Peptidyl Fluoromethyl Ketones and Their Applications in Medicinal Chemistry



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
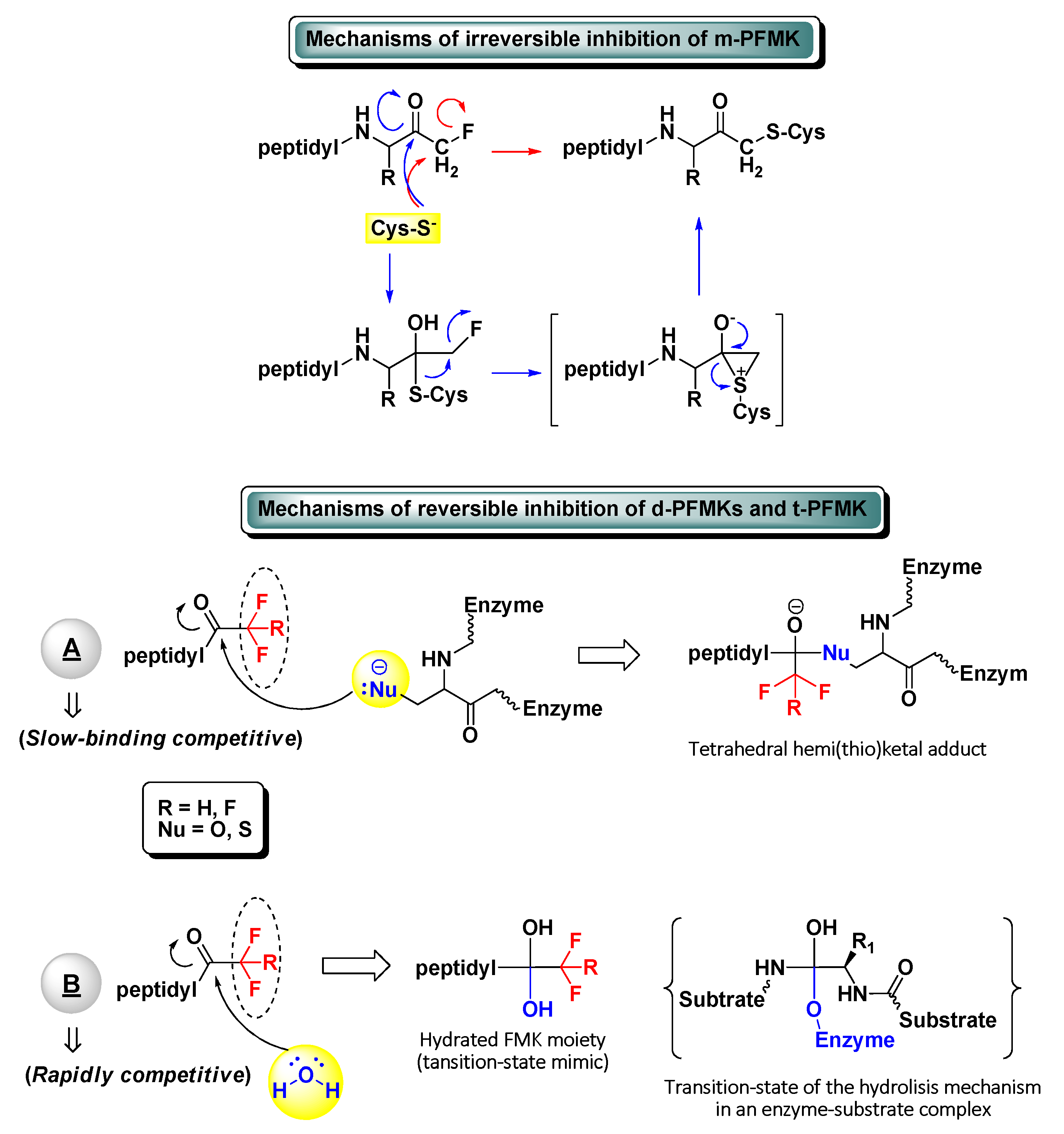
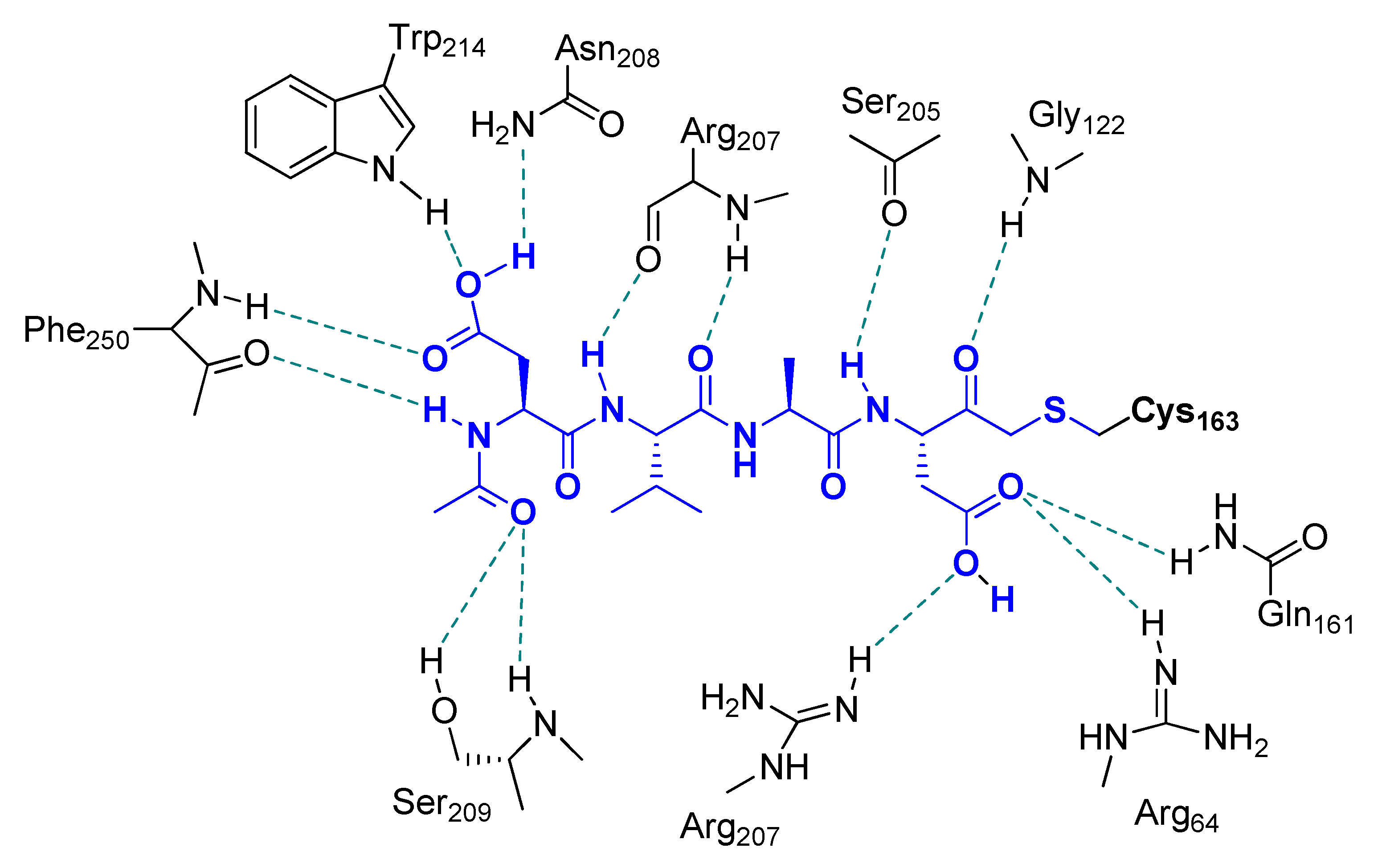
2. Peptidyl Mono-Fluoromethyl Ketones (m-PFMKs)
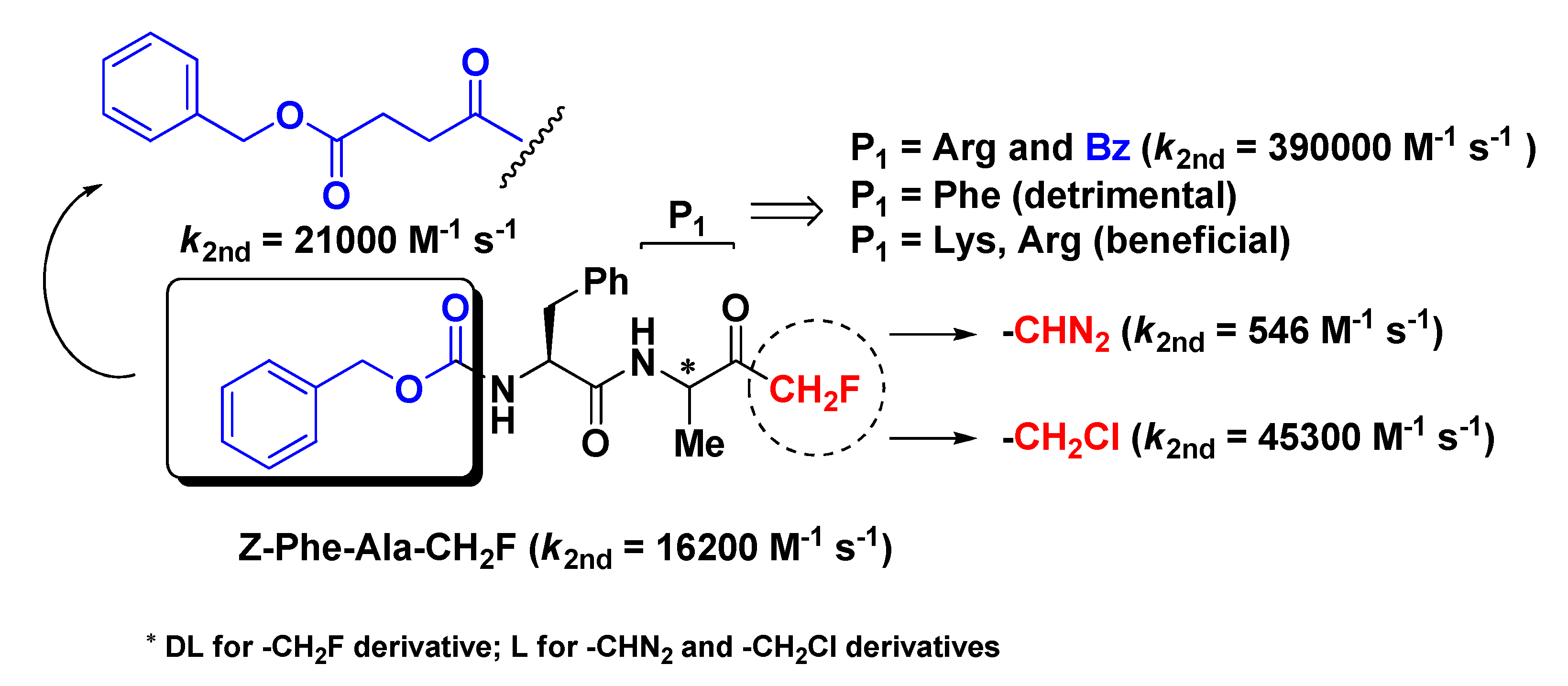
