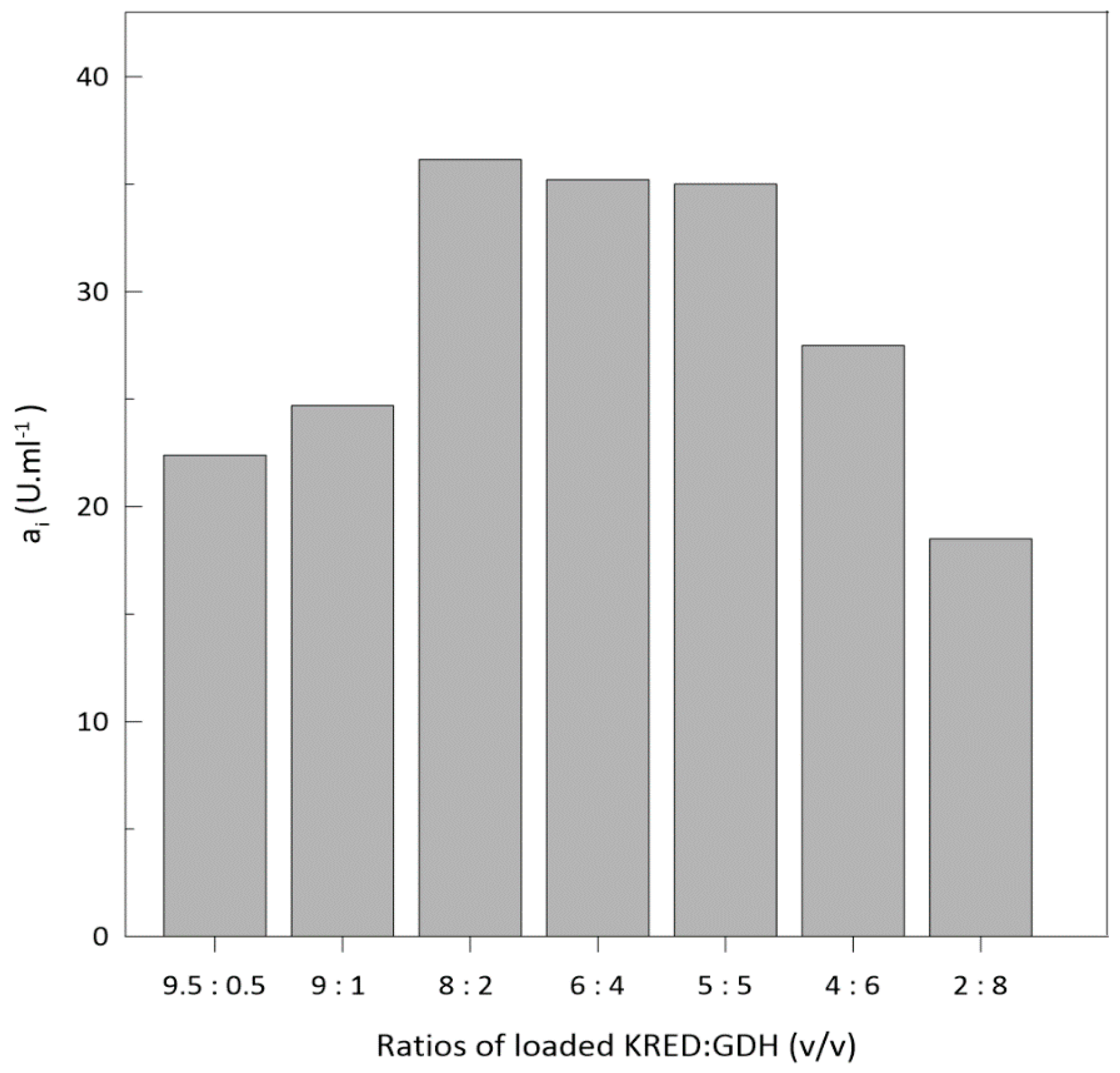
2.3. Ketoreductase and Glucose Dehydrogenase Co-immobilization
2.4. Optimisation of Cofactor (NADP+) Concentration
2.5. Substrate Concentration
Application on Bulky Ketones
2.6. Repeated Biotransformations
2.7. Storage of the Immobilised Biocatalyst
3. Materials and Methods
3.1. Chemicals and Media
3.2. Cloning
3.3. Preparation of Ketoreductase and Glucose Dehydrogenase
3.4. Enzyme Assays
3.5. Co-Immobilisation
Optimization of Enzyme Loading into Column
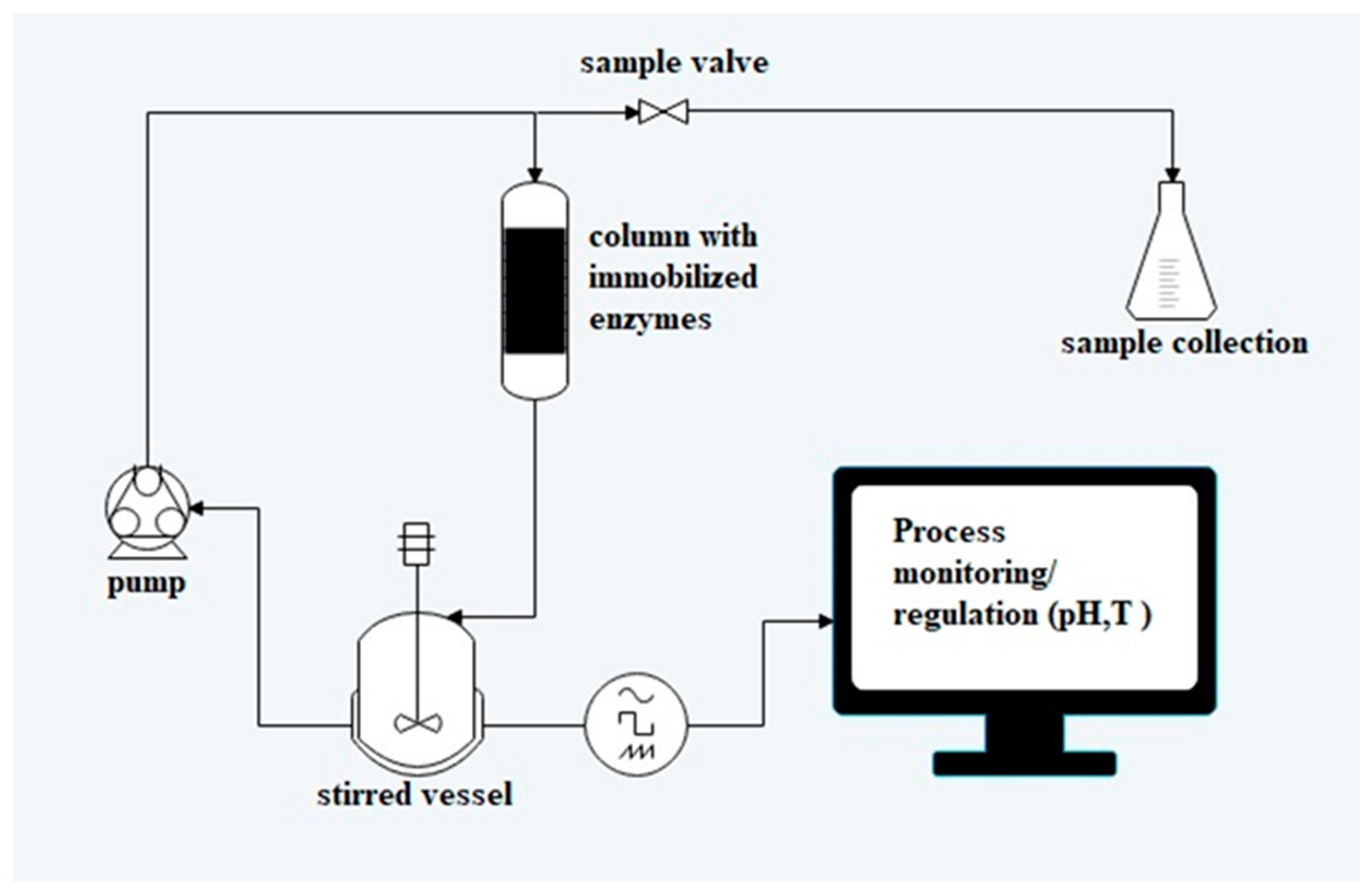
3.6. Biotransformations with Immobilised Enzymes
3.6.1. Substrate 1: ethyl-2-methylacetoacetate
3.6.2. Substrates 2; 3: 4-phenyl-2-butanone; 3′-hydroxyacetophenone
3.7. Biotransformations with Free Enzymes
3.7.1. Substrate 1: ethyl-2-methylacetoacetate
3.7.2. Substrates 2; 3: 4-phenyl-2-butanone; 3′-hydroxyacetophenone
3.8. Analytics
3.8.1. Substrate 1: ethyl-2-methylacetoacetate
3.8.2. Substrates 2; 3: 4-phenyl-2-butanone; 3′-hydroxyacetophenone
3.9. Storage Solution for Immobilized Biocatalyst
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bilal, M.; Zhao, Y.; Noreen, S.; Shah, S.Z.H.; Bharagava, R.N.; Iqbal, H.M.N. Modifying bio-catalytic properties of enzymes for efficient biocatalysis: A review from immobilization strategies viewpoint. Biocatal. Biotransform. 2019, 37, 159–182. [Google Scholar] [CrossRef]
- Hall, M.; Bommarius, A.S. Enantioenriched Compounds via Enzyme-Catalyzed Redox Reactions. Chem. Rev. 2011, 111, 4088–4110. [Google Scholar] [CrossRef]
- Hollmann, F.; Arends, I.W.C.E.; Holtmann, D. Enzymatic reductions for the chemist. Green Chem. 2011, 13, 2285–2314. [Google Scholar] [CrossRef]
- Matsuda, T.; Yamanaka, R.; Nakamura, K. Recent progress in biocatalysis for asymmetric oxidation and reduction. Tetrahedron Asymmetry 2009, 20, 513–557. [Google Scholar] [CrossRef]
- Zhu, D.; Yang, Y.; Buynak, J.D.; Hua, L. Stereoselective ketone reduction by a carbonyl reductase from Sporobolomyces salmonicolor. Substrate specificity, enantioselectivity and enzyme-substrate docking studies. Org. Biomol. Chem. 2006, 4, 2690–2695. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, M.; Yamazaki, T.; Fuhshuku, K.-I.; Sugai, T. First total synthesis of modiolide A, based on the whole-cell yeast-catalyzed asymmetric reduction of a propargyl ketone. Tetrahedron 2007, 63, 8752–8760. [Google Scholar] [CrossRef]
