
Structural Heterogeneities of the Ribosome: New Frontiers and Opportunities for Cryo-EM


Abstract
1. Introduction
2. The Multiple Sources of Heterogeneity in Ribosome Structures
2.1. Sequence and Structural Divergence across Species and Domains of Life
2.2. Consequences of Modifications at Single Sites
2.3. Heterogeneity within Cells and across Cell Types
2.4. Conformational Heterogeneity and Molecular Motion
3. Computational Challenges for Quantifying Heterogeneity from Cryo-EM Structures
3.1. Data Integration for Structural Comparison
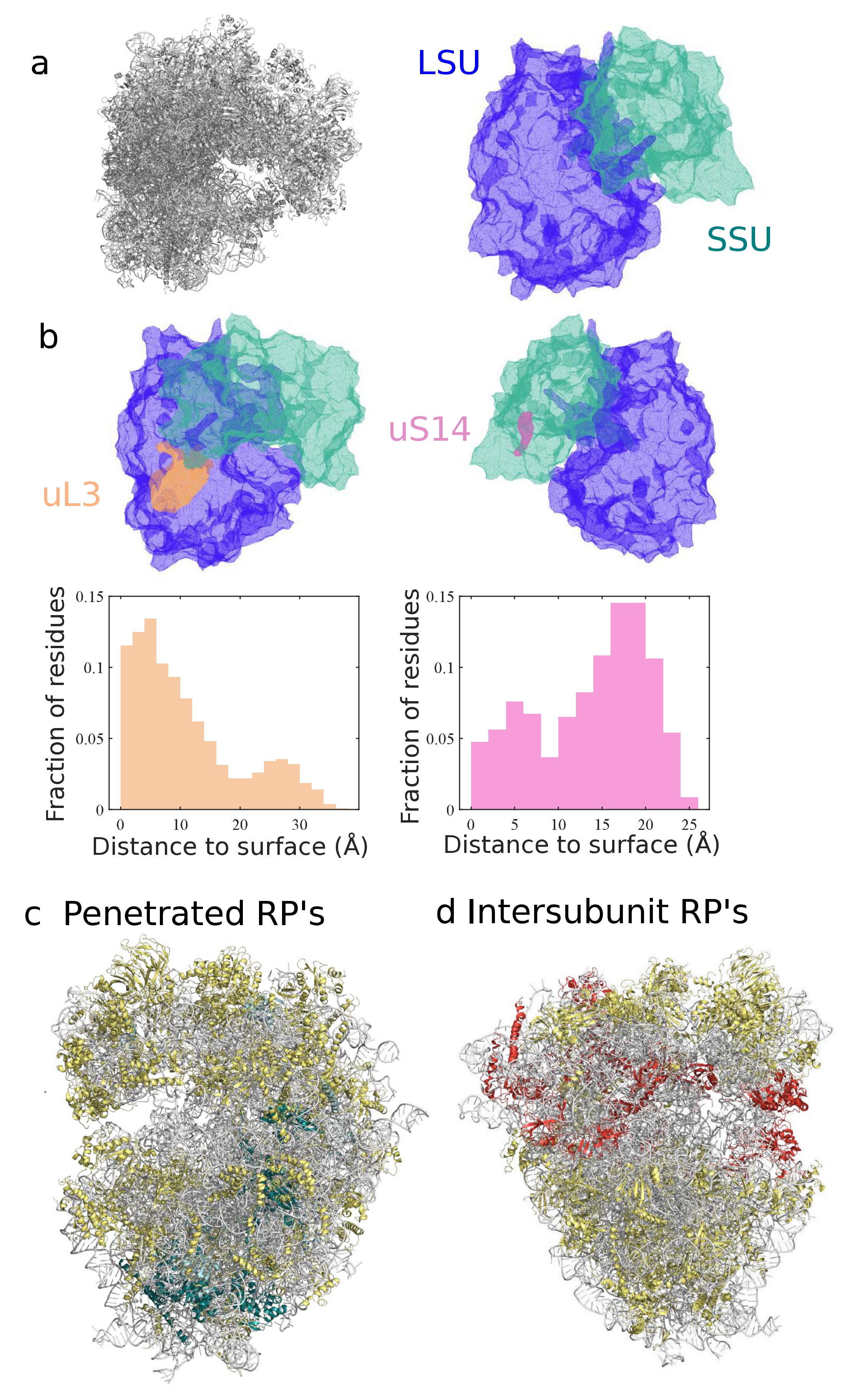
3.2. Classification and Comparison of Ribosomal Components
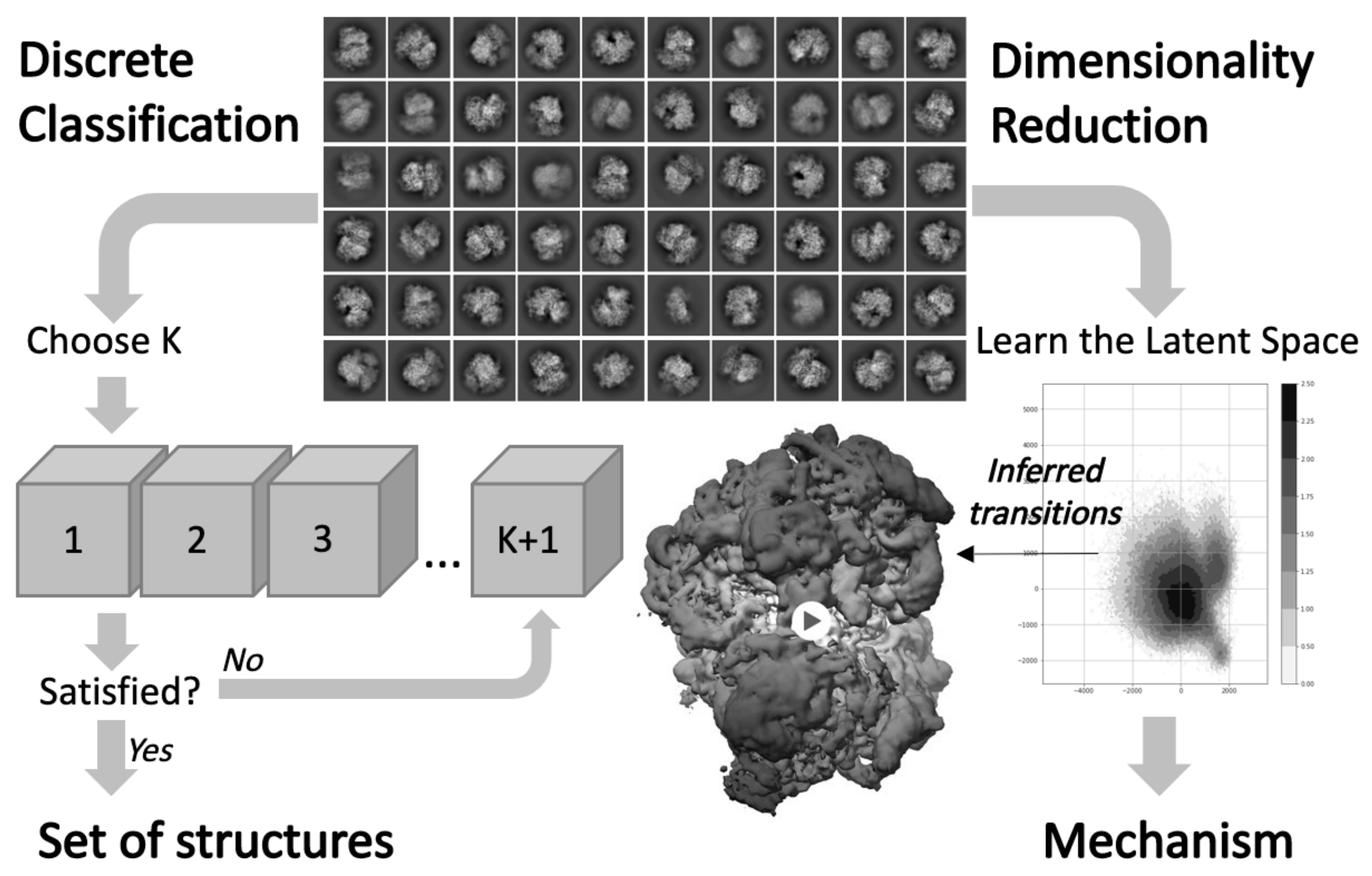
3.3. Investigating Conformational Heterogeneity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species/Organelles | Resolution | Reference | PDB Codename |
|---|---|---|---|
| E. coli (b) | 2.2 Å (EM) | Stojkovic et al. (2020) [119] | 6PJ6 |
| S. aureus (b) | 2.3 Å (EM) | Halfon et al. (2019) [50] | 6S0Z |
| T. cruzi (e) | 2.5 Å (EM) | Liu et al. (2016) [120] | 5T5H |
| S. cerevisea (e) | 2.6 Å (EM) | Tesina et al. (2020) [121] | 6T4Q |
| P. aeruginosa (b) | 2.8 Å (EM) | Halfon et al. (2019) [122] | 6SPB |
| H. sapiens (e) | 2.9 Å (EM) | Natchiar et al. (2017) [20] | 6EK0 |
| A. baumanii (b) | 2.9 Å (EM) | Morgan et al. (2020) [51] | 6V3D |
| L. donovani (e) | 2.9 Å (EM) | Zhang et al. (2016) [24] | 5T2A |
| Mitochondria (H. Sapiens) | 3.1 Å (EM) | Amunts et al. (2015) [33] | 3J9M |
| M. smegmatis (b) | 3.2 Å (EM) | Hentschel et al. (2017) [123] | 5O60 |
| P. falciparum (e) | 3.2 Å (EM) | Wong et al. (2014) [124] | 3J79 |
| T. gondii (e) | 3.2 Å (EM) | Li et al. (2017) [21] | 5XXB |
| T. b. brucei (e) | 3.3 Å (EM) | Saurer et al. (2019) [125] | 6SGB |
| M. tuberculosis (b) | 3.4 Å (EM) | Yang et al. (2017) [126] | 5V7Q |
| T. vaginalis (e) | 3.4 Å (EM) | Li et al. (2017) [21] | 5XY3 |
| C. thermophilum (e) | 3.5 Å (EM) | Cheng et al. (2019) [127] | 6RXU |
| K. lactis (e) | 3.6 Å (EM) | Huang et al. (2020) [128] | 6UZ7 |
| O. cuniculus (e) | 3.7 Å (EM) | Shanmuganathan et al. (2019) [129] | 6R6G |
| Chloroplast (Spinacia) | 3.8 Å (EM) | Ahmed et al. (2017) [130] | 5X8T |
| B. subtilis (b) | 3.8 Å (EM) | Beckert et al. (2017) [131] | 5NJT |
Author Contributions
Funding
Conflicts of Interest
References
- Wimberly, B.T.; Brodersen, D.E.; Clemons, W.M.; Morgan-Warren, R.J.; Carter, A.P.; Vonrhein, C.; Hartsch, T.; Ramakrishnan, V. Structure of the 30S ribosomal subunit. Nature 2000, 407, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Schluenzen, F.; Tocilj, A.; Zarivach, R.; Harms, J.; Gluehmann, M.; Janell, D.; Bashan, A.; Bartels, H.; Agmon, I.; Franceschi, F. Structure of functionally activated small ribosomal subunit at 3.3 Å resolution. Cell 2000, 102, 615–623. [Google Scholar] [CrossRef]
- Ban, N.; Nissen, P.; Hansen, J.; Moore, P.B.; Steitz, T.A. The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science 2000, 289, 905–920. [Google Scholar] [CrossRef] [PubMed]
- Earl, L.A.; Falconieri, V.; Milne, J.L.; Subramaniam, S. Cryo-EM: Beyond the microscope. Curr. Opin. Struct. Biol. 2017, 46, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Genuth, N.R.; Barna, M. The discovery of ribosome heterogeneity and its implications for gene regulation and organismal life. Mol. Cell 2018, 71, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Lyumkis, D. Challenges and opportunities in cryo-EM single-particle analysis. J. Biol. Chem. 2019, 294, 5181–5197. [Google Scholar] [CrossRef]
- Scheres, S.H. RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012, 180, 519–530. [Google Scholar] [CrossRef]
- Punjani, A.; Rubinstein, J.L.; Fleet, D.J.; Brubaker, M.A. cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 2017, 14, 290. [Google Scholar] [CrossRef]
- Sigworth, F.J. Principles of cryo-EM single-particle image processing. Microscopy 2016, 65, 57–67. [Google Scholar] [CrossRef]
- Melnikov, S.; Ben-Shem, A.; De Loubresse, N.G.; Jenner, L.; Yusupova, G.; Yusupov, M. One core, two shells: Bacterial and eukaryotic ribosomes. Nat. Struct. Mol. Biol. 2012, 19, 560. [Google Scholar] [CrossRef]