2.1. Cathepsins Inhibitors
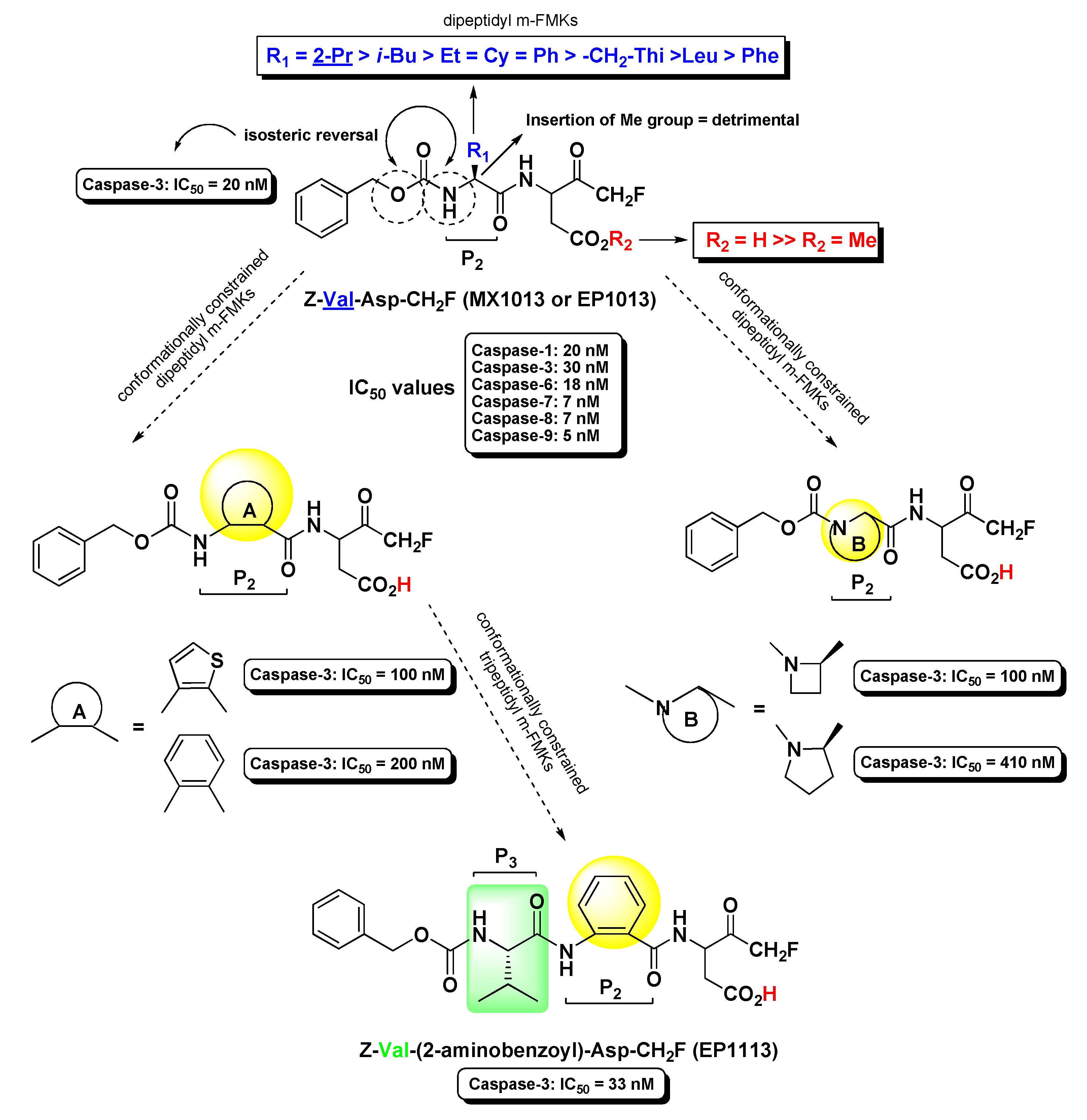
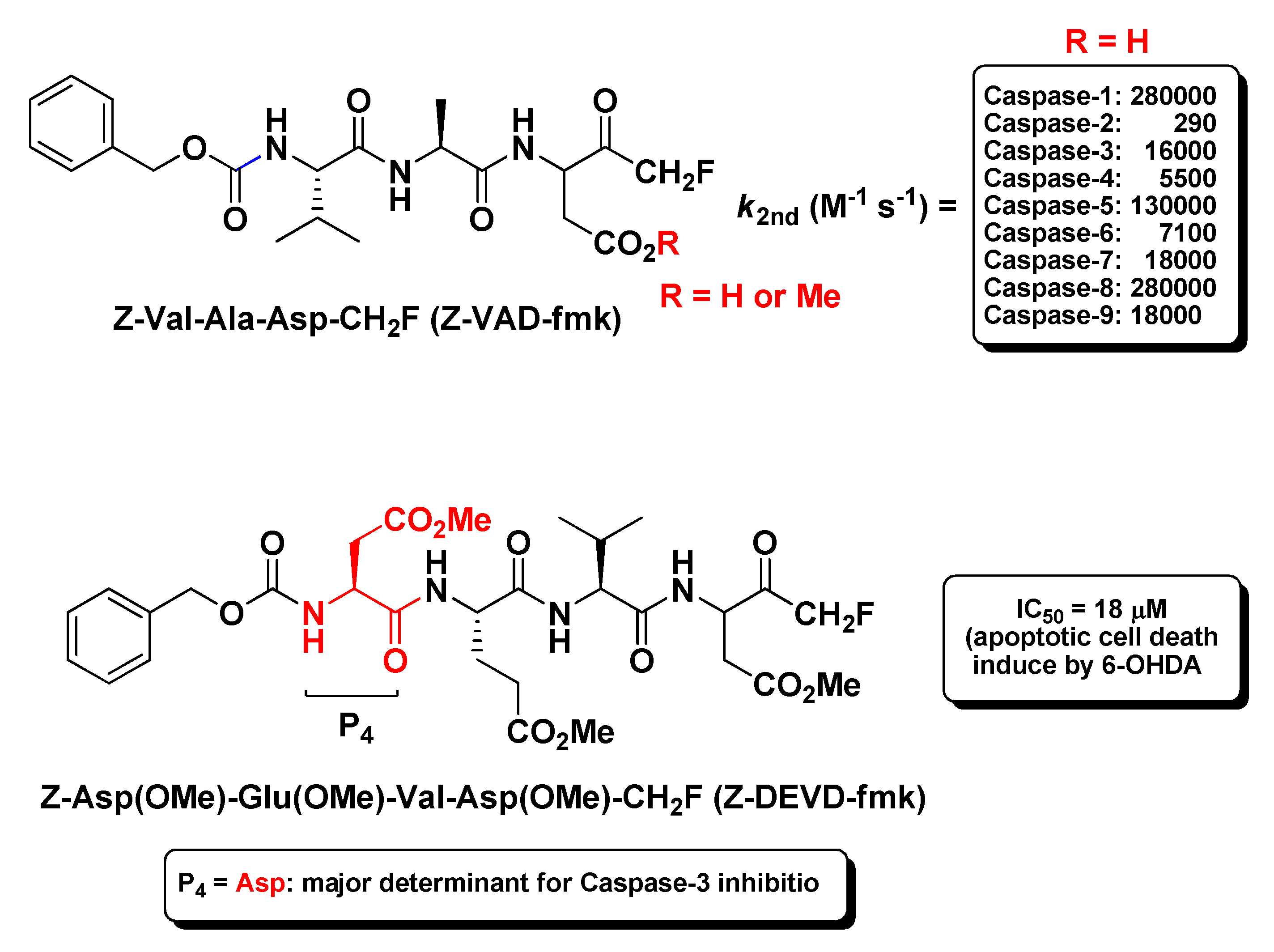
2.2. Caspases Inhibitors