- Contente, M.L.; Serra, I.; Brambilla, M.; Eberini, I.; Gianazza, E.; De Vitis, V.; Molinari, F.; Zambelli, P.; Romano, D. Stereoselective reduction of aromatic ketones by a new ketoreductase from Pichia glucozyma. Appl. Microbiol. Biotechnol. 2016, 100, 193–201. [Google Scholar] [CrossRef]
- Petrovičová, T.; Markošová, K.; Hegyi, Z.; Smonou, I.; Rosenberg, M.; Rebroš, M. Co-Immobilization of Ketoreductase and Glucose Dehydrogenase. Catalysts 2018, 8, 168. [Google Scholar] [CrossRef]
- Han, C.; Savage, S.; Al-Sayah, M.; Yajima, H.; Remarchuk, T.; Reents, R.; Wirz, B.; Iding, H.; Bachmann, S.; Fantasia, S.M.; et al. Asymmetric Synthesis of Akt Kinase Inhibitor Ipatasertib. Org. Lett. 2017, 19, 4806–4809. [Google Scholar] [CrossRef]
- Liang, J.; Lalonde, J.; Borup, B.; Mitchell, V.; Mundorff, E.; Trinh, N.; Kochrekar, D.A.; Nair Cherat, R.; Pai, G.G. Development of a Biocatalytic Process as an Alternative to the (−)-DIP-Cl-Mediated Asymmetric Reduction of a Key Intermediate of Montelukast. Org. Process. Res. Dev. 2010, 14, 193–198. [Google Scholar] [CrossRef]
- Ma, S.K.; Gruber, J.; Davis, C.; Newman, L.; Gray, D.; Wang, A.; Grate, J.; Huisman, G.W.; Sheldon, R.A. A green-by-design biocatalytic process for atorvastatin intermediate. Green Chem. 2010, 12, 81–86. [Google Scholar] [CrossRef]
- Savile, C.K.; Janey, J.M.; Mundorff, E.C.; Moore, J.C.; Tam, S.; Jarvis, W.R.; Colbeck, J.C.; Krebber, A.; Fleitz, F.J.; Brands, J.; et al. Biocatalytic Asymmetric Synthesis of Chiral Amines from Ketones Applied to Sitagliptin Manufacture. Science 2010, 329, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Krasňan, V.; Stloukal, R.; Rosenberg, M.; Rebroš, M. Immobilization of cells and enzymes to LentiKats®. Appl. Microbiol. Biotechnol. 2016, 100, 2535–2553. [Google Scholar] [CrossRef]
- Nagayama, K.; Spiess, A.C.; Büchs, J. Gas phase enantioselective reduction catalyzed by immobilized ketoreductase: Effects of water activity and reaction temperature. Biochem. Eng. J. 2010, 52, 301–303. [Google Scholar] [CrossRef]
- Li, H.; Moncecchi, J.; Truppo, M.D. Development of an Immobilized Ketoreductase for Enzymatic (R)-1-(3,5-Bis(trifluoromethyl)phenyl)ethanol Production. Org. Process. Res. Dev. 2015, 19, 695–700. [Google Scholar] [CrossRef]
