
Identification of Novel Src Inhibitors: Pharmacophore-Based Virtual Screening, Molecular Docking and Molecular Dynamics Simulations


Abstract
1. Introduction
2. Results and Discussion
2.1. Preparation of Chemical Database
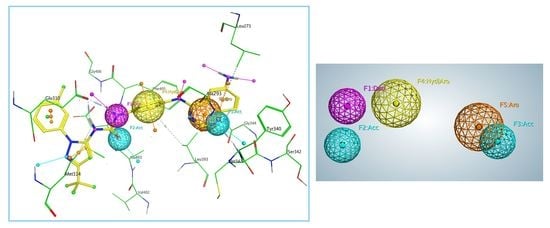
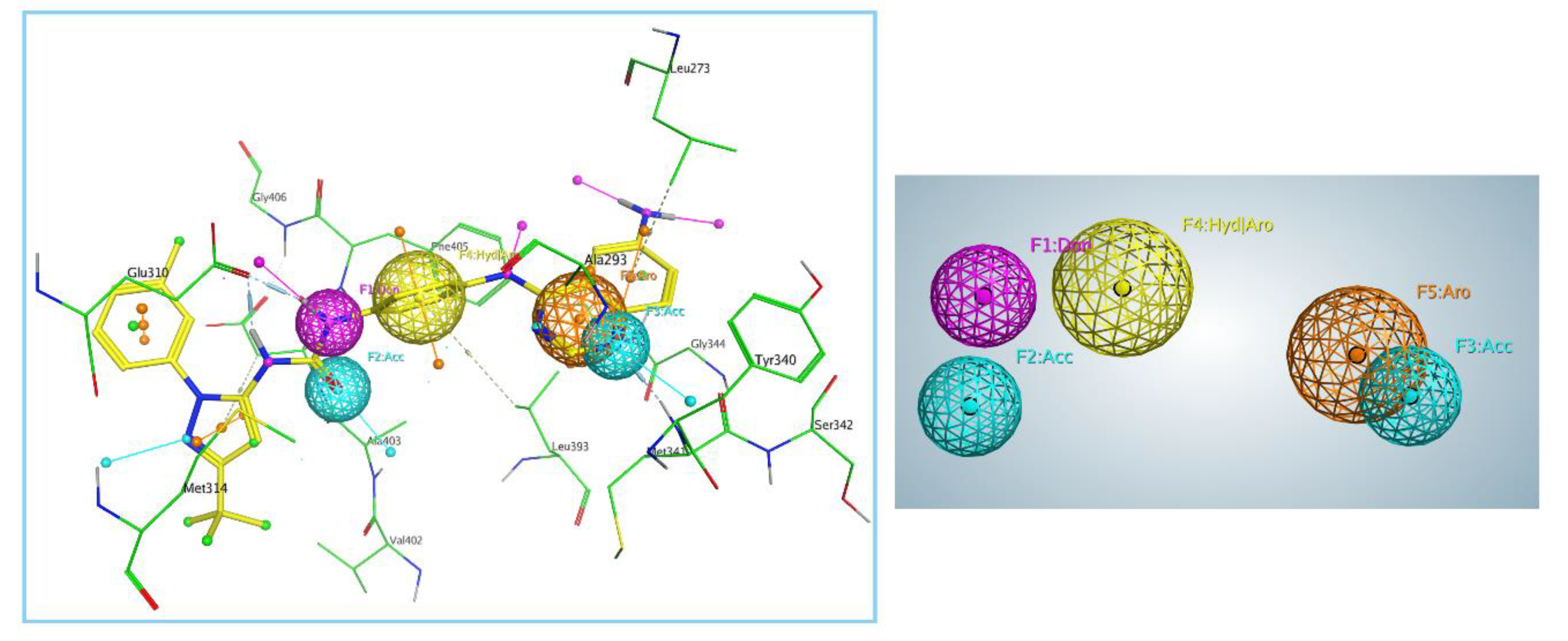
2.2. Generation and Validation of Pharmacophore Model
2.3. Pharmacophore-Based Virtual Screening
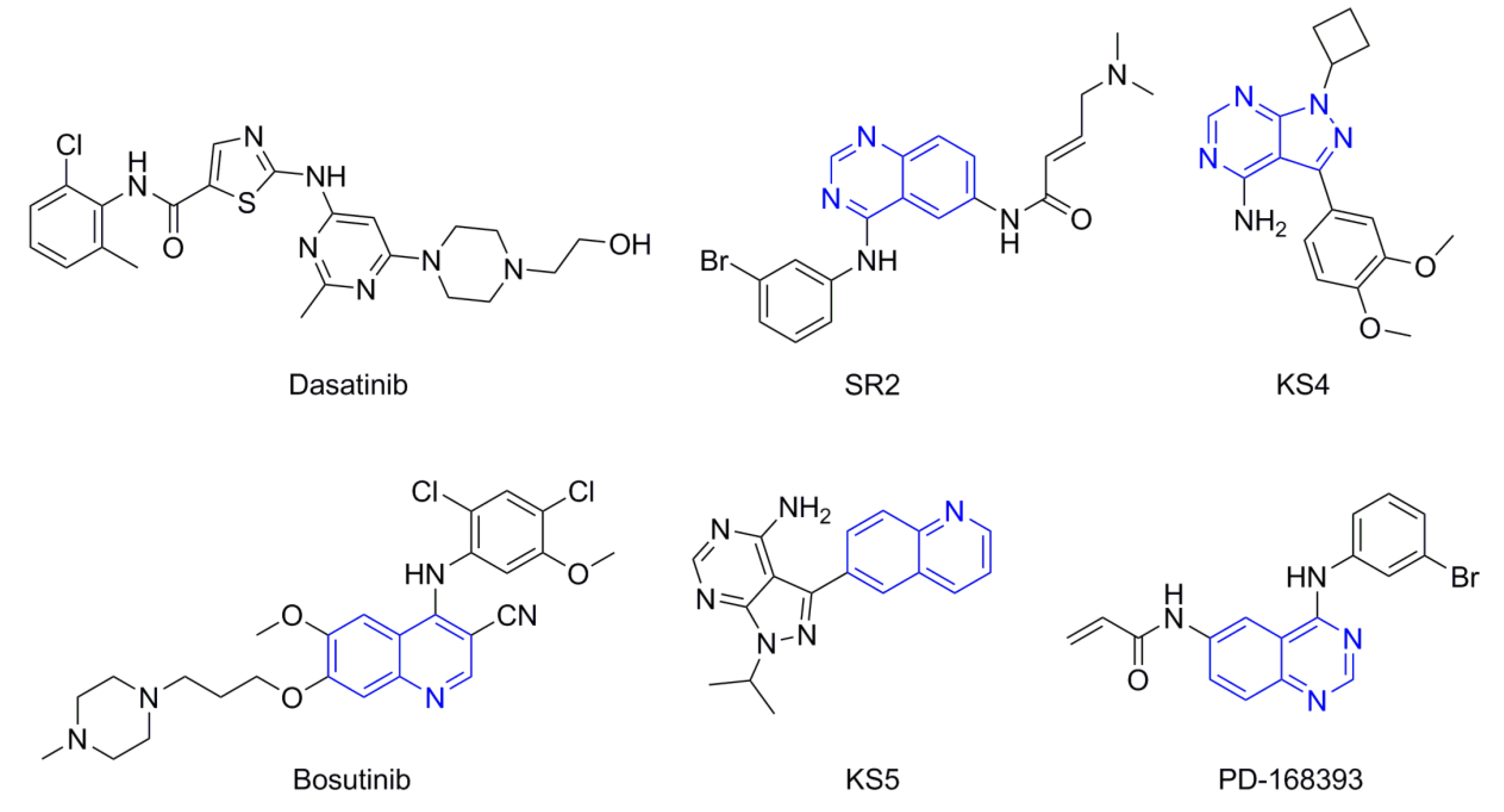
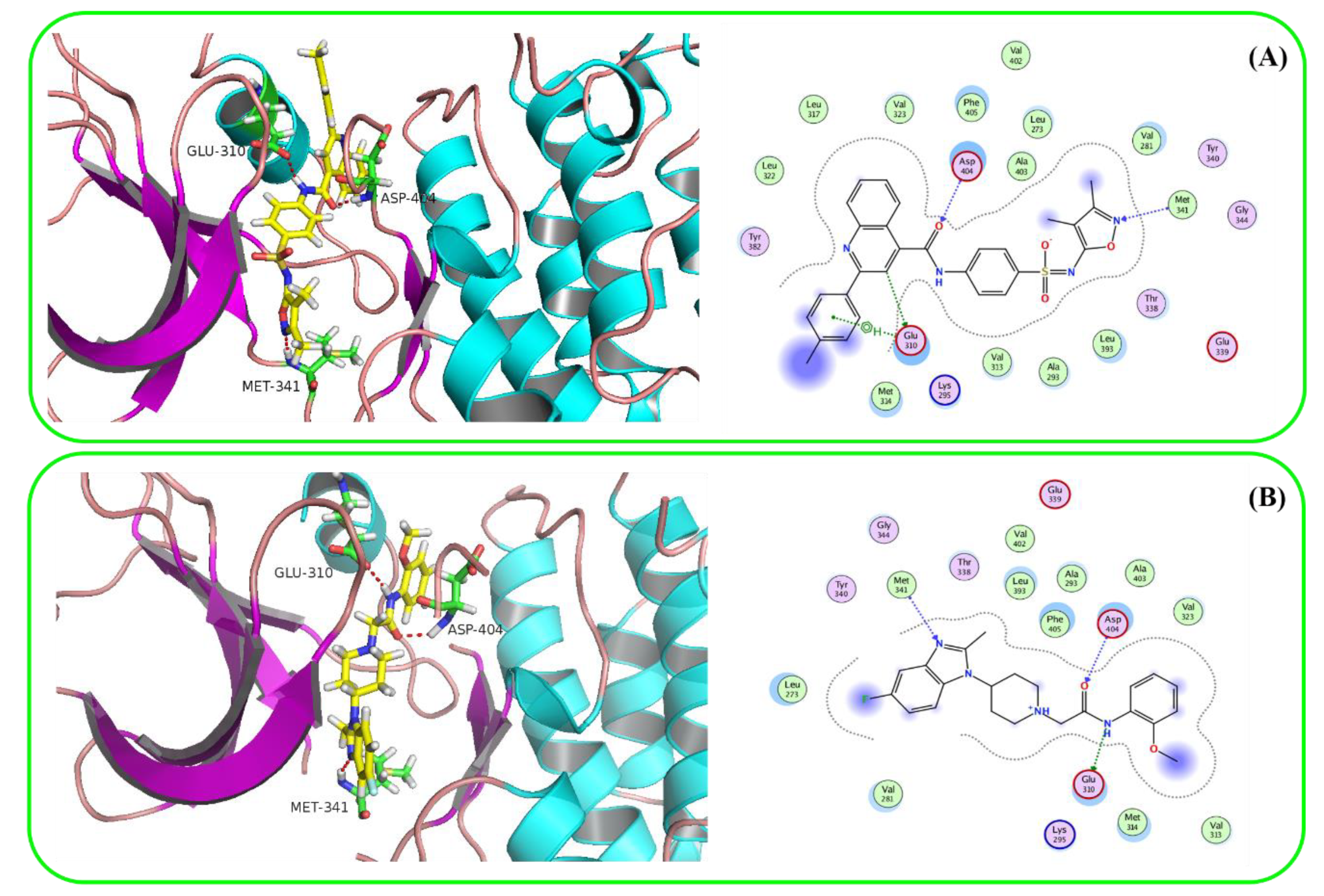
2.4. Molecular Docking
2.5. ADMET Prediction
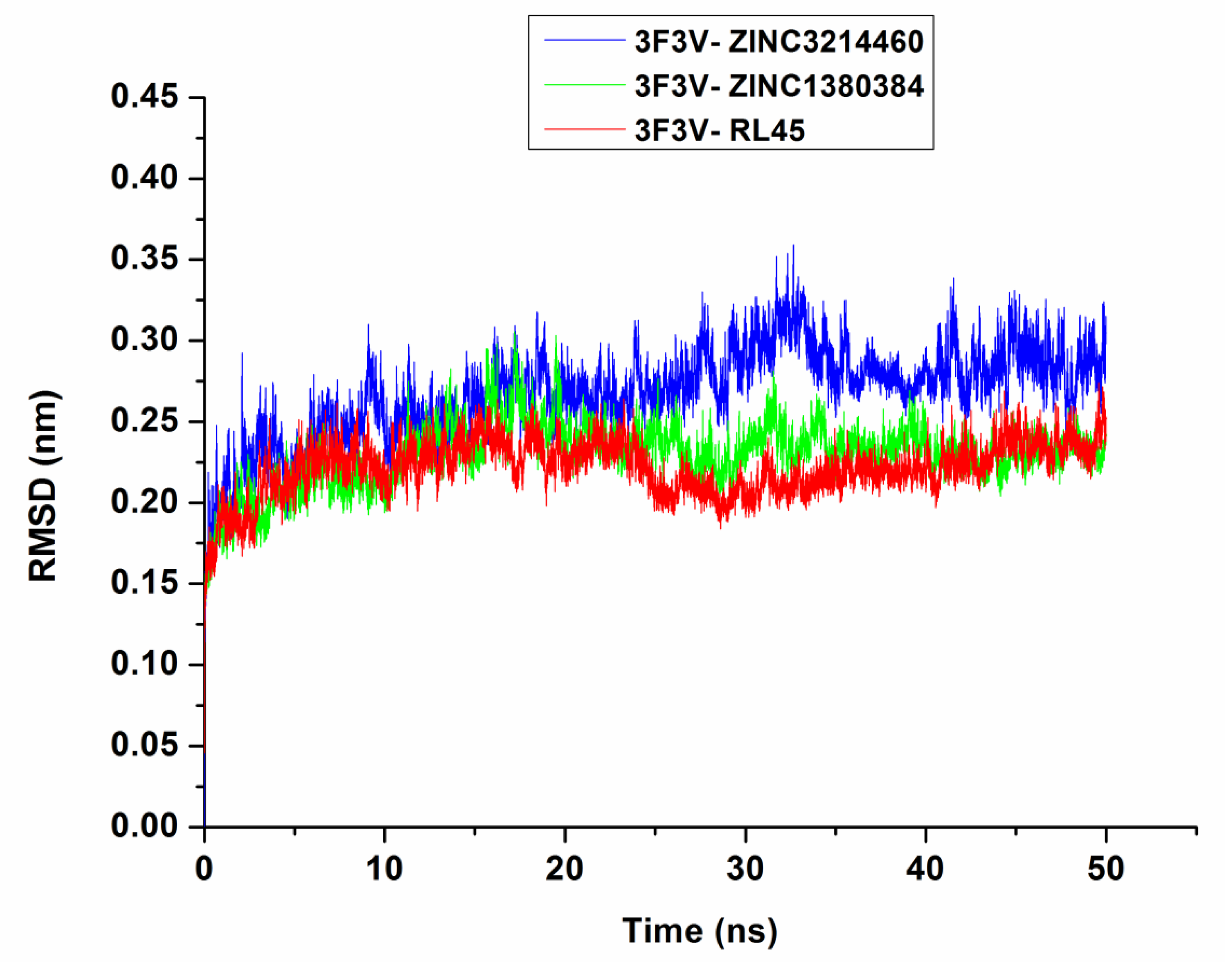
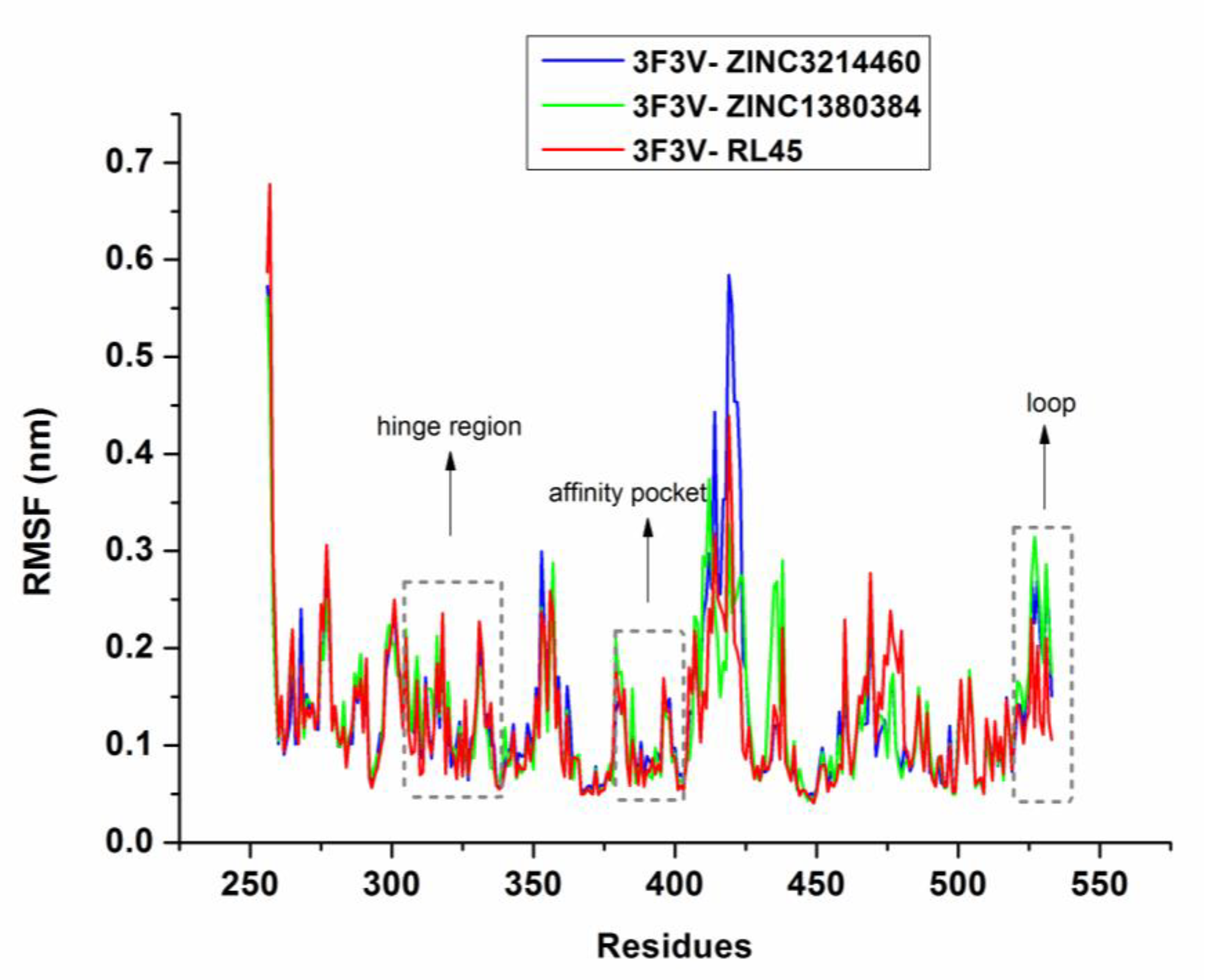
2.6. Molecular Dynamics Simulations
3. Materials and Methods
3.1. Generation and Validation of Pharmacophore Model
3.2. Pharmacophore-Based Virtual Screening
3.3. Molecular Docking
3.4. ADMET Prediction
3.5. Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Roskoski, R., Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Olgen, S. Design strategies, structures and molecular interactions of small molecule Src inhibitors. Anti-Cancer Agents Med. Chem. 2016, 16, 992–1002. [Google Scholar] [CrossRef]
- Zhang, H.; Forman, H.J. 4-Hydroxynonenal activates Src through a non-canonical pathway that involves EGFR/PTP1B. Free Radic. Biol. Med. 2015, 89, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Le Roux, A.L.; Mohammad, I.L.; Mateos, B.; Arbesú, M.; Gairí, M.; Khan, F.A.; Teixeira, J.M.C.; Pons, M. A myristoyl-binding site in the SH3 domain modulates c-Src membrane