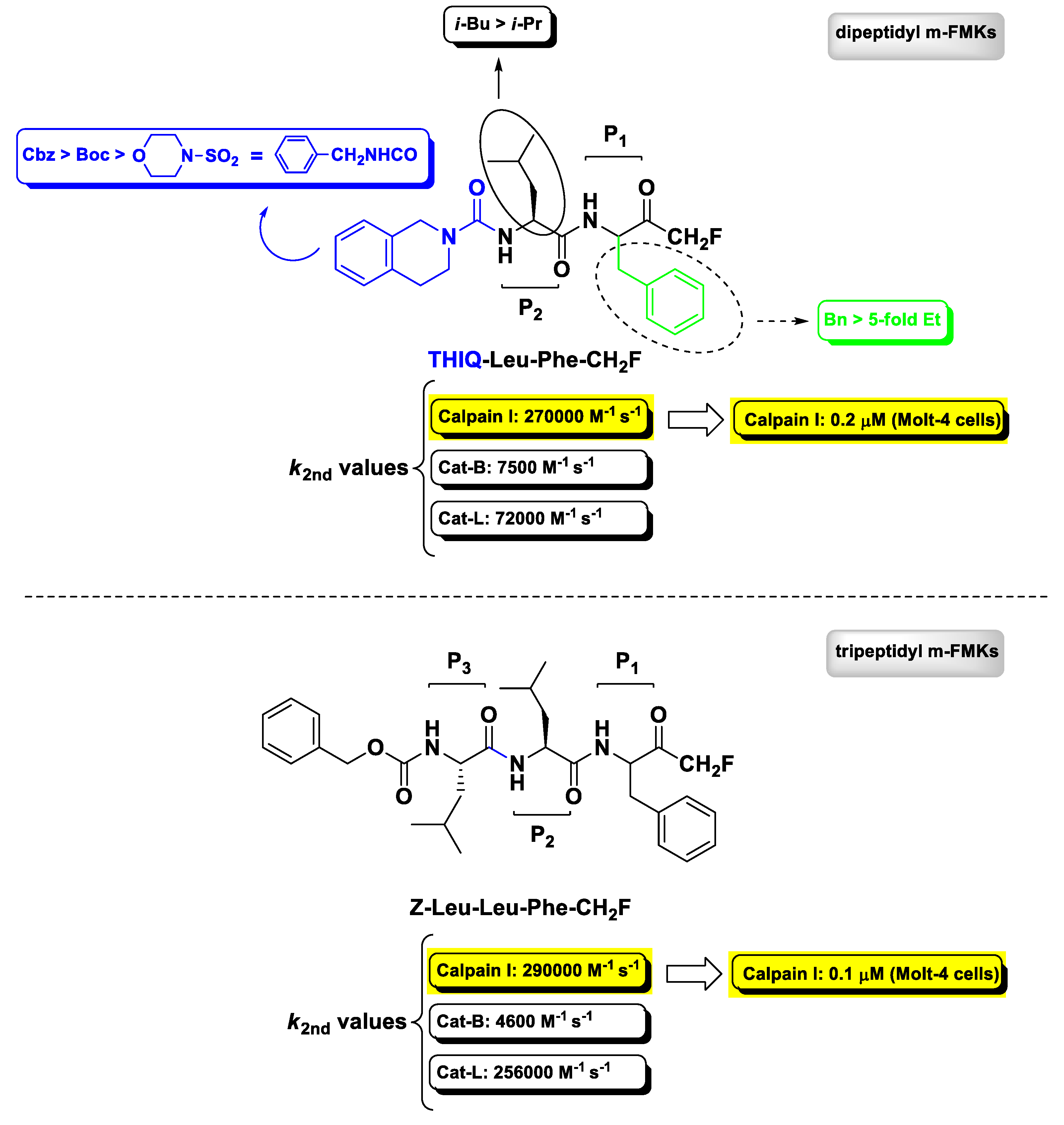
2.3. Calpain(s) Inhibitors
2.4. N-Glycanase Inhibitors
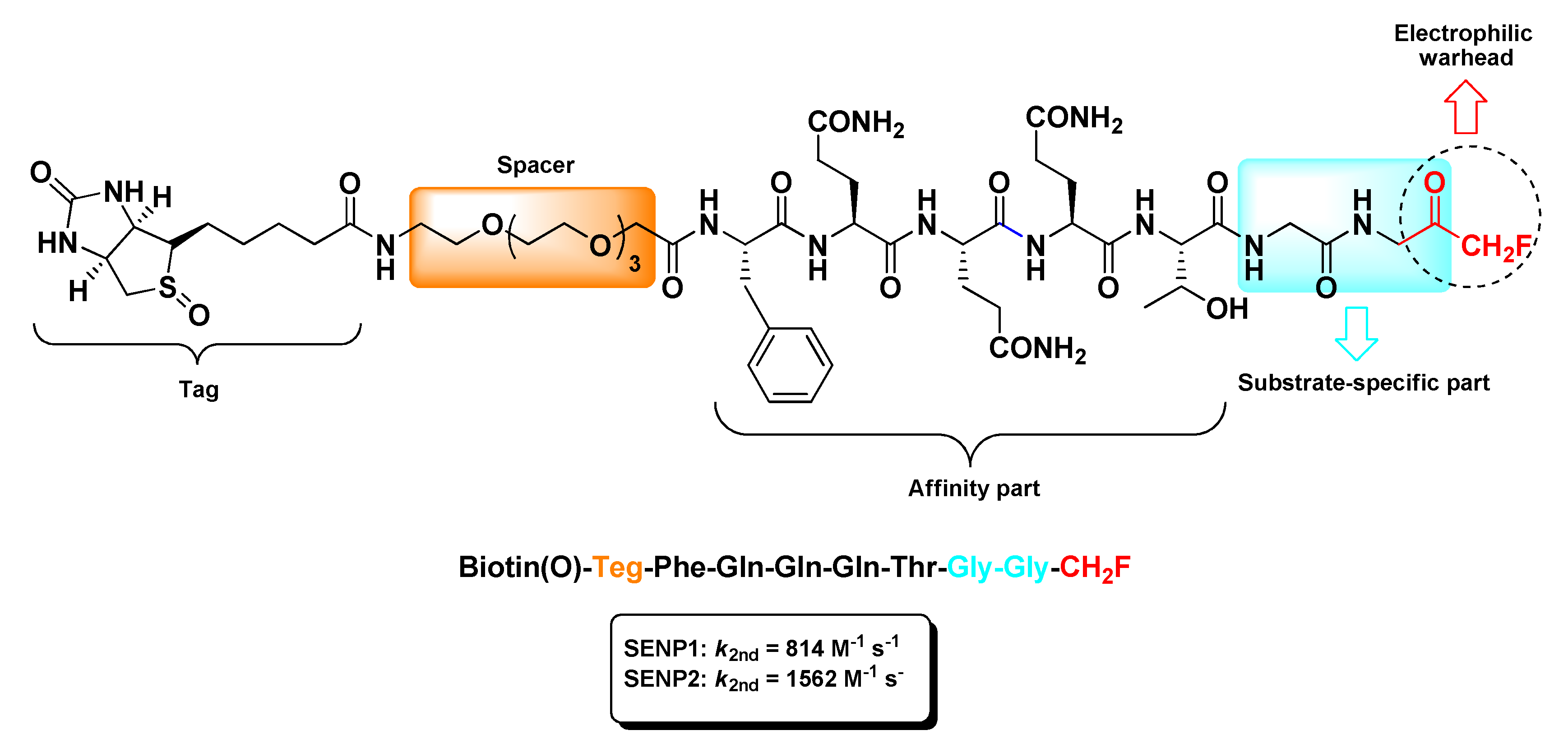
