
Characterizing Epitope Binding Regions of Entire Antibody Panels by Combining Experimental and Computational Analysis of Antibody: Antigen Binding Competition

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Understanding the Structures of Epitope:Paratope Interactions Guides the Design of Superior Immunotherapies and Vaccines
1.2. High-Throughput Epitope Characterization Assays Provide Valuable Epitopic Information
1.3. Integrating High-Throughput Experiment and Computation Enables Characterization of Ab-Specific Epitopes
2. Results
2.1. Experimental Binning Identified Four Communities of Anti-gD Abs
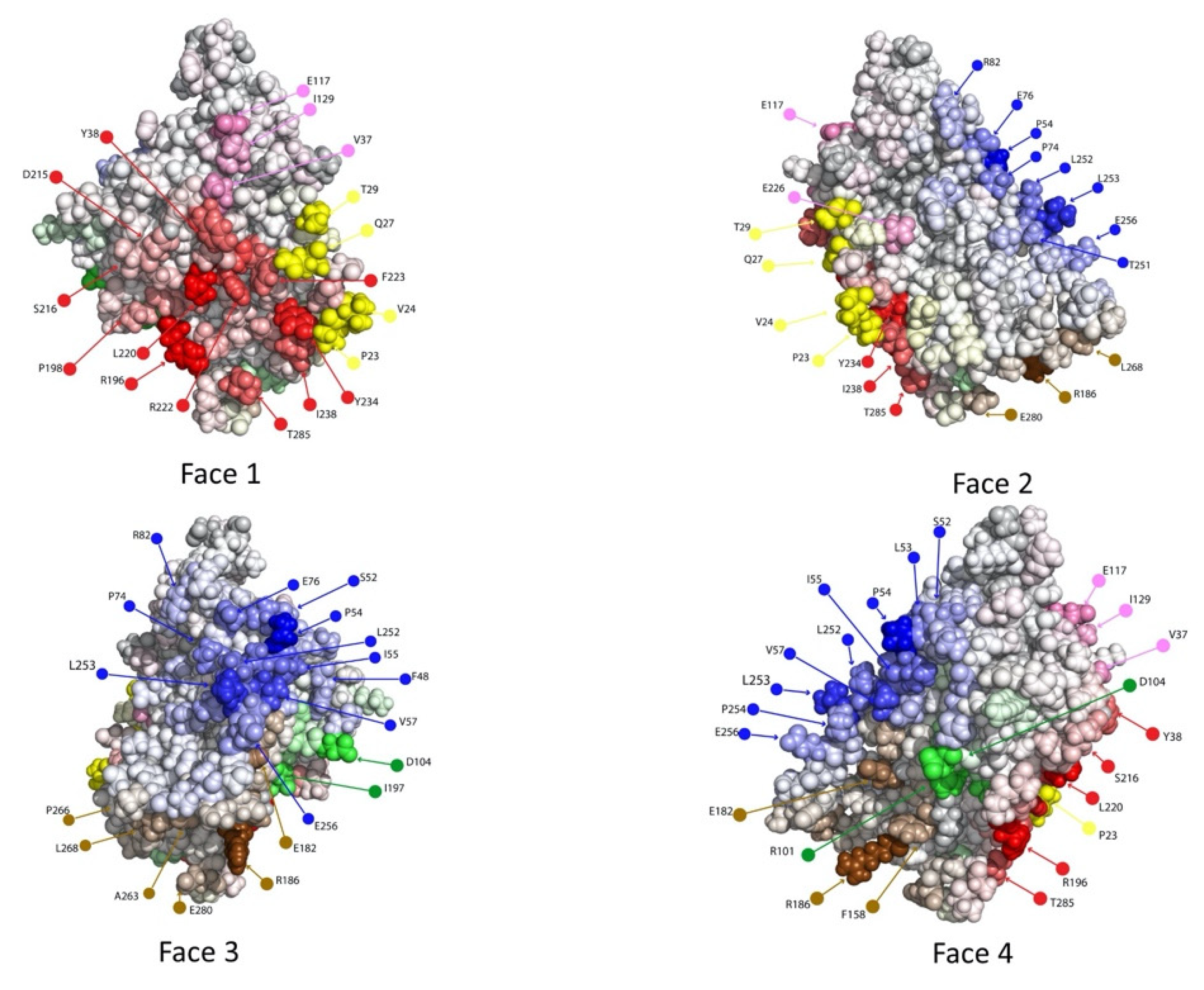
2.2. Computational Epitope Prediction Characterized the Putative Epitope Binding Regions
2.3. Dock Binning Grouped Models and Identified Broadly Antigenic Regions
2.4. Dock Binning Enabled Selection of Experimental Assays to Evaluate Antigenic Regions
2.5. Experimental Data Allowed Re-Docking to Focus Ab:Ag Models Based on Experimental Binding Data, thereby Localizing Each Ab’s Epitope Region
3. Discussion
3.1. The Integration of Experimental and Computational Binning Provides Important Epitope Information That Can Inform Discovery
3.2. While Binning Is Still Limited, Further Computational Advances Will Improve Its Accuracy and Informativeness
- Experimental epitope binning. The fundamental question here is how to cluster the Abs into communities. and new and emerging techniques from network/community analysis, especially in genomics [62,63,64], may prove beneficial. These metrics can be used to identify communities that require further refinement and/or changes to the clustering. Furthermore, the clustering need not be discrete, i.e., the partition into communities can be overlapping, and this information can potentially be leveraged throughout the rest of the process. Such analysis could be particularly important in cases of partial or aberrant competition. While the “sentinel” antibodies used here were selected as most representative of clear-cut communities, future approaches could include algorithmic selection of representatives based on properties of the communities (e.g., ensuring adequate coverage of ambiguous clusters) and of the individual antibodies (e.g., based on sequence and structural analysis, favorability for development, etc.).