- Greber, B.J.; Boehringer, D.; Godinic-Mikulcic, V.; Crnkovic, A.; Ibba, M.; Weygand-Durasevic, I.; Ban, N. Cryo-EM structure of the archaeal 50S ribosomal subunit in complex with initiation factor 6 and implications for ribosome evolution. J. Mol. Biol. 2012, 418, 145–160. [Google Scholar] [CrossRef]
- Armache, J.P.; Anger, A.M.; Marquez, V.; Franckenberg, S.; Fröhlich, T.; Villa, E.; Berninghausen, O.; Thomm, M.; Arnold, G.J.; Beckmann, R. Promiscuous behaviour of archaeal ribosomal proteins: Implications for eukaryotic ribosome evolution. Nucleic Acids Res. 2013, 41, 1284–1293. [Google Scholar] [CrossRef]
- Ito, K. Regulatory Nascent Polypeptides; Springer Japan: Tokyo, Japan, 2014. [Google Scholar]
- Dao Duc, K.; Batra, S.S.; Bhattacharya, N.; Cate, J.H.; Song, Y.S. Differences in the path to exit the ribosome across the three domains of life. Nucleic Acids Res. 2019, 47, 4198–4210. [Google Scholar] [CrossRef]
- Watson, Z.L.; Ward, F.R.; Méheust, R.; Ad, O.; Schepartz, A.; Banfield, J.F.; Cate, J.H. Structure of the Bacterial Ribosome at 2 Å Resolution. bioRxiv 2020. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gillil, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Bernier, C.R.; Petrov, A.S.; Waterbury, C.C.; Jett, J.; Li, F.; Freil, L.E.; Xiong, X.; Wang, L.; Migliozzi, B.L.; Hershkovits, E. RiboVision suite for visualization and analysis of ribosomes. Faraday Discuss. 2014, 169, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Doris, S.M.; Smith, D.R.; Beamesderfer, J.N.; Raphael, B.J.; Nathanson, J.A.; Gerbi, S.A. Universal and domain-specific sequences in 23S–28S ribosomal RNA identified by computational phylogenetics. RNA 2015, 21, 1719–1730. [Google Scholar] [CrossRef] [PubMed]
- Fischer, N.; Neumann, P.; Konevega, A.L.; Bock, L.V.; Ficner, R.; Rodnina, M.V.; Stark, H. Structure of the E. coli ribosome–EF-Tu complex at <3 Å resolution by C s-corrected cryo-EM. Nature 2015, 520, 567. [Google Scholar]
- Natchiar, S.K.; Myasnikov, A.G.; Kratzat, H.; Hazemann, I.; Klaholz, B.P. Visualization of chemical modifications in the human 80S ribosome structure. Nature 2017, 551, 472–477. [Google Scholar] [CrossRef]
- Li, Z.; Guo, Q.; Zheng, L.; Ji, Y.; Xie, Y.T.; Lai, D.H.; Lun, Z.R.; Suo, X.; Gao, N. Cryo-EM structures of the 80S ribosomes from human parasites Trichomonas vaginalis and Toxoplasma gondii. Cell Res. 2017, 27, 1275. [Google Scholar] [CrossRef]
- Shalev-Benami, M.; Zhang, Y.; Matzov, D.; Halfon, Y.; Zackay, A.; Rozenberg, H.; Zimmerman, E.; Bashan, A.; Jaffe, C.L.; Yonath, A. 2.8-Å cryo-EM structure of the large ribosomal subunit from the eukaryotic parasite Leishmania. Cell Rep. 2016, 16, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Hashem, Y.; Des Georges, A.; Fu, J.; Buss, S.N.; Jossinet, F.; Jobe, A.; Zhang, Q.; Liao, H.Y.; Grassucci, R.A.; Bajaj, C. High-resolution cryo-electron microscopy structure of the Trypanosoma brucei ribosome. Nature 2013, 494, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lai, M.; Chang, W.; Yu, I.; Ding, K.; Mrazek, J.; Ng, H.L.; Yang, O.O.; Maslov, D.A.; Zhou, Z.H. Structures and stabilization of kinetoplastid-specific split rRNAs revealed by comparing leishmanial and human ribosomes. Nat. Commun. 2016, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Barandun, J.; Hunziker, M.; Vossbrinck, C.R.; Klinge, S. Evolutionary compaction and adaptation visualized by the structure of the dormant microsporidian ribosome. Nat. Microbiol. 2019, 4, 1798–1804. [Google Scholar] [CrossRef] [PubMed]
- Nikolaeva, D.D.; Gelfand, M.S.; Garushyants, S.K. Simplification of ribosomes in bacteria with tiny genomes. bioRxiv 2019, 755876. [Google Scholar] [CrossRef]
- Greber, B.J.; Ban, N. Structure and function of the mitochondrial ribosome. Annu. Rev. Biochem. 2016, 85, 103–132. [Google Scholar] [CrossRef]
- Soufari, H.; Waltz, F.; Parrot, C.; Durrieu, S.; Bochler, A.; Kuhn, L.; Sissler, M.; Hashem, Y. Structure of the full kinetoplastids mitoribosome and insight on its large subunit maturation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Tomal, A.; Kwasniak-Owczarek, M.; Janska, H. An Update on Mitochondrial Ribosome Biology: The Plant Mitoribosome in the Spotlight. Cells 2019, 8, 1562. [Google Scholar] [CrossRef]