- Xu, M.-Q.; Wang, S.-S.; Li, L.-N.; Gao, J.; Zhang, Y.-W. Combined Cross-Linked Enzyme Aggregates as Biocatalysts. Catalysts 2018, 8, 460. [Google Scholar] [CrossRef]
- Ning, C.; Su, E.; Tian, Y.; Wei, D. Combined cross-linked enzyme aggregates (combi-CLEAs) for efficient integration of a ketoreductase and a cofactor regeneration system. J. Biotechnol. 2014, 184, 7–10. [Google Scholar] [CrossRef]
- Puig, R.T.; Junghanns, C.; Demarche, P.; Moreira, M.T.; Feijoo, G.; Lema, J.; Agathos, S.N. Combined cross-linked enzyme aggregates from versatile peroxidase and glucose oxidase: Production, partial characterization and application for the elimination of endocrine disruptors. Bioresour. Technol. 2011, 102, 6593–6599. [Google Scholar] [CrossRef]
- Jung, D.-H.; Jung, J.-H.; Seo, D.-H.; Ha, S.-J.; Kweon, D.-K.; Park, C.-S. One-pot bioconversion of sucrose to trehalose using enzymatic sequential reactions in combined cross-linked enzyme aggregates. Bioresour. Technol. 2013, 130, 801–804. [Google Scholar] [CrossRef]
- Stressler, T.; Ewert, J.; Eisele, T.; Fischer, L. Cross-linked enzyme aggregates (CLEAs) of PepX and PepN—Production, partial characterization and application of combi-CLEAs for milk protein hydrolysis. Biocatal. Agric. Biotechnol. 2015, 4, 752–760. [Google Scholar] [CrossRef]
- Sheldon, R.A. Cross-Linked Enzyme Aggregates as Industrial Biocatalysts. Org. Process. Res. Dev. 2011, 15, 213–223. [Google Scholar] [CrossRef]
- Jia, F.; Narasimhan, B.; Mallapragada, S.K. Materials-based strategies for multi-enzyme immobilization and co-localization: A review. Biotechnol. Bioeng. 2013, 111, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Kazenwadel, F.; Franzreb, M.; Rapp, B.E. Synthetic enzyme supercomplexes: Co-immobilization of enzyme cascades. Anal. Methods 2015, 7, 4030–4037. [Google Scholar] [CrossRef]
- Kuo, W.-H.K.; Chase, H.A. Exploiting the interactions between poly-histidine fusion tags and immobilized metal ions. Biotechnol. Lett. 2011, 33, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Boehm, C.R.; Freemont, P.S.; Ces, O. Design of a prototype flow microreactor for synthetic biology in vitro. Lab. Chip 2013, 13, 3426–3432. [Google Scholar] [CrossRef]