- Ab modeling and Ab–Ag docking. We used representative high-quality Ab modeling and Ab–Ag docking methods, but others could be employed; given sufficient data points for different targets, the impacts of these choices could be quantitatively assessed. The complete set of thousands of docking models, instead of cluster representatives, could be considered, thereby potentially leveraging redundancy in weighting region importance. As discussed above regarding gD, accounting for protein flexibility (both Ag and Ab) may change the models substantially in some cases. Docking scores/ranks could be taken into account in order to determine the most important regions. In addition to physically-based docking methods, additional data-driven epitope prediction methods could be used to identify putative antigenic regions [65,66,67].
- Dock binning. The same issues with clustering for experimental binning hold here and the same potential solutions can be explored. Machine learning methods could be incorporated in order to train models to integrate the upstream predictions and experimental data, e.g., in a consensus or weighted fashion. New data-driven models that directly seek to predict competition/bins (instead of general-purpose epitope prediction) could be developed.
- Experimental epitope probing. As presented, mutations are selected to assess the predicted antigenic hot spots and deconvolve which hot spots are associated with which Abs. While this has been done before based simply on maximizing binding disruption according to the models [9,61], more refined metrics and optimization techniques could be developed, e.g., using an information-theoretic approach to maximize what is learned about the communities and hotspots for a given experimental “budget” (e.g., maximum-relevance, minimum-redundancy) [68].
- Revised dock bins and antigenic hotspots. We employed the approach of docking with affinity/repulsion in order to focus docking according to the experimental data. Docking models could be subsequently refined (e.g., via energy minimization and conformational sampling) and scored not only for physical modeling, but also for the consistency of the experimental data with the predicted effects of the mutations [69].
4. Materials and Methods
4.1. Antibodies
4.2. Proteins
4.3. Epitope Binning
4.4. Protein Modeling and Docking Prediction
4.5. Dock Binning
4.5.1. Competition
4.5.2. Clustering
4.5.3. Antigenic Heatmap
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gura, T. Therapeutic antibodies: Magic bullets hit the target. Nature 2002, 417, 584–586. [Google Scholar] [CrossRef] [PubMed]
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Abdiche, Y.N.; Miles, A.; Eckman, J.; Foletti, D.; Van Blarcom, T.J.; Yeung, Y.A.; Pons, J.; Rajpal, A. High-Throughput Epitope Binning Assays on Label-Free Array-Based Biosensors Can Yield Exquisite Epitope Discrimination That Facilitates the Selection of Monoclonal Antibodies with Functional Activity. PLoS ONE 2014, 9, e92451. [Google Scholar] [CrossRef] [PubMed]
- Abdiche, Y.N.; Harriman, R.; Deng, X.; Yeung, Y.A.; Miles, A.; Morishige, W.; Boustany, L.; Zhu, L.; Izquierdo, S.M.; Harriman, W. Assessing kinetic and epitopic diversity across orthogonal monoclonal antibody generation platforms. Mabs 2016, 8, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Ditto, N.T.; Brooks, B.D. The emerging role of biosensor-based epitope binning and mapping in antibody-based drug discovery. Expert Opin. Drug Discov. 2016, 11, 925–937. [Google Scholar] [CrossRef]
- Lonberg, N. Fully human antibodies from transgenic mouse and phage display platforms. Curr. Opin. Immunol. 2008, 20, 450–459. [Google Scholar] [CrossRef]