anchoring. iScience 2019, 12, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Ge, M.M.; Zhou, Y.Q.; Tian, X.B.; Manyande, A.; Tian, Y.K.; Ye, D.W.; Yang, H. Src-family protein tyrosine kinases: A promising target for treating chronic pain. Biomed. Pharmacother. 2020, 125, 110017. [Google Scholar] [CrossRef]
- Tintori, C.; Fallacara, A.L.; Radi, M.; Zamperini, C.; Dreassi, E.; Crespan, E.; Maga, G.; Schenone, S.; Musumeci, F.; Brullo, C.; et al. Combining X-ray crystallography and molecular modeling toward the optimization of pyrazolo[3,4-d]pyrimidines as potent c-Src inhibitors active in vivo against neuroblastoma. J. Med. Chem. 2015, 58, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, W.; Gao, S.; Wang, X. MicroRNA-208a directly targets Src kinase signaling inhibitor 1 to facilitate cell proliferation and invasion in non-small cell lung cancer. Mol. Med. Rep. 2019, 20, 3140–3148. [Google Scholar] [CrossRef]
- Poli, G.; Martinelli, A.; Tuccinardi, T. Computational approaches for the identification and optimization of Src family kinases inhibitors. Curr. Med. Chem. 2014, 21, 3281–3293. [Google Scholar] [CrossRef]
- Kadife, E.; Chan, E.; Luwor, R.; Kannourakis, G.; Findlay, J.; Ahmed, N. Paclitaxel-induced Src activation is inhibited by dasatinib treatment, independently of cancer stem cell properties, in a mouse model of ovarian cancer. Cancers 2019, 11, 243. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, Z.; Wang, J.; Zhang, M.; Li, Z.; Wang, S.; Dong, B.; Zhang, C.; Gao, J.; Shen, L. Mouse avatar models of esophageal squamous cell carcinoma proved the potential for EGFR-TKI afatinib and uncovered Src family kinases involved in acquired resistance. J. Hematol. Oncol. 2018, 11, 109. [Google Scholar] [CrossRef]
- Da Costa, J.D.E.F.F.B.; Sant’Anna, C.D.; Muniz, J.A.P.C.; Da Rocha, C.A.M.; Lamarao, L.M.; Nunes, C.D.F.A.M.; De Assumpcao, P.P.; Burbano, R.R. Deregulation of the SRC family tyrosine kinases in gastric carcinogenesis in non-human primates. Anticancer Res. 2018, 38, 6317–6320. [Google Scholar] [CrossRef]
- Feddersen, C.R.; Schillo, J.L.; Varzavand, A.; Vaughn, H.R.; Wadsworth, L.S.; Voigt, A.P.; Zhu, E.Y.; Jennings, B.M.; Mullen, S.A.; Bobera, J.; et al. Src-dependent DBL family members drive resistance to vemurafenib in human melanoma. Cancer Res. 2019, 79, 5074–5087. [Google Scholar] [CrossRef] [PubMed]
- Abere, B.; Samarina, N.; Gramolelli, S.; Rückert, J.; Gerold, G.; Pich, A.; Schulz, T.F. Kaposi’s sarcoma-associated herpesvirus nonstructural membrane protein pK15 recruits the class II phosphatidylinositol 3-kinase PI3K-C2 alpha to activate productive viral replication. J. Virol. 2018, 92, e00544-18. [Google Scholar] [CrossRef] [PubMed]
- Wadhawan, A.; Smith, C.; Nicholson, R.I.; Barrett-Lee, P.; Hiscox, S. Src-mediated regulation of homotypic cell adhesion: Implications for cancer progression and opportunities for therapeutic intervention. Cancer Treat. Rev. 2011, 37, 234–241. [Google Scholar] [CrossRef]