2.5. Sentrin/SUMO-Specific Proteases Inhibitors
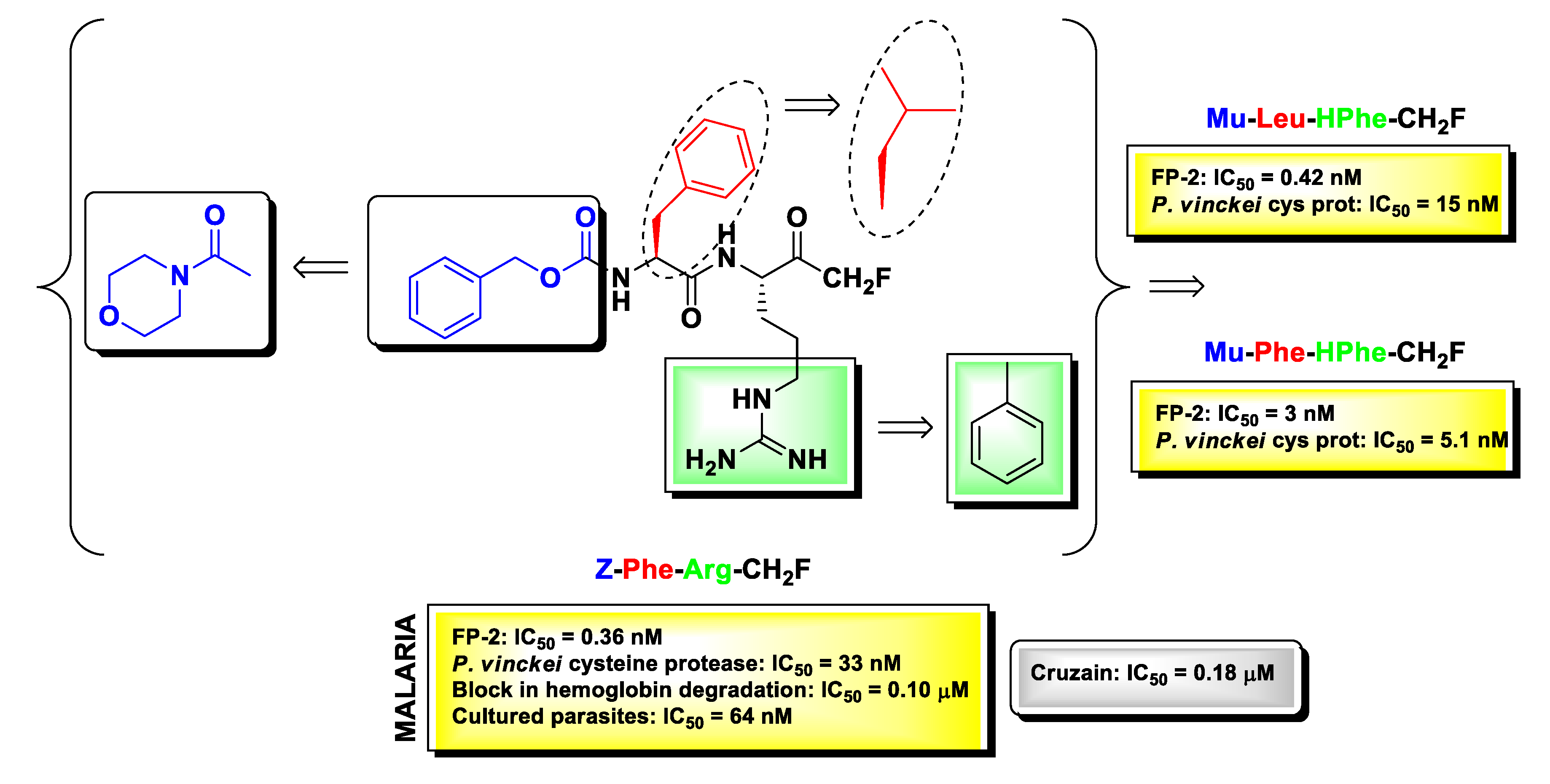
2.6. Protozoan Cysteine Proteases Inhibitors
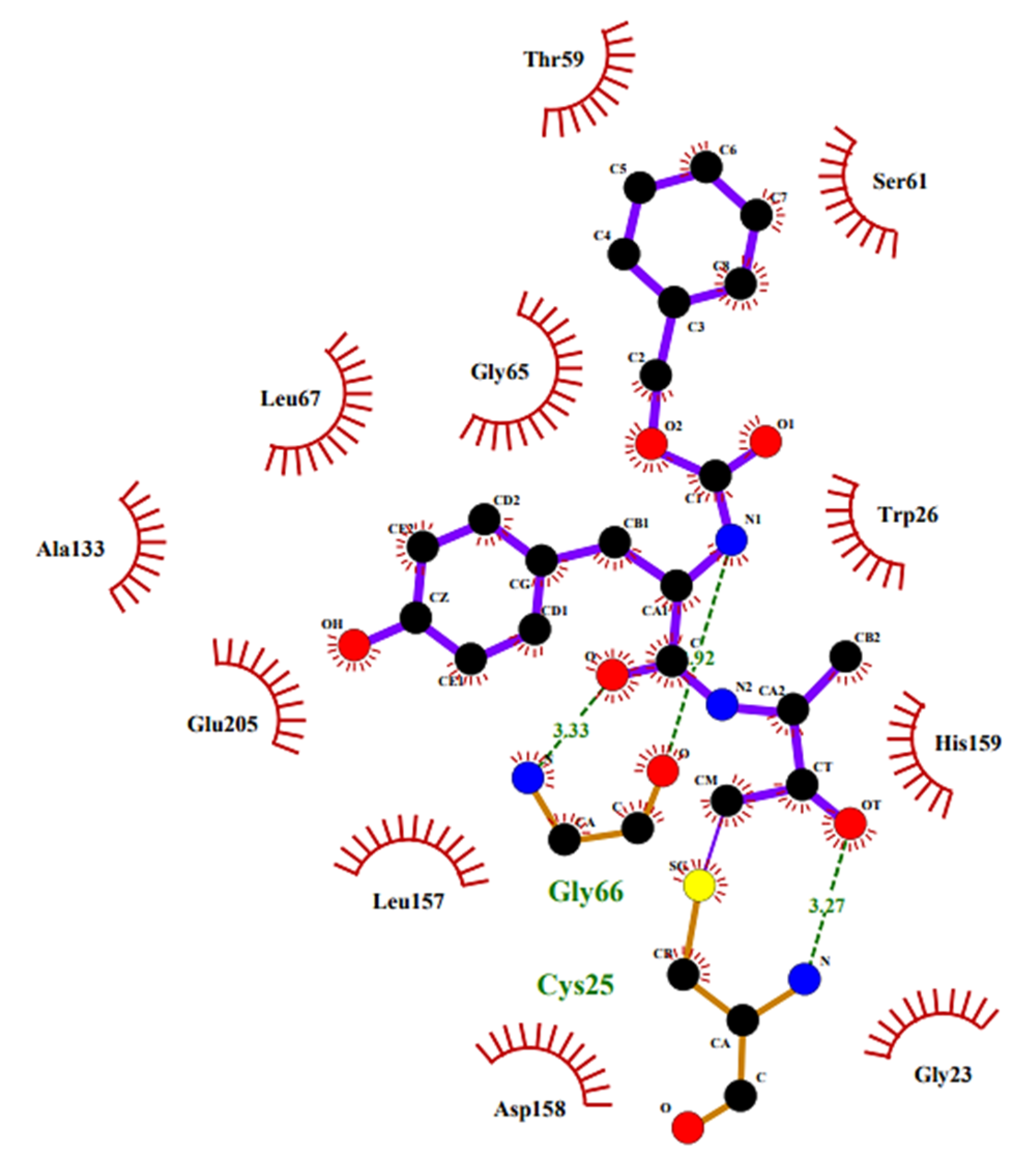