- Clementi, N.; Mancini, N.; Solforosi, L.; Castelli, M.; Clementi, M.; Burioni, R. Phage display-based strategies for cloning and optimization of monoclonal antibodies directed against human pathogens. Int. J. Mol. Sci. 2012, 13, 8273–8292. [Google Scholar] [CrossRef]
- Murphy, A.J.; Macdonald, L.E.; Stevens, S.; Karow, M.; Dore, A.T.; Pobursky, K.; Huang, T.T.; Poueymirou, W.T.; Esau, L.; Meola, M.; et al. Mice with megabase humanization of their immunoglobulin genes generate antibodies as efficiently as normal mice. Proc. Natl. Acad. Sci. USA 2014, 111, 5153–5158. [Google Scholar] [CrossRef]
- Hua, C.K.; Gacerez, A.T.; Sentman, C.L.; Ackerman, M.E.; Choi, Y.; Bailey-Kellogg, C. Computationally-driven identification of antibody epitopes. Elife 2017, 6, e29023. [Google Scholar] [CrossRef]
- Gan, H.K.; Burgess, A.W.; Clayton, A.H.; Scott, A.M. Targeting of a conformationally exposed, tumor-specific epitope of EGFR as a strategy for cancer therapy. Cancer Res. 2012, 72, 2924–2930. [Google Scholar] [CrossRef]
- Garrett, T.P.; Burgess, A.W.; Gan, H.K.; Luwor, R.B.; Cartwright, G.; Walker, F.; Orchard, S.G.; Clayton, A.H.; Nice, E.C.; Rothacker, J.; et al. Antibodies specifically targeting a locally misfolded region of tumor associated EGFR. Proc. Natl. Acad. Sci. USA 2009, 106, 5082–5087. [Google Scholar] [CrossRef] [PubMed]
- Zolla-Pazner, S.; deCamp, A.; Gilbert, P.B.; Williams, C.; Yates, N.L.; Williams, W.T.; Howington, R.; Fong, Y.; Morris, D.E.; Soderberg, K.A.; et al. Vaccine-Induced IgG Antibodies to V1V2 Regions of Multiple HIV-1 Subtypes Correlate with Decreased Risk of HIV-1 Infection. PLoS ONE 2014, 9, e87572. [Google Scholar] [CrossRef] [PubMed]
- Gottardo, R.; Bailer, R.T.; Korber, B.T.; Gnanakaran, S.; Phillips, J.; Shen, X.; Tomaras, G.D.; Turk, E.; Imholte, G.; Eckler, L.; et al. Plasma IgG to Linear Epitopes in the V2 and V3 Regions of HIV-1 gp120 Correlate with a Reduced Risk of Infection in the RV144 Vaccine Efficacy Trial. PLoS ONE 2013, 8, e75665. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Coljee, V.W.; Maynard, J.A. Back to the future: Recombinant polyclonal antibody therapeutics. Curr. Opin. Chem. Eng. 2013, 2, 405–415. [Google Scholar] [CrossRef]
- Steel, J.; Lowen, A.C.; Wang, T.T.; Yondola, M.; Gao, Q.; Haye, K.; García-Sastre, A.; Palese, P. Influenza Virus Vaccine Based on the Conserved Hemagglutinin Stalk Domain. Mbio 2010, 1, e00018-10. [Google Scholar] [CrossRef]
- Gocnik, M.; Fislova, T.; Sladkova, T.; Mucha, V.; Kostolansky, F.; Vareckova, E. Antibodies specific to the HA2 glycopolypeptide of influenza A virus haemagglutinin with fusion-inhibition activity contribute to the protection of mice against lethal infection. J. Gen. Virol. 2007, 88, 951–955. [Google Scholar] [CrossRef]
- Liu, W.-C.; Jan, J.-T.; Huang, Y.-J.; Chen, T.-H.; Wu, S.-C. Unmasking Stem-Specific Neutralizing Epitopes by Abolishing N-Linked Glycosylation Sites of Influenza Virus Hemagglutinin Proteins for Vaccine Design. J. Virol. 2016, 90, 8496–8508. [Google Scholar] [CrossRef]
- Eggink, D.; Goff, P.H.; Palese, P. Guiding the Immune Response against Influenza Virus Hemagglutinin toward the Conserved Stalk Domain by Hyperglycosylation of the Globular Head Domain. J. Virol. 2014, 88, 699–704. [Google Scholar] [CrossRef]
- Margine, I.; Krammer, F.; Hai, R.; Heaton, N.S.; Tan, G.S.; Andrews, S.A.; Runstadler, J.A.; Wilson, P.C.; Albrecht, R.A.; García-Sastre, A.; et al. Hemagglutinin Stalk-Based Universal Vaccine Constructs Protect against Group 2 Influenza A Viruses. J. Virol. 2013, 87, 10435–10446. [Google Scholar] [CrossRef]