- Ungefroren, H.; Sebens, S.; Groth, S.; Gieseler, F.; Fändrich, F. Differential roles of Src in transforming growth factor-ß regulation of growth arrest, epithelial-to-mesenchymal transition and cell migration in pancreatic ductal adenocarcinoma cells. Int. J. Oncol. 2011, 38, 797–805. [Google Scholar] [CrossRef]
- Du, G.; Rao, S.; Gurbani, D.; Henning, N.J.; Jiang, J.; Che, J.; Yang, A.; Ficarro, S.B.; Marto, J.A.; Aguirre, A.J.; et al. Structure-based design of a potent and selective covalent inhibitor for SRC kinase that targets a P-Loop cysteine. J. Med. Chem. 2020, 63, 1624–1641. [Google Scholar] [CrossRef]
- Li, J.; Rix, U.; Fang, B.; Bai, Y.; Edwards, A.; Colinge, J.; Bennett, K.L.; Gao, J.; Song, L.; Eschrich, S.; et al. A chemical and phosphoproteomic characterization of dasatinib action in lung cancer. Nat. Chem. Biol. 2010, 6, 291–299. [Google Scholar] [CrossRef]
- Remsing Rix, L.L.; Rix, U.; Colinge, J.; Hantschel, O.; Bennett, K.L.; Stranzl, T.; Müller, A.; Baumgartner, C.; Valent, P.; Augustin, M.; et al. Global target profile of the kinase inhibitor bosutinib in primary chronic myeloid leukemia cells. Leukemia 2009, 23, 477–485. [Google Scholar] [CrossRef]
- Gentile, F.; Barakat, K.H.; Tuszynski, J.A. Computational characterization of small molecules binding to the human XPF active site and virtual screening to identify potential new DNA repair inhibitors targeting the ERCC1-XPF endonuclease. Int. J. Mol. Sci. 2018, 19, 1328. [Google Scholar] [CrossRef]