- Fornera, S.; Kuhn, P.; Lombardi, D.; Schlüter, A.D.; Dittrich, P.S.; Walde, P. Sequential Immobilization of Enzymes in Microfluidic Channels for Cascade Reactions. ChemPlusChem 2012, 77, 98–101. [Google Scholar] [CrossRef]
- Keefe, A.D.; Wilson, D.S.; Seelig, B.; Szostak, J.W. One-Step Purification of Recombinant Proteins Using a Nanomolar-Affinity Streptavidin-Binding Peptide, the SBP-Tag. Protein Expr. Purif. 2001, 23, 440–446. [Google Scholar] [CrossRef]
- Los, G.V.; Encell, L.P.; McDougall, M.G.; Hartzell, D.D.; Karassina, N.; Zimprich, C.; Wood, M.G.; Learish, R.; Ohana, R.F.; Urh, M.; et al. HaloTag: A Novel Protein Labeling Technology for Cell Imaging and Protein Analysis. ACS Chem. Biol. 2008, 3, 373–382. [Google Scholar] [CrossRef]
- Peschke, T.; Rabe, K.S.; Niemeyer, C.M. Orthogonal Surface Tags for Whole-Cell Biocatalysis. Angew. Chem. Int. Ed. 2017, 56, 2183–2186. [Google Scholar] [CrossRef]
- Khan, F.; He, M.; Taussig, M.J. Double-Hexahistidine Tag with High-Affinity Binding for Protein Immobilization, Purification, and Detection on Ni−Nitrilotriacetic Acid Surfaces. Anal. Chem. 2006, 78, 3072–3079. [Google Scholar] [CrossRef]
- Dorn, I.T.; Neumaier, K.R.; Tampé, R. Molecular Recognition of Histidine-Tagged Molecules by Metal-Chelating Lipids Monitored by Fluorescence Energy Transfer and Correlation Spectroscopy. J. Am. Chem. Soc. 1998, 120, 2753–2763. [Google Scholar] [CrossRef]
- Bolanos-Garcia, V.M.; Davies, O.R. Structural analysis and classification of native proteins from E. coli commonly co-purified by immobilised metal affinity chromatography. Biochim. Biophys. Acta (BBA) Gen. Subj. 2006, 1760, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Terpe, K. Overview of tag protein fusions: From molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 2003, 60, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Peschke, T.; Bitterwolf, P.; Rabe, K.S.; Niemeyer, C.M. Self-Immobilizing Oxidoreductases for Flow Biocatalysis in Miniaturized Packed-Bed Reactors. Chem. Eng. Technol. 2019, 42, 2009–2017. [Google Scholar] [CrossRef]
- Wu, J.; Filutowicz, M. Hexahistidine (His6)-tag dependent protein dimerization: A cautionary tale. Acta Biochim. Pol. 1999, 46, 591–599. [Google Scholar] [CrossRef]