- Brooks, B.D.; Miles, A.; Abdiche, Y. High-throughput epitope binning of therapeutic monoclonal antibodies: Why you need to bin the fridge. Drug Discov. Today 2014, 19, 1040–1044. [Google Scholar] [CrossRef]
- Abdiche, Y.N.; Yeung, A.Y.; Ni, I.; Stone, D.; Miles, A.; Morishige, W.; Rossi, A.; Strop, P. Antibodies Targeting Closely Adjacent or Minimally Overlapping Epitopes Can Displace One Another. PLoS ONE 2017, 12, e0169535. [Google Scholar] [CrossRef] [PubMed]
- Estep, P.; Reid, F.; Nauman, C.; Liu, Y.; Sun, T.; Sun, J.; Xu, Y. High throughput solution-based measurement of antibody-antigen affinity and epitope binning. Mabs 2013, 5, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, D.S. Therapeutic antibodies, vaccines and antibodyomes. Mabs 2010, 2, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Renaud, J.-P.; Chung, C.; Danielson, U.H.; Egner, U.; Hennig, M.; Hubbard, R.E.; Nar, H. Biophysics in drug discovery: Impact, challenges and opportunities. Nat. Rev. Drug Discov. 2016, 15, 679–698. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Handing, K.B.; Zimmerman, M.D.; Shabalin, I.G.; Almo, S.C.; Minor, W. X-ray crystallography over the past decade for novel drug discovery–where are we heading next? Expert Opin. Drug Discov. 2015, 10, 975–989. [Google Scholar] [CrossRef]
- Abbott, W.M.; Damschroder, M.M.; Lowe, D.C. Current approaches to fine mapping of antigen–antibody interactions. Immunology 2014, 142, 526–535. [Google Scholar] [CrossRef]
- Weiss, G.A.; Watanabe, C.K.; Zhong, A.; Goddard, A.; Sidhu, S.S. Rapid mapping of protein functional epitopes by combinatorial alanine scanning. Proc. Natl. Acad. Sci. USA 2000, 97, 8950–8954. [Google Scholar] [CrossRef]
- Greenspan, N.S.; Di Cera, E. Defining epitopes: It’s not as easy as it seems. Nat. Biotechnol. 1999, 17, 936–937. [Google Scholar] [CrossRef]
- Arkin, M.R.; Randal, M.; DeLano, W.L.; Hyde, J.; Luong, T.N.; Oslob, J.D.; Raphael, D.R.; Taylor, L.; Wang, J.; McDowell, R.S.; et al. Binding of small molecules to an adaptive protein-protein interface. Proc. Natl. Acad. Sci. USA 2003, 100, 1603–1608. [Google Scholar] [CrossRef]
- Novel Inhibitors of DNA Gyrase: 3D Structure Based Biased Needle Screening, Hit Validation by Biophysical Methods, and 3D Guided Optimization. A Promising Alternative to Random Screening-Journal of Medicinal Chemistry (ACS Publications). Available online: https://pubs-acs-org.ezproxy.lib.utah.edu/doi/abs/10.1021/jm000017s (accessed on 28 February 2019).
- Davidoff, S.N.; Ditto, N.T.; Brooks, A.E.; Eckman, J.; Brooks, B.D. Surface Plasmon Resonance for Therapeutic Antibody Characterization. In Label-Free Biosensor Methods in Drug Discovery; Humana Press: New York, NY, USA, 2015; pp. 35–76. [Google Scholar]
- Brooks, B. The Importance of Epitope Binning for Biological Drug Discovery. Curr. Drug Discov. Technol. 2013, 11, 109–112. [Google Scholar] [CrossRef]
- Abdiche, Y.N.; Lindquist, K.C.; Pinkerton, A.; Pons, J.; Rajpal, A. Expanding the ProteOn XPR36 biosensor into a 36-ligand array expedites protein interaction analysis. Anal. Biochem. 2011, 411, 139–151. [Google Scholar] [CrossRef]
- Abdiche, Y.N.; Malashock, D.S.; Pons, J. Probing the binding mechanism and affinity of tanezumab, a recombinant humanized anti-NGF monoclonal antibody, using a repertoire of biosensors. Protein Sci. 2008, 17, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Abdiche, Y.N.; Malashock, D.S.; Pinkerton, A.; Pons, J. Exploring blocking assays using Octet, ProteOn, and Biacore biosensors. Anal. Biochem. 2009, 386, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Sela-Culang, I.; Kunik, V.; Ofran, Y. The Structural Basis of Antibody-Antigen Recognition. Front. Immunol. 2013, 4, 302. [Google Scholar] [CrossRef] [PubMed]