- Braga, R.C.; Andrade, C.H. Assessing the performance of 3D pharmacophore models in virtual screening: How good are they? Curr. Top. Med. Chem. 2013, 13, 1127–1138. [Google Scholar] [CrossRef]
- Dhanjal, J.K.; Sharma, S.; Grover, A.; Das, A. Use of ligand-based pharmacophore modeling and docking approach to find novel acetylcholinesterase inhibitors for treating Alzheimer’s. Biomed. Pharmacother. 2015, 71, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Deibler, K.K.; Mishra, R.K.; Clutter, M.R.; Antanasijevic, A.; Bergan, R.; Caffrey, M.; Scheidt, K.A. A chemical probe strategy for interrogating inhibitor selectivity across the MEK kinase family. Acs Chem. Biol. 2017, 12, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Gattelli, A.; García Solá, M.E.; Roloff, T.C.; Cardiff, R.D.; Kordon, E.C.; Chodosh, L.A.; Hynes, N.E. Chronic expression of wild-type Ret receptor in the mammary gland induces luminal tumors that are sensitive to Ret inhibition. Oncogene 2018, 37, 4046–4054. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.P.; Koutsoukas, A.; Mohd Fauzi, F.; Drakakis, G.; Maciejewski, M.; Glen, R.C.; Bender, A. Diversity selection of compounds based on ‘protein affinity fingerprints’ improves sampling of bioactive chemical space. Chem. Biol. Drug Des. 2013, 82, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Lill, M.A.; Danielson, M.L. Computer-aided drug design platform using PyMOL. J. Comput. Aided Mol. Des. 2011, 25, 13–19. [Google Scholar] [CrossRef]