- Block, H.; Kubicek, J.; Labahn, J.; Roth, U.; Schäfer, F. Production and comprehensive quality control of recombinant human Interleukin-1β: A case study for a process development strategy. Protein Expr. Purif. 2008, 57, 244–254. [Google Scholar] [CrossRef]
- Gasparini, G.; Archer, I.; Jones, E.; Ashe, R. Scaling Up Biocatalysis Reactions in Flow Reactors. Org. Process. Res. Dev. 2012, 16, 1013–1016. [Google Scholar] [CrossRef]
- Gutmann, B.; Cantillo, D.; Kappe, C.O. Continuous-Flow Technology-A Tool for the Safe Manufacturing of Active Pharmaceutical Ingredients. Angew. Chem. Int. Ed. 2015, 54, 6688–6728. [Google Scholar] [CrossRef]
- Junior, I.I.; Flores, M.C.; Sutili, F.K.; Leite, S.G.F.; de M. Miranda, L.S.; Leal, I.C.R.; de Souza, R.O.M.A. Lipase-Catalyzed Monostearin Synthesis under Continuous Flow Conditions. Org. Process. Res. Dev. 2012, 16, 1098–1101. [Google Scholar] [CrossRef]
- Tamborini, L.; Romano, D.; Pinto, A.; Bertolani, A.; Molinari, F.; Conti, P. An efficient method for the lipase-catalysed resolution and in-line purification of racemic flurbiprofen in a continuous-flow reactor. J. Mol. Catal. B Enzym. 2012, 84, 78–82. [Google Scholar] [CrossRef]
- Lawrence, J.; O’Sullivan, B.; Lye, G.J.; Wohlgemuth, R.; Szita, N. Microfluidic multi-input reactor for biocatalytic synthesis using transketolase. J. Mol. Catal. B Enzym. 2013, 95, 111–117. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Babich, L.; Hartog, A.F.; van Hemert, L.J.C.; Rutjes, F.P.J.T.; Wever, R. Synthesis of Carbohydrates in a Continuous Flow Reactor by Immobilized Phosphatase and Aldolase. ChemSusChem 2012, 5, 2348–2353. [Google Scholar] [CrossRef] [PubMed]
- Zambelli, P.; Tamborini, L.; Cazzamalli, S.; Pinto, A.; Arioli, S.; Balzaretti, S.; Plou, F.J.; Fernandez-Arrojo, L.; Molinari, F.; Conti, P.; et al. An efficient continuous flow process for the synthesis of a non-conventional mixture of fructooligosaccharides. Food Chem. 2016, 190, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Tamborini, L.; Fernandes, P.; Paradisi, F.; Molinari, F. Flow Bioreactors as Complementary Tools for Biocatalytic Process Intensification. Trends Biotechnol. 2018, 36, 73–88. [Google Scholar] [CrossRef]
- Dall’Oglio, F.; Contente, M.L.; Conti, P.; Molinari, F.; Monfredi, D.; Pinto, A.; Romano, D.; Ubiali, D.; Tamborini, L.; Serra, I. Flow-based stereoselective reduction of ketones using an immobilized ketoreductase/glucose dehydrogenase mixed bed system. Catal. Commun. 2017, 93, 29–32. [Google Scholar] [CrossRef]