- Sela-Culang, I.; Ofran, Y.; Peters, B. Antibody specific epitope prediction—Emergence of a new paradigm. Curr. Opin. Virol. 2015, 11, 98–102. [Google Scholar] [CrossRef]
- Zhao, L.; Li, J. Mining for the antibody-antigen interacting associations that predict the B cell epitopes. BMC Struct. Biol. 2010, 10, S6. [Google Scholar] [CrossRef]
- Sircar, A.; Gray, J.J. SnugDock: Paratope Structural Optimization during Antibody-Antigen Docking Compensates for Errors in Antibody Homology Models. PLoS Comput. Biol. 2010, 6, e1000644. [Google Scholar] [CrossRef]
- Brenke, R.; Hall, D.R.; Chuang, G.-Y.; Comeau, S.R.; Bohnuud, T.; Beglov, D.; Schueler-Furman, O.; Vajda, S.; Kozakov, D. Application of asymmetric statistical potentials to antibody–protein docking. Bioinformatics 2012, 28, 2608–2614. [Google Scholar] [CrossRef]
- Wollacott, A.M.; Robinson, L.N.; Ramakrishnan, B.; Tissire, H.; Viswanathan, K.; Shriver, Z.; Babcock, G.J. Structural prediction of antibody-APRIL complexes by computational docking constrained by antigen saturation mutagenesis library data. J. Mol. Recognit. 2019, 32, e2778. [Google Scholar] [CrossRef]
- Long, D.; Madara, T.J.; De Leon, M.P.; Cohen, G.H.; Montgomery, P.C.; Eisenberg, R.J. Glycoprotein D protects mice against lethal challenge with herpes simplex virus types 1 and 2. Infect. Immun. 1984, 43, 761–764. [Google Scholar] [CrossRef]
- Schrier, R.D.; Pizer, L.I.; Moorhead, J.W. Type-specific delayed hypersensitivity and protective immunity induced by isolated herpes simplex virus glycoprotein. J. Immunol. 1983, 130, 1413–1418. [Google Scholar] [PubMed]
- Berman, P.W.; Gregory, T.; Crase, D.; Lasky, L.A. Protection from genital herpes simplex virus type 2 infection by vaccination with cloned type 1 glycoprotein D. Science 1985, 227, 1490–1492. [Google Scholar] [CrossRef] [PubMed]
- Rajčáni, J.; Bánáti, F.; Szenthe, K.; Szathmary, S. The potential of currently unavailable herpes virus vaccines. Expert Rev. Vaccines 2018, 17, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Cairns, T.M.; Ditto, N.T.; Lou, H.; Brooks, B.D.; Atanasiu, D.; Eisenberg, R.J.; Cohen, G.H. Global sensing of the antigenic structure of herpes simplex virus gD using high-throughput array-based SPR imaging. PLoS Pathog. 2017, 13, e1006430. [Google Scholar] [CrossRef] [PubMed]
- Ditto, N.T.; Cairns, T.M.; Lou, H.; Closmore, A.; Eisenberg, R.J.; Cohen, G.C.; Brooks, B.D. Understanding antibody: Antigen Relationships using Antigenic Variants with Array-Based SPRi Epitope Mapping. J. Drug Res. Dev. 2016, 2, 2470-1009. [Google Scholar]
- Hook, L.M.; Cairns, T.M.; Awasthi, S.; Brooks, B.D.; Ditto, N.T.; Eisenberg, R.J.; Cohen, G.H.; Friedman, H.M. Vaccine-induced antibodies to herpes simplex virus glycoprotein D epitopes involved in virus entry and cell-to-cell spread correlate with protection against genital disease in guinea pigs. PLoS Pathog. 2018, 14, e1007095. [Google Scholar] [CrossRef]
- Whitbeck, J.C.; Huang, Z.-Y.; Cairns, T.M.; Gallagher, J.R.; Lou, H.; Ponce-de-Leon, M.; Belshe, R.B.; Eisenberg, R.J.; Cohen, G.H. Repertoire of epitopes recognized by serum IgG from humans vaccinated with herpes simplex virus 2 glycoprotein D. J. Virol. 2014, 88, 7786–7795. [Google Scholar] [CrossRef][Green Version]
- Whitbeck, J.C.; Muggeridge, M.I.; Rux, A.H.; Hou, W.; Krummenacher, C.; Lou, H.; van Geelen, A.; Eisenberg, R.J.; Cohen, G.H. The major neutralizing antigenic site on herpes simplex virus glycoprotein D overlaps a receptor-binding domain. J. Virol. 1999, 73, 9879–9890. [Google Scholar] [CrossRef]