- Clark, R.D. Predicting mammalian metabolism and toxicity of pesticides in silico. Pest Manag. Sci. 2018, 74, 1992–2003. [Google Scholar] [CrossRef]
- Takagi, Y.; Matsui, K.; Nobori, H.; Maeda, H.; Sato, A.; Kurosu, T.; Orba, Y.; Sawa, H.; Hattori, K.; Higashino, K.; et al. Discovery of novel cyclic peptide inhibitors of dengue virus NS2B-NS3 protease with antiviral activity. Bioorg. Med. Chem. Lett. 2017, 27, 3586–3590. [Google Scholar] [CrossRef]
- Wadood, A.; Riaz, M.; Uddin, R.; Ul-Haq, Z. In silico identification and evaluation of leads for the simultaneous inhibition of protease and helicase activities of HCV NS3/4A protease using complex based pharmacophore mapping and virtual screening. PLoS ONE 2014, 9, e89109. [Google Scholar] [CrossRef]
- Schuster, D. 3D pharmacophores as tools for activity profiling. Drug Discov. Today. Technol. 2010, 7, e205–e211. [Google Scholar] [CrossRef]
- Drwal, M.N.; Griffith, R. Combination of ligand- and structure-based methods in virtual screening. Drug Discov. Today. Technol. 2013, 10, e395–e401. [Google Scholar] [CrossRef]
- Courcot, B.; Bridgeman, A.J. Modeling the interactions between polyoxometalates and their environment. J. Comput. Chem. 2011, 32, 3143–3153. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J. Comput. Aided Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.Q.; Liu, Y.Y.; Feng, X.Y.; Xu, W.R.; Cheng, X.C. Discovery of novel and highly selective PI3Kδ inhibitors based on the p110δ crystal structure. J. Biomol. Struct. Dyn. 2020, 38, 2499–2508. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Lu, X.; Xiao, M.; Li, Z.; Zou, Y.; Ren, S.; Cheng, Y.; Luo, G.; Xiang, H. Discovery of potent and highly selective covalent inhibitors of Bruton’s tyrosine kinase bearing triazine scaffold. Eur. J. Med. Chem. 2020, 199, 112339. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. Softwarex 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Dolezal, R.; Soukup, O.; Malinak, D.; Savedra, R.M.L.; Marek, J.; Dolezalova, M.; Pasdiorova, M.; Salajkova, S.; Korabecny, J.; Honegr, J.; et al. Towards understanding the mechanism of action of antibacterial N-alkyl-3-hydroxypyridinium salts: Biological activities, molecular modeling and QSAR studies. Eur. J. Med. Chem. 2016, 121, 699–711. [Google Scholar] [CrossRef]
- Childers, M.C.; Daggett, V. Validating molecular dynamics simulations against experimental observables in light of underlying conformational ensembles. J. Phys. Chem. B 2018, 122, 6673–6689. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ZINC ID | Structure | Src Docking Score (kcal/mol) |
|---|---|---|
| ZINC3214460 |  | −9.6287 |
| ZINC61925676 |  | −9.1879 |
| ZINC58158745 |  | −9.1320 |
| ZINC12075400 |  | −8.9992 |
| ZINC1380384 |  | −8.9096 |
| ZINC12853028 |  | −8.7889 |
| ZINC23247639 |  | −8.7219 |
| ZINC949873 |  | −8.5887 |
| ZINC36389462 |  | −8.5816 |
| ZINC10479320 |  | −8.5090 |
| Compound | Buffer Solubility 1 | BBB 2 | Caco-2 3 | HIA 4 | PPB 5 | CYP2D6 Inhibition | hERG Inhibition |