2.7. SARS-CoV Mpro and Other Viral Proteases Inhibitors
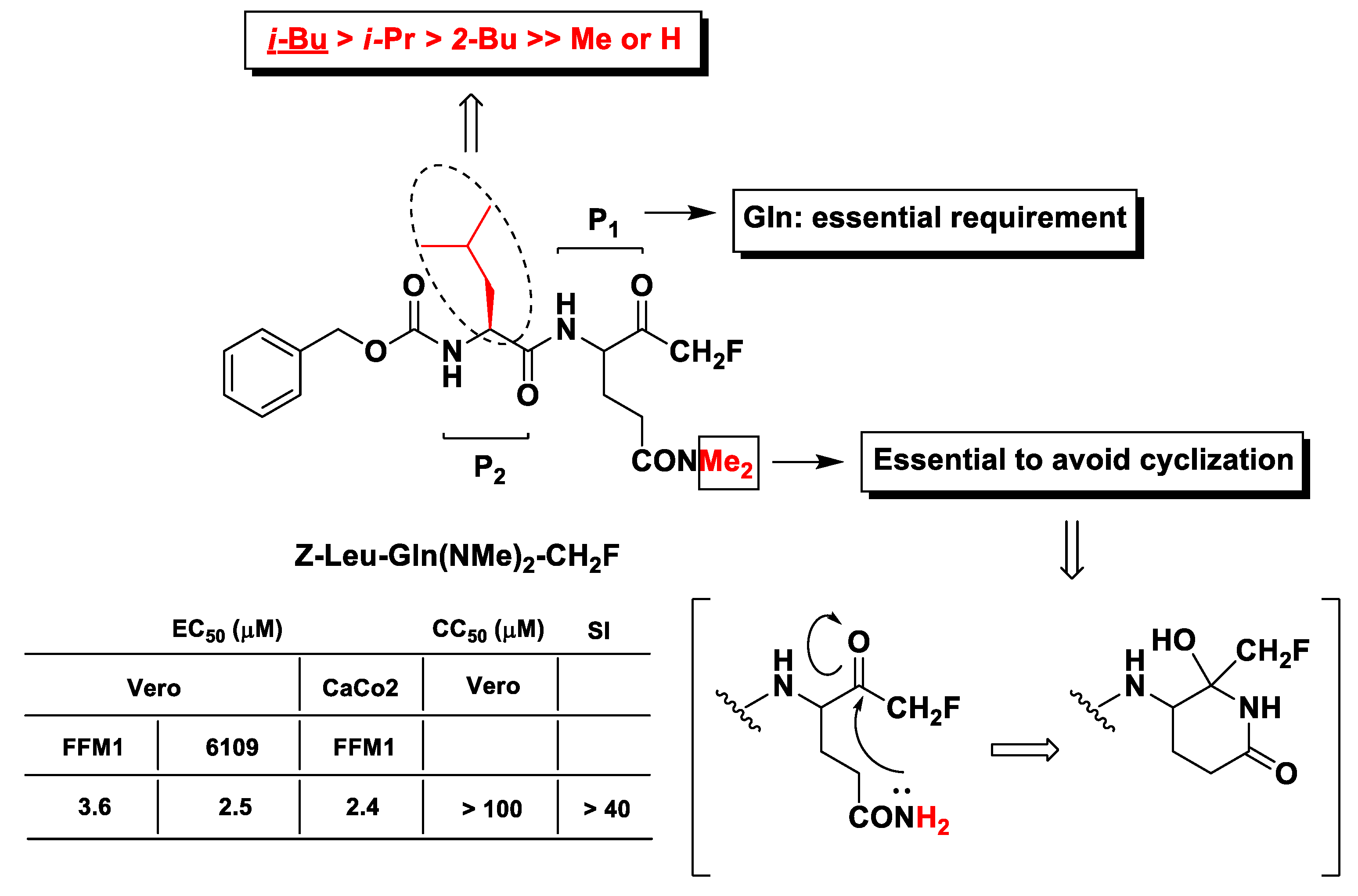
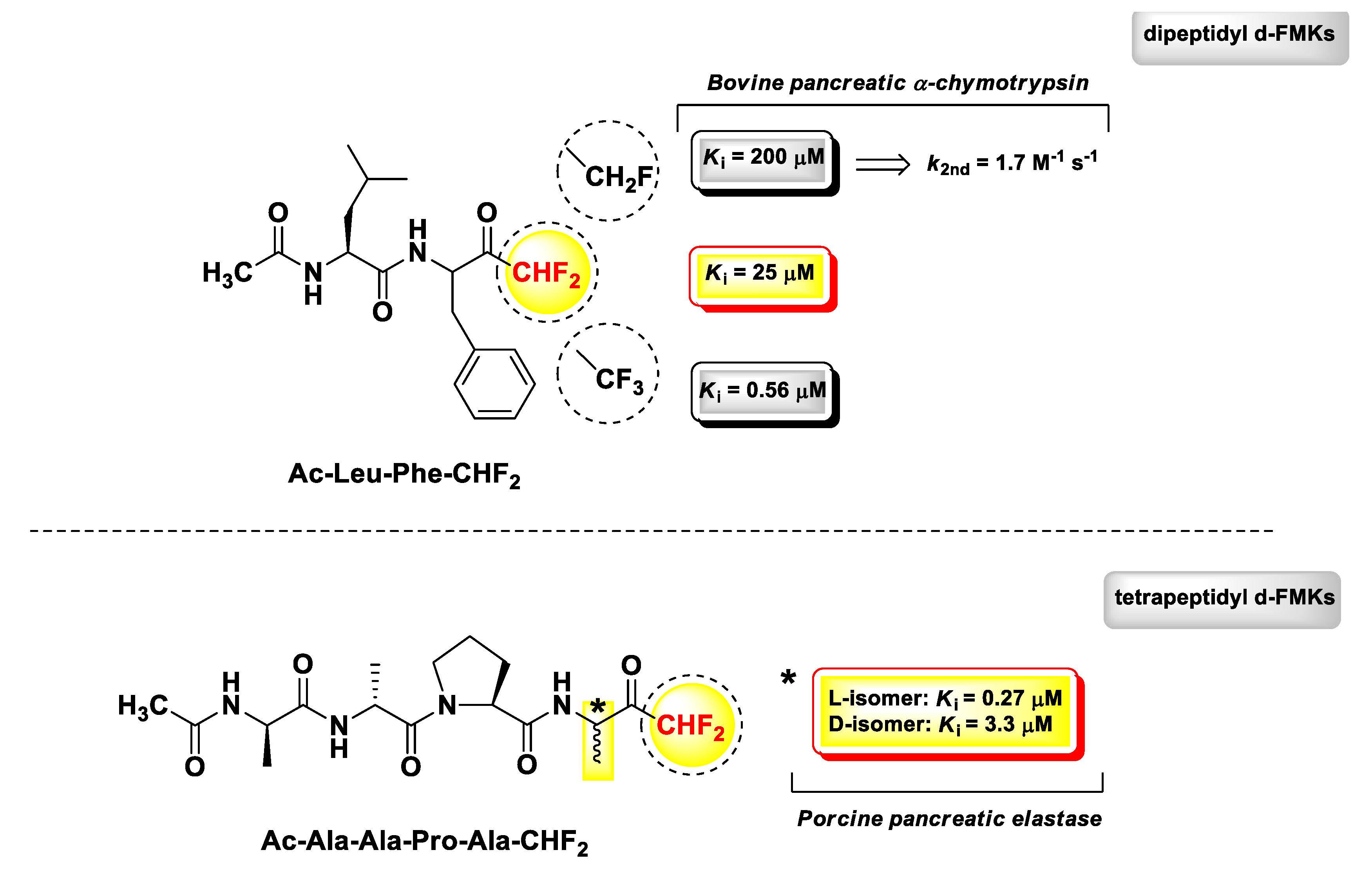