- Lee, C.-C.; Lin, L.-L.; Chan, W.-E.; Ko, T.-P.; Lai, J.-S.; Wang, A.H.-J. Structural basis for the antibody neutralization of Herpes simplex virus. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1935–1945. [Google Scholar] [CrossRef]
- Finn, J.A.; Koehler Leman, J.; Willis, J.R.; Cisneros, A.; Crowe, J.E.; Meiler, J. Improving Loop Modeling of the Antibody Complementarity-Determining Region 3 Using Knowledge-Based Restraints. PLoS ONE 2016, 11, e0154811. [Google Scholar] [CrossRef]
- Messih, M.A.; Lepore, R.; Marcatili, P.; Tramontano, A. Improving the accuracy of the structure prediction of the third hypervariable loop of the heavy chains of antibodies. Bioinformatics 2014, 30, 2733–2740. [Google Scholar] [CrossRef] [PubMed]
- Krummenacher, C.; Supekar, V.M.; Whitbeck, J.C.; Lazear, E.; Connolly, S.A.; Eisenberg, R.J.; Cohen, G.H.; Wiley, D.C.; Carfí, A. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 2005, 24, 4144–4153. [Google Scholar] [CrossRef] [PubMed]
- Kozakov, D.; Brenke, R.; Comeau, S.; Vajda, S. PIPER: An FFT-based protein docking program with pairwise potentials. Proteins Struct. Funct. Bioinform. 2006, 65, 392–406. [Google Scholar] [CrossRef] [PubMed]
- Eddings, M.A.; Johnson, M.A.; Gale, B.K. Determining the optimal PDMS–PDMS bonding technique for microfluidic devices. J. Micromech. Microeng. 2008, 18, 067001. [Google Scholar] [CrossRef]
- Ponomarenko, J.V.; Bourne, P.E. Antibody-protein interactions: Benchmark datasets and prediction tools evaluation. BMC Struct. Biol. 2007, 7, 64. [Google Scholar] [CrossRef]
- Lensink, M.F.; Wodak, S.J. Docking, scoring, and affinity prediction in CAPRI. Proteins 2013, 81, 2082–2095. [Google Scholar] [CrossRef]
- Atanasiu, D.; Saw, W.T.; Lazear, E.; Whitbeck, J.C.; Cairns, T.M.; Lou, H.; Eisenberg, R.J.; Cohen, G.H. Using antibodies and mutants to localize the presumptive gH/gL binding site on HSV gD. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Connolly, S.A.; Jackson, J.O.; Jardetzky, T.S.; Longnecker, R. Fusing structure and function: A structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 2011, 9, 369–381. [Google Scholar] [CrossRef]
- Choi, Y.; Furlon, J.M.; Amos, R.B.; Griswold, K.E.; Bailey-Kellogg, C. DisruPPI: Structure-based computational redesign algorithm for protein binding disruption. Bioinformatics 2018, 34, i245–i253. [Google Scholar] [CrossRef]
- Eisen, M.B.; Spellman, P.T.; Brown, P.O.; Botstein, D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 1998, 95, 14863–14868. [Google Scholar] [CrossRef]
- Jiang, D.; Tang, C.; Zhang, A. Cluster analysis for gene expression data: A survey. IEEE Trans. Knowl. Data Eng. 2004, 16, 1370–1386. [Google Scholar] [CrossRef]
- Hartuv, E.; Shamir, R. A clustering algorithm based on graph connectivity. Inf. Process. Lett. 2000, 76, 175–181. [Google Scholar] [CrossRef]
- Pittala, S.; Bailey-Kellogg, C. Learning Context-aware Structural Representations to Predict Antigen and Antibody Binding Interfaces. Bioinformatics 2020, 36, 3996–4003. [Google Scholar] [CrossRef]
- Krawczyk, K.; Liu, X.; Baker, T.; Shi, J.; Deane, C.M. Improving B-cell epitope prediction and its application to global antibody-antigen docking. Bioinformatics 2014, 30, 2288–2294. [Google Scholar] [CrossRef]
- Kringelum, J.V.; Lundegaard, C.; Lund, O.; Nielsen, M. Reliable B Cell Epitope Predictions: Impacts of Method Development and Improved Benchmarking. PLoS Comput. Biol. 2012, 8, e1002829. [Google Scholar] [CrossRef] [PubMed]
- Feature Selection Based on Mutual Information: Criteria of Max-Dependency, Max-Relevance, and Min-Redundancy. Available online: https://www.computer.org/csdl/journal/tp/2005/08/i1226/13rRUy3xY96 (accessed on 16 March 2020).