- Šalić, A.; Zelić, B. ADH-catalysed hexanol oxidation with fully integrated NADH regeneration performed in microreactors connected in series. RSC Adv. 2014, 4, 41714–41721. [Google Scholar] [CrossRef]
- Hanson, R.L.; Goldberg, S.; Goswami, A.; Tully, T.P.; Patel, R.N. Purification and Cloning of a Ketoreductase used for the Preparation of Chiral Alcohols. Adv. Synth. Catal. 2005, 347, 1073–1080. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Brady, D.; Bode, M.L. The Hitchhiker’s guide to biocatalysis: Recent advances in the use of enzymes in organic synthesis. Chem. Sci. 2020, 11, 2587–2605. [Google Scholar] [CrossRef]
- Zhu, D.; Yang, Y.; Hua, L. Stereoselective Enzymatic Synthesis of Chiral Alcohols with the Use of a Carbonyl Reductase fromCandidamagnoliaewith Anti-Prelog Enantioselectivity. J. Org. Chem. 2006, 71, 4202–4205. [Google Scholar] [CrossRef]
- Peschke, T.; Skoupi, M.; Burgahn, T.; Gallus, S.; Ahmed, I.; Rabe, K.S.; Niemeyer, C.M. Self-Immobilizing Fusion Enzymes for Compartmentalized Biocatalysis. ACS Catal. 2017, 7, 7866–7872. [Google Scholar] [CrossRef]
- Kalaitzakis, D.; Rozzell, J.D.; Kambourakis, S.; Smonou, I. Highly Stereoselective Reductions of α-Alkyl-1,3-diketones and α-Alkyl-β-keto Esters Catalyzed by Isolated NADPH-Dependent Ketoreductases. Org. Lett. 2005, 7, 4799–4801. [Google Scholar] [CrossRef] [PubMed]
- Nagy-Győr, L.; Abaházi, E.; Bódai, V.; Sátorhelyi, P.; Erdélyi, B.; Balogh-Weiser, D.; Paizs, C.; Hornyánszky, G.; Poppe, L. Co-immobilized Whole Cells with ω-Transaminase and Ketoreductase Activities for Continuous-Flow Cascade Reactions. ChemBioChem 2018, 19, 1845–1848. [Google Scholar] [CrossRef] [PubMed]
- Gröger, H.; Chamouleau, F.; Orologas, N.; Rollmann, C.; Drauz, K.; Hummel, W.; Weckbecker, A.; May, O. Enantioselective Reduction of Ketones with “Designer Cells” at High Substrate Concentrations: Highly Efficient Access to Functionalized Optically Active Alcohols. Angew. Chem. Int. Ed. 2006, 45, 5677–5681. [Google Scholar] [CrossRef] [PubMed]
- Rebroš, M.; Rosenberg, M.; Mlichová, Z.; Krištofíková, Ľ. Hydrolysis of sucrose by invertase entrapped in polyvinyl alcohol hydrogel capsules. Food Chem. 2007, 102, 784–787. [Google Scholar] [CrossRef]