- Waltz, F.; Soufari, H.; Bochler, A.; Giegé, P.; Hashem, Y. Cryo-EM structure of the RNA-rich plant mitochondrial ribosome. Nat. Plants 2020, 6, 377–383. [Google Scholar] [CrossRef]
- Desai, N.; Brown, A.; Amunts, A.; Ramakrishnan, V. The structure of the yeast mitochondrial ribosome. Science 2017, 355, 528–531. [Google Scholar] [CrossRef]
- Bieri, P.; Greber, B.J.; Ban, N. High-resolution structures of mitochondrial ribosomes and their functional implications. Curr. Opin. Struct. Biol. 2018, 49, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Amunts, A.; Brown, A.; Toots, J.; Scheres, S.H.; Ramakrishnan, V. The structure of the human mitochondrial ribosome. Science 2015, 348, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Waltz, F.; Nguyen, T.T.; Arrivé, M.; Bochler, A.; Chicher, J.; Hammann, P.; Kuhn, L.; Quadrado, M.; Mireau, H.; Hashem, Y. Small is big in Arabidopsis mitochondrial ribosome. Nat. Plants 2019, 5, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Petrov, A.S.; Wood, E.C.; Bernier, C.R.; Norris, A.M.; Brown, A.; Amunts, A. Structural patching fosters divergence of mitochondrial ribosomes. Mol. Biol. Evol. 2019, 36, 207–219. [Google Scholar] [CrossRef] [PubMed]
- Sagan, L. On the origin of mitosing cells. J. Theor. Biol. 1967, 14, 225–274. [Google Scholar] [CrossRef]
- D’Aquino, A.E.; Azim, T.; Aleksashin, N.A.; Hockenberry, A.J.; Krüger, A.; Jewett, M.C. Mutational characterization and mapping of the 70S ribosome active site. Nucleic Acids Res. 2020, 48, 2777–2789. [Google Scholar] [CrossRef]
- Kampen, K.R.; Sulima, S.O.; Vereecke, S.; De Keersmaecker, K. Hallmarks of ribosomopathies. Nucleic Acids Res. 2020, 48, 1013–1028. [Google Scholar] [CrossRef]
- Goudarzi, K.M.; Lindström, M.S. Role of ribosomal protein mutations in tumor development. Int. J. Oncol. 2016, 48, 1313–1324. [Google Scholar] [CrossRef]
- Subramaniam, S.; Earl, L.A.; Falconieri, V.; Milne, J.L.; Egelman, E.H. Resolution advances in cryo-EM enable application to drug discovery. Curr. Opin. Struct. Biol. 2016, 41, 194–202. [Google Scholar] [CrossRef]
- Gilles, A.; Frechin, L.; Natchiar, K.; Biondani, G.; Loeffelholz, O.V.; Holvec, S.; Malaval, J.L.; Winum, J.Y.; Klaholz, B.P.; Peyron, J.F. Targeting the human 80S ribosome in cancer: From structure to function and drug design for innovative adjuvant therapeutic strategies. Cells 2020, 9, 629. [Google Scholar] [CrossRef]
- Li, W.; Ward, F.R.; McClure, K.F.; Chang, S.T.L.; Montabana, E.; Liras, S.; Dullea, R.G.; Cate, J.H. Structural basis for selective stalling of human ribosome nascent chain complexes by a drug-like molecule. Nat. Struct. Mol. Biol. 2019, 26, 501–509. [Google Scholar] [CrossRef]
- Myasnikov, A.G.; Natchiar, S.K.; Nebout, M.; Hazemann, I.; Imbert, V.; Khatter, H.; Peyron, J.F.; Klaholz, B.P. Structure–function insights reveal the human ribosome as a cancer target for antibiotics. Nat. Commun. 2016, 7, 1–8. [Google Scholar] [CrossRef]
- De Loubresse, N.G.; Prokhorova, I.; Holtkamp, W.; Rodnina, M.V.; Yusupova, G.; Yusupov, M. Structural basis for the inhibition of the eukaryotic ribosome. Nature 2014, 513, 517–522. [Google Scholar] [CrossRef]
- Polikanov, Y.S.; Aleksashin, N.A.; Beckert, B.; Wilson, D.N. The mechanisms of action of ribosome-targeting peptide antibiotics. Front. Mol. Biosci. 2018, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Laslop, N.; Mankin, A.S. How macrolide antibiotics work. Trends Biochem. Sci. 2018, 43, 668–684. [Google Scholar] [CrossRef]
- Cocozaki, A.I.; Altman, R.B.; Huang, J.; Buurman, E.T.; Kazmirski, S.L.; Doig, P.; Prince, D.B.; Blanchard, S.C.; Cate, J.H.; Ferguson, A.D. Resistance mutations generate divergent antibiotic susceptibility profiles against translation inhibitors. Proc. Natl. Acad. Sci. USA 2016, 113, 8188–8193. [Google Scholar] [CrossRef]
- Long, K.S.; Poehlsgaard, J.; Hansen, L.H.; Hobbie, S.N.; Böttger, E.C.; Vester, B. Single 23S rRNA mutations at the ribosomal peptidyl transferase centre confer resistance to valnemulin and other antibiotics in Mycobacterium smegmatis by perturbation of the drug binding pocket. Mol. Microbiol. 2009, 71, 1218–1227. [Google Scholar] [CrossRef]