- Moretti, R.; Lyskov, S.; Das, R.; Meiler, J.; Gray, J.J. Web-accessible molecular modeling with Rosetta: The Rosetta Online Server that Includes Everyone (ROSIE). Protein Sci. 2018, 27, 259–268. [Google Scholar] [CrossRef]
- Rux, A.H.; Willis, S.H.; Nicola, A.V.; Hou, W.; Peng, C.; Lou, H.; Cohen, G.H.; Eisenberg, R.J. Functional Region IV of Glycoprotein D from Herpes Simplex Virus Modulates Glycoprotein Binding to the Herpesvirus Entry Mediator. J. Virol. 1998, 72, 7091–7098. [Google Scholar] [CrossRef]
- Lazear, E.; Whitbeck, J.C.; Ponce-de-Leon, M.; Cairns, T.M.; Willis, S.H.; Zuo, Y.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J. Antibody-Induced Conformational Changes in Herpes Simplex Virus Glycoprotein gD Reveal New Targets for Virus Neutralization. J. Virol. 2012, 86, 1563–1576. [Google Scholar] [CrossRef]
- Friedman, H.M.; Cohen, G.H.; Eisenberg, R.J.; Seidel, C.A.; Cines, D.B. Glycoprotein C of herpes simplex virus 1 acts as a receptor for the C3b complement component on infected cells. Nature 1984, 309, 633–635. [Google Scholar] [CrossRef]
- Eisenberg, R.J.; Long, D.; Pereira, L.; Hampar, B.; Zweig, M.; Cohen, G.H. Effect of monoclonal antibodies on limited proteolysis of native glycoprotein gD of herpes simplex virus type 1. J. Virol. 1982, 41, 478–488. [Google Scholar] [CrossRef]
- Cohen, G.H.; Isola, V.J.; Kuhns, J.; Berman, P.W.; Eisenberg, R.J. Localization of discontinuous epitopes of herpes simplex virus glycoprotein D: Use of a nondenaturing (“native” gel) system of polyacrylamide gel electrophoresis coupled with Western blotting. J. Virol. 1986, 60, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Muggeridge, M.I.; Wu, T.T.; Johnson, D.C.; Glorioso, J.C.; Eisenberg, R.J.; Cohen, G.H. Antigenic and functional analysis of a neutralization site of HSV-1 glycoprotein D. Virology 1990, 174, 375–387. [Google Scholar] [CrossRef]
- Carfí, A.; Willis, S.H.; Whitbeck, J.C.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J.; Wiley, D.C. Herpes Simplex Virus Glycoprotein D Bound to the Human Receptor HveA. Mol. Cell 2001, 8, 169–179. [Google Scholar] [CrossRef]
- Di Giovine, P.; Settembre, E.C.; Bhargava, A.K.; Luftig, M.A.; Lou, H.; Cohen, G.H.; Eisenberg, R.J.; Krummenacher, C.; Carfi, A. Structure of Herpes Simplex Virus Glycoprotein D Bound to the Human Receptor Nectin-1. PLoS Pathog. 2011, 7, e1002277. [Google Scholar] [CrossRef]
- Lu, G.; Zhang, N.; Qi, J.; Li, Y.; Chen, Z.; Zheng, C.; Gao, G.F.; Yan, J. Crystal Structure of Herpes Simplex Virus 2 gD Bound to Nectin-1 Reveals a Conserved Mode of Receptor Recognition. J. Virol. 2014, 88, 13678–13688. [Google Scholar] [CrossRef] [PubMed]
- Lazear, E.; Whitbeck, J.C.; Zuo, Y.; Carfí, A.; Cohen, G.H.; Eisenberg, R.J.; Krummenacher, C. Induction of conformational changes at the N-terminus of herpes simplex virus glycoprotein D upon binding to HVEM and nectin-1. Virology 2014, 448, 185–195. [Google Scholar] [CrossRef]
- van Kooij, A.; Middel, J.; Jakab, F.; Elfferich, P.; Koedijk, D.G.; Feijlbrief, M.; Scheffer, A.J.; Degener, J.E.; The, T.H.; Scheek, R.M.; et al. High level expression and secretion of truncated forms of herpes simplex virus type 1 and type 2 glycoprotein D by the methylotrophic yeast Pichia pastoris. Protein Expr. Purif. 2002, 25, 400–408. [Google Scholar] [CrossRef]
- Minson, A.C.; Hodgman, T.C.; Digard, P.; Hancock, D.C.; Bell, S.E.; Buckmaster, E.A. An Analysis of the Biological Properties of Monoclonal Antibodies against Glycoprotein D of Herpes Simplex Virus and Identification of Amino Acid Substitutions that Confer Resistance to Neutralization. J. Gen. Virol. 1986, 67, 1001–1013. [Google Scholar] [CrossRef]
- Pereira, L.; Klassen, T.; Baringer, J.R. Type-common and type-specific monoclonal antibody to herpes simplex virus type 1. Infect. Immun. 1980, 29, 724–732. [Google Scholar]