3. Peptidyl di-Fluoromethyl Ketones (d-PFMKs)
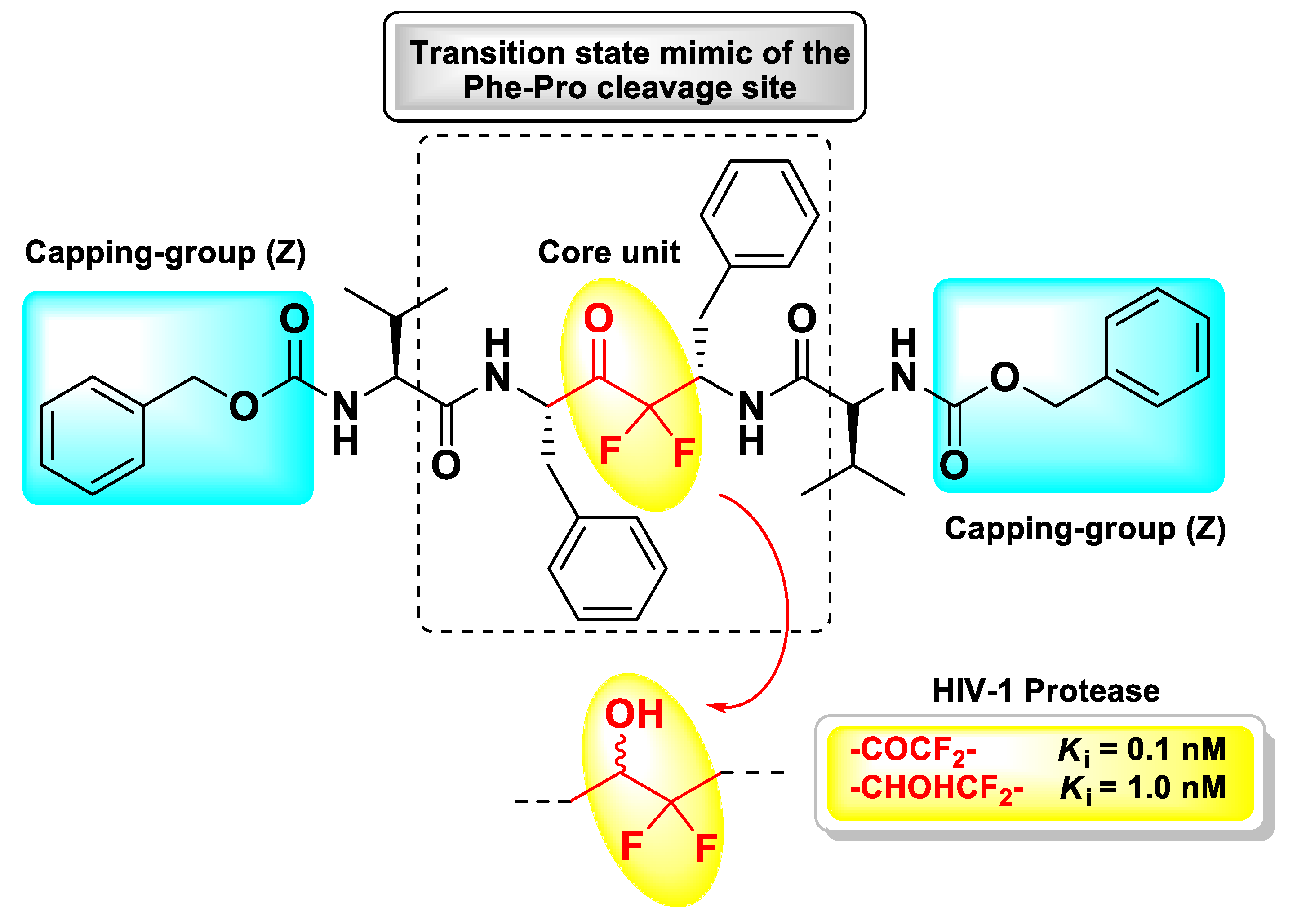
4. Peptidyl tri-Fluoromethyl Ketones (t-PFMKs)