- Wilson, D.N. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 2014, 12, 35–48. [Google Scholar] [CrossRef]
- Halfon, Y.; Matzov, D.; Eyal, Z.; Bashan, A.; Zimmerman, E.; Kjeldgaard, J.; Ingmer, H.; Yonath, A. Exit tunnel modulation as resistance mechanism of S. aureus erythromycin resistant mutant. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef]
- Morgan, C.E.; Huang, W.; Rudin, S.D.; Taylor, D.J.; Kirby, J.E.; Bonomo, R.A.; Edward, W.Y. Cryo-electron Microscopy Structure of the Acinetobacter baumannii 70S Ribosome and Implications for New Antibiotic Development. Mbio 2020, 11, e03117–e03119. [Google Scholar] [CrossRef]
- Pichkur, E.B.; Paleskava, A.; Tereshchenkov, A.G.; Kasatsky, P.; Komarova, E.S.; Shiriaev, D.I.; Bogdanov, A.A.; Dontsova, O.A.; Osterman, I.A.; Sergiev, P.V. Insights into the improved macrolide inhibitory activity from the high-resolution cryo-EM structure of dirithromycin bound to the E. coli 70S ribosome. RNA 2020, 26, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Travin, D.Y.; Watson, Z.L.; Metelev, M.; Ward, F.R.; Osterman, I.A.; Khven, I.M.; Khabibullina, N.F.; Serebryakova, M.; Mergaert, P.; Polikanov, Y.S. Structure of ribosome-bound azole-modified peptide phazolicin rationalizes its species-specific mode of bacterial translation inhibition. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Khabibullina, N.F.; Tereshchenkov, A.G.; Komarova, E.S.; Syroegin, E.A.; Shiriaev, D.I.; Paleskava, A.; Kartsev, V.G.; Bogdanov, A.A.; Konevega, A.L.; Dontsova, O.A. Structure of dirithromycin bound to the bacterial ribosome suggests new ways for rational improvement of macrolides. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [PubMed]
- Sauert, M.; Temmel, H.; Moll, I. Heterogeneity of the translational machinery: Variations on a common theme. Biochimie 2015, 114, 39–47. [Google Scholar] [CrossRef]
- Ferretti, M.B.; Karbstein, K. Does functional specialization of ribosomes really exist? RNA 2019, 25, 521–538. [Google Scholar]
- Slavov, N.; Semrau, S.; Airoldi, E.; Budnik, B.; van Oudenaarden, A. Differential stoichiometry among core ribosomal proteins. Cell Rep. 2015, 13, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.W.; Brandl, C.; Haubenreisser, O.; Wimmer, B.; Weber, M.; Karl, T.; Klausegger, A.; Breitenbach, M.; Hintner, H.; von der Haar, T. Specialized yeast ribosomes: A customized tool for selective mRNA translation. PLoS ONE 2013, 8, e67609. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Fujii, K.; Kovary, K.M.; Genuth, N.R.; Röst, H.L.; Teruel, M.N.; Barna, M. Heterogeneous ribosomes preferentially translate distinct subpools of mRNAs genome-wide. Mol. Cell 2017, 67, 71–83. [Google Scholar] [CrossRef]
- Farley-Barnes, K.I.; Ogawa, L.M.; Baserga, S.J. Ribosomopathies: Old concepts, new controversies. Trends Genet. 2019, 35, 754–767. [Google Scholar] [CrossRef]
- Lilleorg, S.; Reier, K.; Pulk, A.; Liiv, A.; Tammsalu, T.; Peil, L.; Cate, J.H.; Remme, J. Bacterial ribosome heterogeneity: Changes in ribosomal protein composition during transition into stationary growth phase. Biochimie 2019, 156, 169–180. [Google Scholar] [CrossRef]
- Middleton, S.A.; Eberwine, J.; Kim, J. Comprehensive catalog of dendritically localized mRNA isoforms from sub-cellular sequencing of single mouse neurons. BMC Biol. 2019, 17, 5. [Google Scholar] [CrossRef] [PubMed]
- Mazaré, N.; Oudart, M.; Moulard, J.; Cheung, G.; Tortuyaux, R.; Mailly, P.; Mazaud, D.; Bemelmans, A.P.; Boulay, A.C.; Blugeon, C. Local translation in perisynaptic astrocytic processes is specific and regulated by fear conditioning. bioRxiv 2020. [Google Scholar] [CrossRef]
- Shigeoka, T.; Koppers, M.; Wong, H.H.W.; Lin, J.Q.; Cagnetta, R.; Dwivedy, A.; de Freitas Nascimento, J.; van Tartwijk, F.W.; Ströhl, F.; Cioni, J.M. On-site ribosome remodeling by locally synthesized ribosomal proteins in axons. Cell Rep. 2019, 29, 3605–3619. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.; Baumeister, W. Cryo-electron tomography: Can it reveal the molecular sociology of cells in atomic detail? Trends Cell Biol. 2016, 26, 825–837. [Google Scholar] [CrossRef]