|---|---|---|---|---|---|---|---|
| ZINC3214460 | 81.69 | 0.01036 | 18.87 | 97.41 | 100 | None | Low risk |
| ZINC1380384 | 3735.39 | 0.4491 | 26.62 | 92.11 | 34.90 | None | High risk |
| Dasatinib | 0.3113 | 0.03504 | 32.01 | 93.59 | 70.29 | None | Medium risk |
| Bosutinib | 5.500 | 0.06055 | 50.35 | 97.23 | 85.07 | None | Medium risk |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Zhang, T.-j.; Tu, S.; Zhang, Z.-h.; Meng, F.-h. Identification of Novel Src Inhibitors: Pharmacophore-Based Virtual Screening, Molecular Docking and Molecular Dynamics Simulations. Molecules 2020, 25, 4094. https://doi.org/10.3390/molecules25184094
Zhang Y, Zhang T-j, Tu S, Zhang Z-h, Meng F-h. Identification of Novel Src Inhibitors: Pharmacophore-Based Virtual Screening, Molecular Docking and Molecular Dynamics Simulations. Molecules. 2020; 25(18):4094. https://doi.org/10.3390/molecules25184094
Chicago/Turabian StyleZhang, Yi, Ting-jian Zhang, Shun Tu, Zhen-hao Zhang, and Fan-hao Meng. 2020. "Identification of Novel Src Inhibitors: Pharmacophore-Based Virtual Screening, Molecular Docking and Molecular Dynamics Simulations" Molecules 25, no. 18: 4094. https://doi.org/10.3390/molecules25184094
APA StyleZhang, Y., Zhang, T.-j., Tu, S., Zhang, Z.-h., & Meng, F.-h. (2020). Identification of Novel Src Inhibitors: Pharmacophore-Based Virtual Screening, Molecular Docking and Molecular Dynamics Simulations. Molecules, 25(18), 4094. https://doi.org/10.3390/molecules25184094