- Ellis, R.J.; Minton, A.P. Join the crowd. Nature 2003, 425, 27–28. [Google Scholar] [CrossRef]
- Miyawaki, O.; Tatsuno, M. Thermodynamic analysis of alcohol effect on thermal stability of proteins. J. Biosci. Bioeng. 2011, 111, 198–203. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhuang, W.; Niu, H.; Ge, L.; Hernandez, B.V.; Wu, J.; Wang, K.; Liu, N.; Chen, Y.; Zhu, C.; et al. Affinity induced immobilization of adenylate cyclase from the crude cell lysate for ATP conversion. Colloids Surfaces B Biointerfaces 2018, 164, 155–164. [Google Scholar] [CrossRef]
- Vahidi, A.K.; Yang, Y.; Ngo, T.P.N.; Li, Z. Simple and Efficient Immobilization of Extracellular His-Tagged Enzyme Directly from Cell Culture Supernatant as Active and Recyclable Nanobiocatalyst: High-Performance Production of Biodiesel from Waste Grease. ACS Catal. 2015, 5, 3157–3161. [Google Scholar] [CrossRef]
- Vahidi, A.K.; Wang, Z.; Wong, W.S.Y.; Li, Z. Immobilization of O-acetylserine sulfhydrylase as a highly active and recyclable nanobiocatalyst: Efficient synthesis of β-pyrazol-1-yl-l-alanine. Catal. Sci. Technol. 2016, 6, 6286–6293. [Google Scholar] [CrossRef]
Sample Availability: Samples of enzymes ketoreductase and glucose dehydrogenase are available from the authors. |













© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plž, M.; Petrovičová, T.; Rebroš, M. Semi-Continuous Flow Biocatalysis with Affinity Co-Immobilized Ketoreductase and Glucose Dehydrogenase. Molecules 2020, 25, 4278. https://doi.org/10.3390/molecules25184278
Plž M, Petrovičová T, Rebroš M. Semi-Continuous Flow Biocatalysis with Affinity Co-Immobilized Ketoreductase and Glucose Dehydrogenase. Molecules. 2020; 25(18):4278. https://doi.org/10.3390/molecules25184278
Chicago/Turabian StylePlž, Michal, Tatiana Petrovičová, and Martin Rebroš. 2020. "Semi-Continuous Flow Biocatalysis with Affinity Co-Immobilized Ketoreductase and Glucose Dehydrogenase" Molecules 25, no. 18: 4278. https://doi.org/10.3390/molecules25184278
APA StylePlž, M., Petrovičová, T., & Rebroš, M. (2020). Semi-Continuous Flow Biocatalysis with Affinity Co-Immobilized Ketoreductase and Glucose Dehydrogenase. Molecules, 25(18), 4278. https://doi.org/10.3390/molecules25184278