- Gold, V.A.; Chroscicki, P.; Bragoszewski, P.; Chacinska, A. Visualization of cytosolic ribosomes on the surface of mitochondria by electron cryo-tomography. EMBO Rep. 2017, 18, 1786–1800. [Google Scholar] [CrossRef]
- O’Reilly, F.J.; Xue, L.; Graziadei, A.; Sinn, L.; Lenz, S.; Tegunov, D.; Blötz, C.; Hagen, W.J.; Cramer, P.; Stülke, J. In-cell architecture of an actively transcribing-translating expressome. bioRxiv 2020, 369, 554–557. [Google Scholar] [CrossRef]
- Robinson, C.V.; Sali, A.; Baumeister, W. The molecular sociology of the cell. Nature 2007, 450, 973–982. [Google Scholar] [CrossRef]
- Schaffer, M.; Pfeffer, S.; Mahamid, J.; Kleindiek, S.; Laugks, T.; Albert, S.; Engel, B.D.; Rummel, A.; Smith, A.J.; Baumeister, W. A cryo-FIB lift-out technique enables molecular-resolution cryo-ET within native Caenorhabditis elegans tissue. Nat. Methods 2019, 16, 757–762. [Google Scholar] [CrossRef]
- Tegunov, D.; Xue, L.; Dienemann, C.; Cramer, P.; Mahamid, J. Multi-particle cryo-EM refinement with M visualizes ribosome-antibiotic complex at 3.7 Å inside cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Brown, A.; Shao, S. Ribosomes and cryo-EM: A duet. Curr. Opin. Struct. Biol. 2018, 52, 1–7. [Google Scholar] [CrossRef]
- Liu, Q.; Fredrick, K. Intersubunit bridges of the bacterial ribosome. J. Mol. Biol. 2016, 428, 2146–2164. [Google Scholar] [CrossRef] [PubMed]
- Noeske, J.; Cate, J.H. Structural basis for protein synthesis: Snapshots of the ribosome in motion. Curr. Opin. Struct. Biol. 2012, 22, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Behrmann, E.; Loerke, J.; Budkevich, T.V.; Yamamoto, K.; Schmidt, A.; Penczek, P.A.; Vos, M.R.; Bürger, J.; Mielke, T.; Scheerer, P. Structural snapshots of actively translating human ribosomes. Cell 2015, 161, 845–857. [Google Scholar] [CrossRef]
- Loveland, A.B.; Demo, G.; Korostelev, A.A. Cryo-EM of elongating ribosome with EF-Tu• GTP elucidates tRNA proofreading. Nature 2020, 584, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Kaledhonkar, S.; Fu, Z.; Caban, K.; Li, W.; Chen, B.; Sun, M.; Gonzalez, R.L.; Frank, J. Late steps in bacterial translation initiation visualized using time-resolved cryo-EM. Nature 2019, 570, 400–404. [Google Scholar] [CrossRef]
- Fu, Z.; Indrisiunaite, G.; Kaledhonkar, S.; Shah, B.; Sun, M.; Chen, B.; Grassucci, R.A.; Ehrenberg, M.; Frank, J. The structural basis for release-factor activation during translation termination revealed by time-resolved cryogenic electron microscopy. Nat. Commun. 2019, 10, 1–7. [Google Scholar] [CrossRef]
- Adio, S.; Sharma, H.; Senyushkina, T.; Karki, P.; Maracci, C.; Wohlgemuth, I.; Holtkamp, W.; Peske, F.; Rodnina, M.V. Dynamics of ribosomes and release factors during translation termination in E. coli. Elife 2018, 7, e34252. [Google Scholar] [CrossRef]
- Razi, A.; Britton, R.A.; Ortega, J. The impact of recent improvements in cryo-electron microscopy technology on the understanding of bacterial ribosome assembly. Nucleic Acids Res. 2017, 45, 1027–1040. [Google Scholar] [CrossRef]
- Klinge, S.; Woolford, J.L. Ribosome assembly coming into focus. Nat. Rev. Mol. Cell Biol. 2019, 20, 116–131. [Google Scholar] [CrossRef]
- Bock, L.V.; Blau, C.; Schröder, G.F.; Davydov, I.I.; Fischer, N.; Stark, H.; Rodnina, M.V.; Vaiana, A.C.; Grubmüller, H. Energy barriers and driving forces in tRNA translocation through the ribosome. Nat. Struct. Mol. Biol. 2013, 20, 1390–1396. [Google Scholar] [CrossRef]
- Agirrezabala, X.; Liao, H.Y.; Schreiner, E.; Fu, J.; Ortiz-Meoz, R.F.; Schulten, K.; Green, R.; Frank, J. Structural characterization of mRNA-tRNA translocation intermediates. Proc. Natl. Acad. Sci. USA 2012, 109, 6094–6099. [Google Scholar] [CrossRef] [PubMed]
- Hussain, T.; Llácer, J.L.; Wimberly, B.T.; Kieft, J.S.; Ramakrishnan, V. Large-scale movements of IF3 and tRNA during bacterial translation initiation. Cell 2016, 167, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Murray, J.; Brown, A.; Taunton, J.; Ramakrishnan, V.; Hegde, R.S. Decoding mammalian ribosome-mRNA states by translational GTPase complexes. Cell 2016, 167, 1229–1240. [Google Scholar] [CrossRef] [PubMed]