- Pereira, L.; Dondero, D.V.; Gallo, D.; Devlin, V.; Woodie, J.D. Serological analysis of herpes simplex virus types 1 and 2 with monoclonal antibodies. Infect. Immun. 1982, 35, 363–367. [Google Scholar] [CrossRef]
- Showalter, S.D.; Zweig, M.; Hampar, B. Monoclonal antibodies to herpes simplex virus type 1 proteins, including the immediate-early protein ICP 4. Infect. Immun. 1981, 34, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Isola, V.J.; Eisenberg, R.J.; Siebert, G.R.; Heilman, C.J.; Wilcox, W.C.; Cohen, G.H. Fine mapping of antigenic site II of herpes simplex virus glycoprotein D. J. Virol. 1989, 63, 2325–2334. [Google Scholar] [CrossRef] [PubMed]
- Muggeridge, M.I.; Roberts, S.R.; Isola, V.J.; Cohen, G.H.; Eisenberg, R.J. Herpes simplex virus. Immunochem. Viruses 1990, 2, 459–481. [Google Scholar]
- Chiang, H.Y.; Cohen, G.H.; Eisenberg, R.J. Identification of functional regions of herpes simplex virus glycoprotein gD by using linker-insertion mutagenesis. J. Virol. 1994, 68, 2529–2543. [Google Scholar] [CrossRef]
- Seigneurin, J.; Desgranges, C.; Seigneurin, D.; Paire, J.; Renversez, J.; Jacquemont, B.; Micouin, C. Herpes simplex virus glycoprotein D: Human monoclonal antibody produced by bone marrow cell line. Science 1983, 221, 173–175. [Google Scholar] [CrossRef]
- Nicola, A.V.; Willis, S.H.; Naidoo, N.N.; Eisenberg, R.J.; Cohen, G.H. Structure-function analysis of soluble forms of herpes simplex virus glycoprotein D. J. Virol. 1996, 70, 3815–3822. [Google Scholar] [CrossRef]
- Connolly, S.A.; Landsburg, D.J.; Carfi, A.; Whitbeck, J.C.; Zuo, Y.; Wiley, D.C.; Cohen, G.H.; Eisenberg, R.J. Potential Nectin-1 Binding Site on Herpes Simplex Virus Glycoprotein D. J. Virol. 2005, 79, 1282–1295. [Google Scholar] [CrossRef]
- Gallagher, J.R.; Saw, W.T.; Atanasiu, D.; Lou, H.; Eisenberg, R.J.; Cohen, G.H. Displacement of the C Terminus of Herpes Simplex Virus gD Is Sufficient To Expose the Fusion-Activating Interfaces on gD. J. Virol. 2013, 87, 12656–12666. [Google Scholar] [CrossRef]
- Charrad, M.; Ghazzali, N.; Boiteau, V.; Niknafs, A. NbClust Package for Determining the Best Number of Clusters. R Package Version 2014, 2. [Google Scholar]
- Charrad, M.; Ghazzali, N.; Boiteau, V.; Niknafs, A.; Charrad, M.M. Package ‘NbClust’. J. Stat. Softw. 2014, 61, 1–36. [Google Scholar]
- Malika, C.; Ghazzali, N.; Boiteau, V.; Niknafs, A. NbClust: An R Package for Determining the Relevant Number of Clusters in a Data Set. J. Stat. Softw. 2014, 61, 1–36. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brooks, B.D.; Closmore, A.; Yang, J.; Holland, M.; Cairns, T.; Cohen, G.H.; Bailey-Kellogg, C. Characterizing Epitope Binding Regions of Entire Antibody Panels by Combining Experimental and Computational Analysis of Antibody: Antigen Binding Competition. Molecules 2020, 25, 3659. https://doi.org/10.3390/molecules25163659
Brooks BD, Closmore A, Yang J, Holland M, Cairns T, Cohen GH, Bailey-Kellogg C. Characterizing Epitope Binding Regions of Entire Antibody Panels by Combining Experimental and Computational Analysis of Antibody: Antigen Binding Competition. Molecules. 2020; 25(16):3659. https://doi.org/10.3390/molecules25163659
Chicago/Turabian StyleBrooks, Benjamin D., Adam Closmore, Juechen Yang, Michael Holland, Tina Cairns, Gary H. Cohen, and Chris Bailey-Kellogg. 2020. "Characterizing Epitope Binding Regions of Entire Antibody Panels by Combining Experimental and Computational Analysis of Antibody: Antigen Binding Competition" Molecules 25, no. 16: 3659. https://doi.org/10.3390/molecules25163659
APA StyleBrooks, B. D., Closmore, A., Yang, J., Holland, M., Cairns, T., Cohen, G. H., & Bailey-Kellogg, C. (2020). Characterizing Epitope Binding Regions of Entire Antibody Panels by Combining Experimental and Computational Analysis of Antibody: Antigen Binding Competition. Molecules, 25(16), 3659. https://doi.org/10.3390/molecules25163659