- Frank, J. Time-resolved cryo-electron microscopy: Recent progress. J. Struct. Biol. 2017, 200, 303–306. [Google Scholar] [CrossRef]
- Sanbonmatsu, K.Y. Large-scale simulations of nucleoprotein complexes: Ribosomes, nucleosomes, chromatin, chromosomes and CRISPR. Curr. Opin. Struct. Biol. 2019, 55, 104–113. [Google Scholar] [CrossRef]
- Bock, L.V.; Kolář, M.H.; Grubmüller, H. Molecular simulations of the ribosome and associated translation factors. Curr. Opin. Struct. Biol. 2018, 49, 27–35. [Google Scholar] [CrossRef]
- Larsen, K.P.; Choi, J.; Prabhakar, A.; Puglisi, E.V.; Puglisi, J.D. Relating structure and dynamics in RNA biology. Cold Spring Harb. Perspect. Biol. 2019, 11, a032474. [Google Scholar] [CrossRef]
- Nakane, T.; Kimanius, D.; Lindahl, E.; Scheres, S.H. Characterisation of molecular motions in cryo-EM single-particle data by multi-body refinement in RELION. Elife 2018, 7, e36861. [Google Scholar] [CrossRef]
- Punjani, A.; Fleet, D.J. 3D Variability Analysis: Directly resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM images. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhong, E.D.; Bepler, T.; Davis, J.H.; Berger, B. Reconstructing continuous distributions of 3D protein structure from cryo-EM images. In Proceedings of the International Conference on Learning Representations (ICLR), Addis Ababa, Ethiopia, 26 April–1 May 2020. [Google Scholar]
- Ban, N.; Beckmann, R.; Cate, J.H.; Dinman, J.D.; Dragon, F.; Ellis, S.R.; Lafontaine, D.L.; Lindahl, L.; Liljas, A.; Lipton, J.M. A new system for naming ribosomal proteins. Curr. Opin. Struct. Biol. 2014, 24, 165–169. [Google Scholar] [CrossRef]
- Van De Waterbeemd, M.; Tamara, S.; Fort, K.L.; Damoc, E.; Franc, V.; Bieri, P.; Itten, M.; Makarov, A.; Ban, N.; Heck, A.J. Dissecting ribosomal particles throughout the kingdoms of life using advanced hybrid mass spectrometry methods. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Jarasch, A.; Dziuk, P.; Becker, T.; Armache, J.P.; Hauser, A.; Wilson, D.N.; Beckmann, R. The DARC site: A database of aligned ribosomal complexes. Nucleic Acids Res. 2012, 40, D495–D500. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Da Silva, W.M.; Wercelens, P.; Walter, M.E.M.; Holanda, M.; Brígido, M. Graph databases in molecular biology. In Proceedings of the Brazilian Symposium on Bioinformatics, Niterói, Brazil, 30 October–1 November 2018; Springer: Cham, Switzerland, 2018; pp. 50–57. [Google Scholar]
- Orengo, C.; Velankar, S.; Wodak, S.; Zoete, V.; Bonvin, A.M.; Elofsson, A.; Feenstra, K.A.; Gerloff, D.L.; Hamelryck, T.; Hancock, J.M. A community proposal to integrate structural bioinformatics activities in ELIXIR (3D-Bioinfo Community). F1000Research 2020, 9, 278. [Google Scholar] [CrossRef] [PubMed]
- Melnikov, S.; Manakongtreecheep, K.; Söll, D. Revising the structural diversity of ribosomal proteins across the three domains of life. Mol. Biol. Evol. 2018, 35, 1588–1598. [Google Scholar] [CrossRef]
- Wilson, D.N.; Cate, J.H.D. The structure and function of the eukaryotic ribosome. Cold Spring Harb. Perspect. Biol. 2012, 4, a011536. [Google Scholar] [CrossRef]
- Kim, D.S.; Ryu, J.; Cho, Y.; Lee, M.; Cha, J.; Song, C.; Kim, S.W.; Laskowski, R.A.; Sugihara, K.; Bhak, J. MGOS: A library for molecular geometry and its operating system. Comput. Phys. Commun. 2019, 251, 107101. [Google Scholar] [CrossRef]
- Manak, M.; Zemek, M.; Szkandera, J.; Kolingerova, I.; Papaleo, E.; Lambrughi, M. Hybrid Voronoi diagrams, their computation and reduction for applications in computational biochemistry. J. Mol. Graph. Model. 2017, 74, 225–233. [Google Scholar] [CrossRef]
- Wilson, J.A.; Bender, A.; Kaya, T.; Clemons, P.A. Alpha shapes applied to molecular shape characterization exhibit novel properties compared to established shape descriptors. J. Chem. Inf. Model. 2009, 49, 2231–2241. [Google Scholar] [CrossRef]
- Seddon, M.P.; Cosgrove, D.A.; Packer, M.J.; Gillet, V.J. Alignment-Free Molecular Shape Comparison Using Spectral Geometry: The Framework. J. Chem. Inf. Model. 2018, 59, 98–116. [Google Scholar] [CrossRef]
- Pravda, L.; Sehnal, D.; Toušek, D.; Navrátilová, V.; Bazgier, V.; Berka, K.; Svobodová Vařeková, R.; Koča, J.; Otyepka, M. MOLEonline: A web-based tool for analyzing channels, tunnels and pores (2018 update). Nucleic Acids Res. 2018, 46, W368–W373. [Google Scholar] [CrossRef]
- Fedyukina, D.V.; Jennaro, T.S.; Cavagnero, S. Charge segregation and low hydrophobicity are key features of ribosomal proteins from different organisms. J. Biol. Chem. 2014, 289, 6740–6750. [Google Scholar] [CrossRef]
- Dao Duc, K.; Song, Y.S. The impact of ribosomal interference, codon usage, and exit tunnel interactions on translation elongation rate variation. PLoS Genet. 2018, 14, e1007166. [Google Scholar] [CrossRef] [PubMed]
- Nissley, D.A.; Vu, Q.V.; Trovato, F.; Ahmed, N.; Jiang, Y.; Li, M.S.; O’Brien, E.P. Electrostatic interactions govern extreme nascent protein ejection times from ribosomes and can delay ribosome recycling. J. Am. Chem. Soc. 2020, 142, 6103–6110. [Google Scholar] [CrossRef]
- Kudva, R.; Tian, P.; Pardo-Avila, F.; Carroni, M.; Best, R.B.; Bernstein, H.D.; Von Heijne, G. The shape of the bacterial ribosome exit tunnel affects cotranslational protein folding. Elife 2018, 7, e36326. [Google Scholar] [CrossRef]
- Lyumkis, D.; Brilot, A.F.; Theobald, D.L.; Grigorieff, N. Likelihood-based classification of cryo-EM images using FREALIGN. J. Struct. Biol. 2013, 183, 377–388. [Google Scholar] [CrossRef]
- Scheres, S.H. Processing of structurally heterogeneous cryo-EM data in RELION. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 579, pp. 125–157. [Google Scholar]
- Penczek, P.A.; Kimmel, M.; Spahn, C.M. Identifying conformational states of macromolecules by eigen-analysis of resampled cryo-EM images. Structure 2011, 19, 1582–1590. [Google Scholar] [CrossRef] [PubMed]
- Andén, J.; Katsevich, E.; Singer, A. Covariance estimation using conjugate gradient for 3D classification in cryo-EM. In Proceedings of the 2015 IEEE 12th International Symposium on Biomedical Imaging (ISBI), Brooklyn Bridge, NY, USA, 16–19 April 2015; pp. 200–204. [Google Scholar]
- Ecoffet, A.; Poitevin, F.; Dao Duc, K. MorphOT: Transport-based interpolation between EM maps with UCSF ChimeraX. bioRxiv 2020. [Google Scholar] [CrossRef]
- Tek, A.; Korostelev, A.A.; Flores, S.C. MMB-GUI: A fast morphing method demonstrates a possible ribosomal tRNA translocation trajectory. Nucleic Acids Res. 2016, 44, 95–105. [Google Scholar] [CrossRef]
- Tagare, H.D.; Kucukelbir, A.; Sigworth, F.J.; Wang, H.; Rao, M. Directly reconstructing principal components of heterogeneous particles from cryo-EM images. J. Struct. Biol. 2015, 191, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Frank, J.; Ourmazd, A. Continuous changes in structure mapped by manifold embedding of single-particle data in cryo-EM. Methods 2016, 100, 61–67. [Google Scholar] [CrossRef]
- Moscovich, A.; Halevi, A.; Andén, J.; Singer, A. Cryo-EM reconstruction of continuous heterogeneity by Laplacian spectral volumes. Inverse Probl. 2020, 36, 024003. [Google Scholar] [CrossRef] [PubMed]
- Miolane, N.; Poitevin, F.; Li, Y.T.; Holmes, S. Estimation of orientation and camera parameters from cryo-electron microscopy images with variational autoencoders and generative adversarial networks. In Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition Workshops, Seattle, WA, USA, 16–18 June 2020; pp. 970–971. [Google Scholar]
- Gupta, H.; McCann, M.T.; Donati, L.; Unser, M. CryoGAN: A New Reconstruction Paradigm for Single-particle Cryo-EM Via Deep Adversarial Learning. BioRxiv 2020. [Google Scholar] [CrossRef]
- Stojković, V.; Myasnikov, A.G.; Young, I.D.; Frost, A.; Fraser, J.S.; Fujimori, D.G. Assessment of the nucleotide modifications in the high-resolution cryo-electron microscopy structure of the Escherichia coli 50S subunit. Nucleic Acids Res. 2020, 48, 2723–2732. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Gutierrez-Vargas, C.; Wei, J.; Grassucci, R.A.; Ramesh, M.; Espina, N.; Sun, M.; Tutuncuoglu, B.; Madison-Antenucci, S.; Woolford, J.L. Structure and assembly model for the Trypanosoma cruzi 60S ribosomal subunit. Proc. Natl. Acad. Sci. USA 2016, 113, 12174–12179. [Google Scholar] [CrossRef] [PubMed]
- Tesina, P.; Lessen, L.N.; Buschauer, R.; Cheng, J.; Wu, C.C.C.; Berninghausen, O.; Buskirk, A.R.; Becker, T.; Beckmann, R.; Green, R. Molecular mechanism of translational stalling by inhibitory codon combinations and poly (A) tracts. EMBO J. 2020, 39, e103365. [Google Scholar] [CrossRef]
- Halfon, Y.; Jimenez-Fernandez, A.; La Rosa, R.; Portero, R.E.; Johansen, H.K.; Matzov, D.; Eyal, Z.; Bashan, A.; Zimmerman, E.; Belousoff, M. Structure of Pseudomonas aeruginosa ribosomes from an aminoglycoside-resistant clinical isolate. Proc. Natl. Acad. Sci. USA 2019, 116, 22275–22281. [Google Scholar] [CrossRef]
- Hentschel, J.; Burnside, C.; Mignot, I.; Leibundgut, M.; Boehringer, D.; Ban, N. The complete structure of the mycobacterium smegmatis 70S ribosome. Cell Rep. 2017, 20, 149–160. [Google Scholar] [CrossRef]
- Wong, W.; Bai, X.c.; Brown, A.; Fernandez, I.S.; Hanssen, E.; Condron, M.; Tan, Y.H.; Baum, J.; Scheres, S.H. Cryo-EM structure of the Plasmodium falciparum 80S ribosome bound to the anti-protozoan drug emetine. Elife 2014, 3, e03080. [Google Scholar] [CrossRef]
- Saurer, M.; Ramrath, D.J.F.; Niemann, M.; Calderaro, S.; Prange, C.; Mattei, S.; Scaiola, A.; Leitner, A.; Bieri, P.; Horn, E.K.; et al. Mitoribosomal small subunit biogenesis in trypanosomes involves an extensive assembly machinery. Science 2019, 365, 1144–1149. [Google Scholar] [CrossRef]
- Yang, K.; Chang, J.Y.; Cui, Z.; Li, X.; Meng, R.; Duan, L.; Thongchol, J.; Jakana, J.; Huwe, C.M.; Sacchettini, J.C. Structural insights into species-specific features of the ribosome from the human pathogen Mycobacterium tuberculosis. Nucleic Acids Res. 2017, 45, 10884–10894. [Google Scholar] [CrossRef]
- Cheng, J.; Baßler, J.; Fischer, P.; Lau, B.; Kellner, N.; Kunze, R.; Griesel, S.; Kallas, M.; Berninghausen, O.; Strauss, D. Thermophile 90S pre-ribosome structures reveal the reverse order of co-transcriptional 18S rRNA subdomain integration. Mol. Cell 2019, 75, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.Y.; Fernández, I.S. Long-range interdomain communications in eIF5B regulate GTP hydrolysis and translation initiation. Proc. Natl. Acad. Sci. USA 2020, 117, 1429–1437. [Google Scholar] [CrossRef]
- Shanmuganathan, V.; Schiller, N.; Magoulopoulou, A.; Cheng, J.; Braunger, K.; Cymer, F.; Berninghausen, O.; Beatrix, B.; Kohno, K.; von Heijne, G.; et al. Structural and mutational analysis of the ribosome-arresting human XBP1u. Elife 2019, 8, e46267. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.; Shi, J.; Bhushan, S. Unique localization of the plastid-specific ribosomal proteins in the chloroplast ribosome small subunit provides mechanistic insights into the chloroplastic translation. Nucleic Acids Res. 2017, 45, 8581–8595. [Google Scholar] [CrossRef] [PubMed]
- Beckert, B.; Abdelshahid, M.; Schäfer, H.; Steinchen, W.; Arenz, S.; Berninghausen, O.; Beckmann, R.; Bange, G.; Turgay, K.; Wilson, D.N. Structure of the Bacillus subtilis hibernating 100S ribosome reveals the basis for 70S dimerization. EMBO J. 2017, 36, 2061–2072. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poitevin, F.; Kushner, A.; Li, X.; Dao Duc, K. Structural Heterogeneities of the Ribosome: New Frontiers and Opportunities for Cryo-EM. Molecules 2020, 25, 4262. https://doi.org/10.3390/molecules25184262
Poitevin F, Kushner A, Li X, Dao Duc K. Structural Heterogeneities of the Ribosome: New Frontiers and Opportunities for Cryo-EM. Molecules. 2020; 25(18):4262. https://doi.org/10.3390/molecules25184262
Chicago/Turabian StylePoitevin, Frédéric, Artem Kushner, Xinpei Li, and Khanh Dao Duc. 2020. "Structural Heterogeneities of the Ribosome: New Frontiers and Opportunities for Cryo-EM" Molecules 25, no. 18: 4262. https://doi.org/10.3390/molecules25184262
APA StylePoitevin, F., Kushner, A., Li, X., & Dao Duc, K. (2020). Structural Heterogeneities of the Ribosome: New Frontiers and Opportunities for Cryo-EM. Molecules, 25(18), 4262. https://doi.org/10.3390/molecules25184262