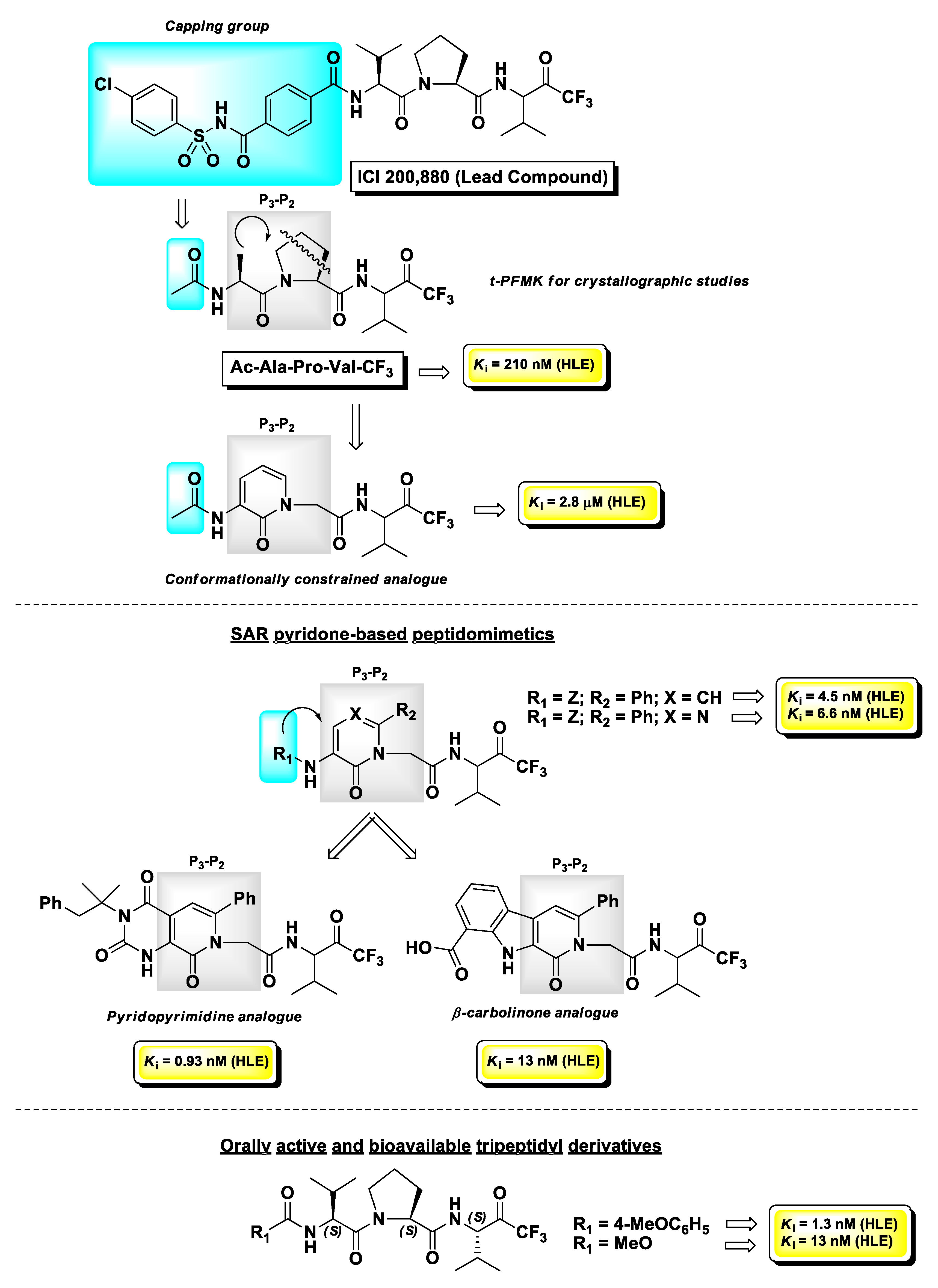
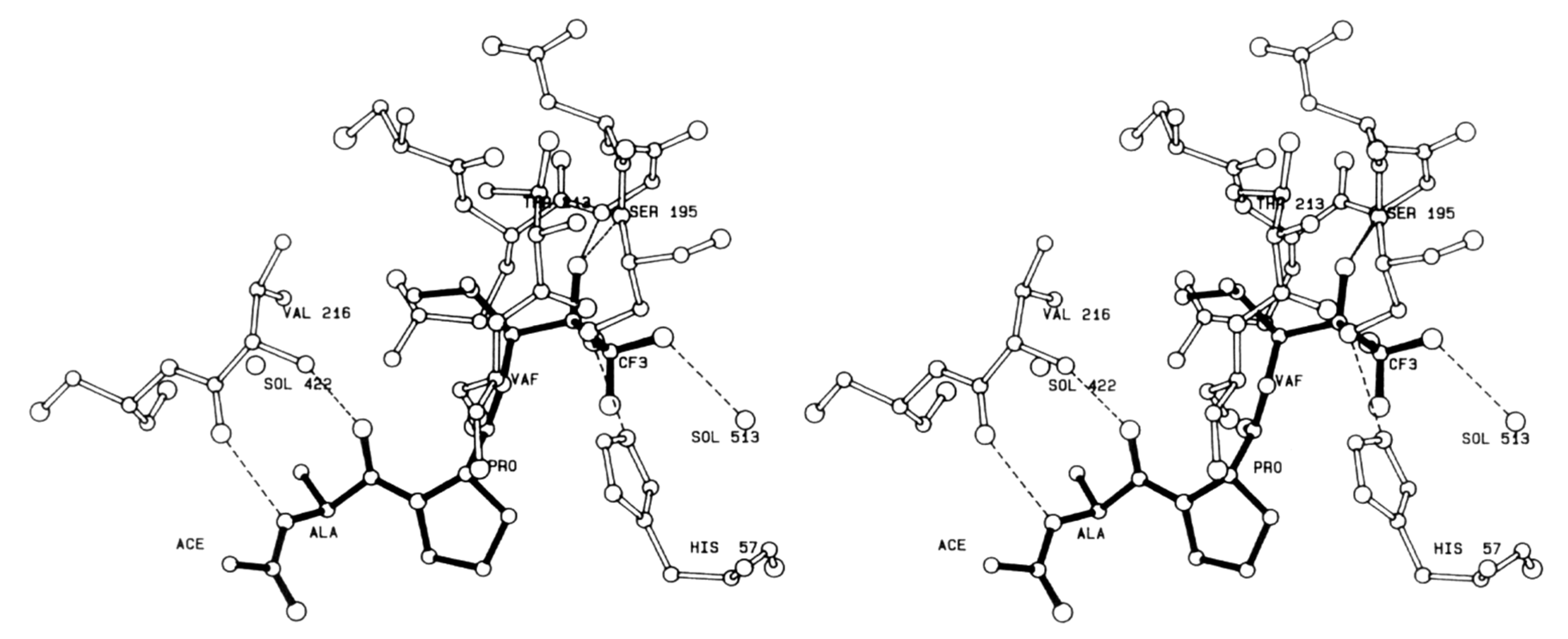
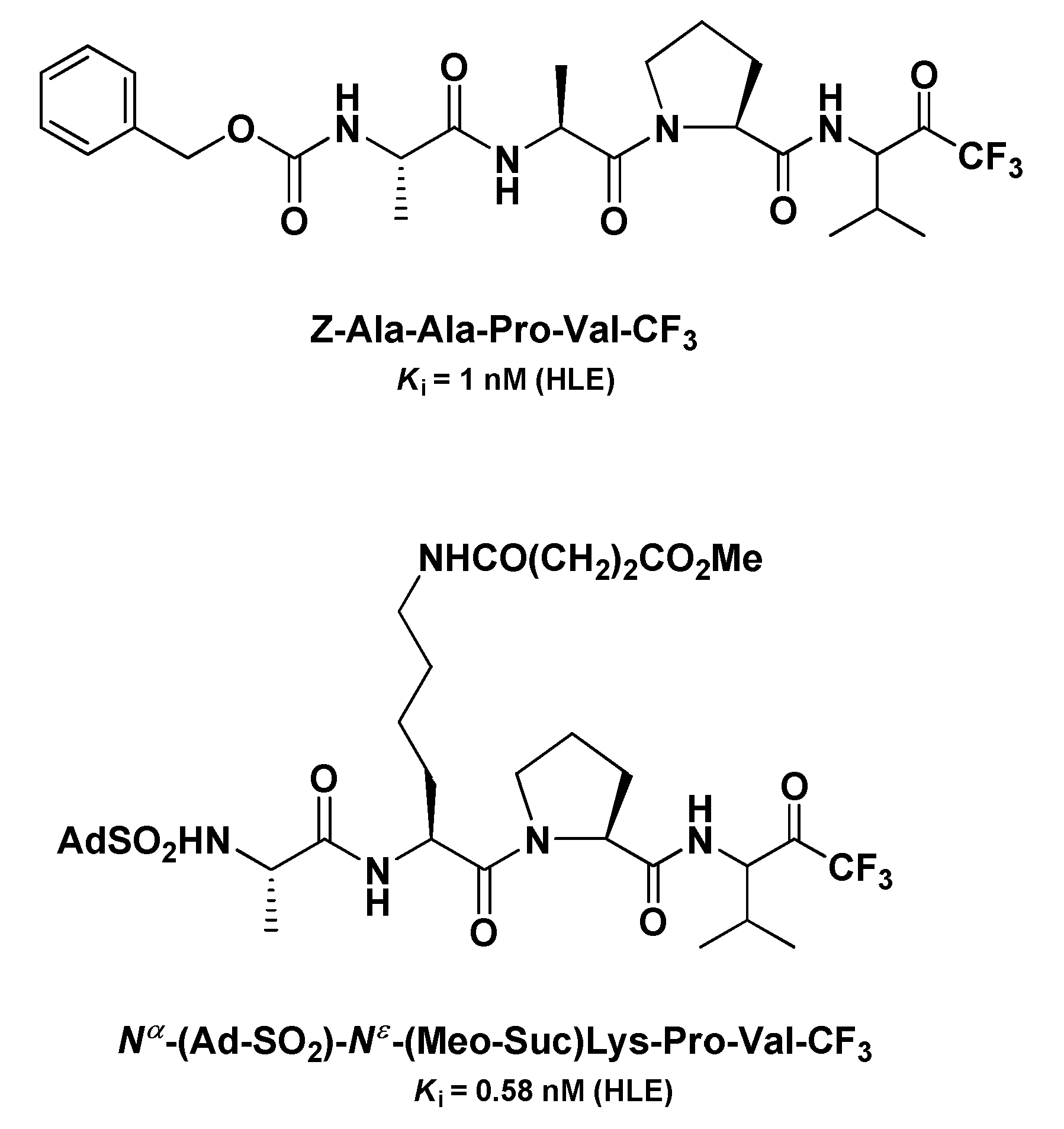
4.1. Elastase Inhibitors
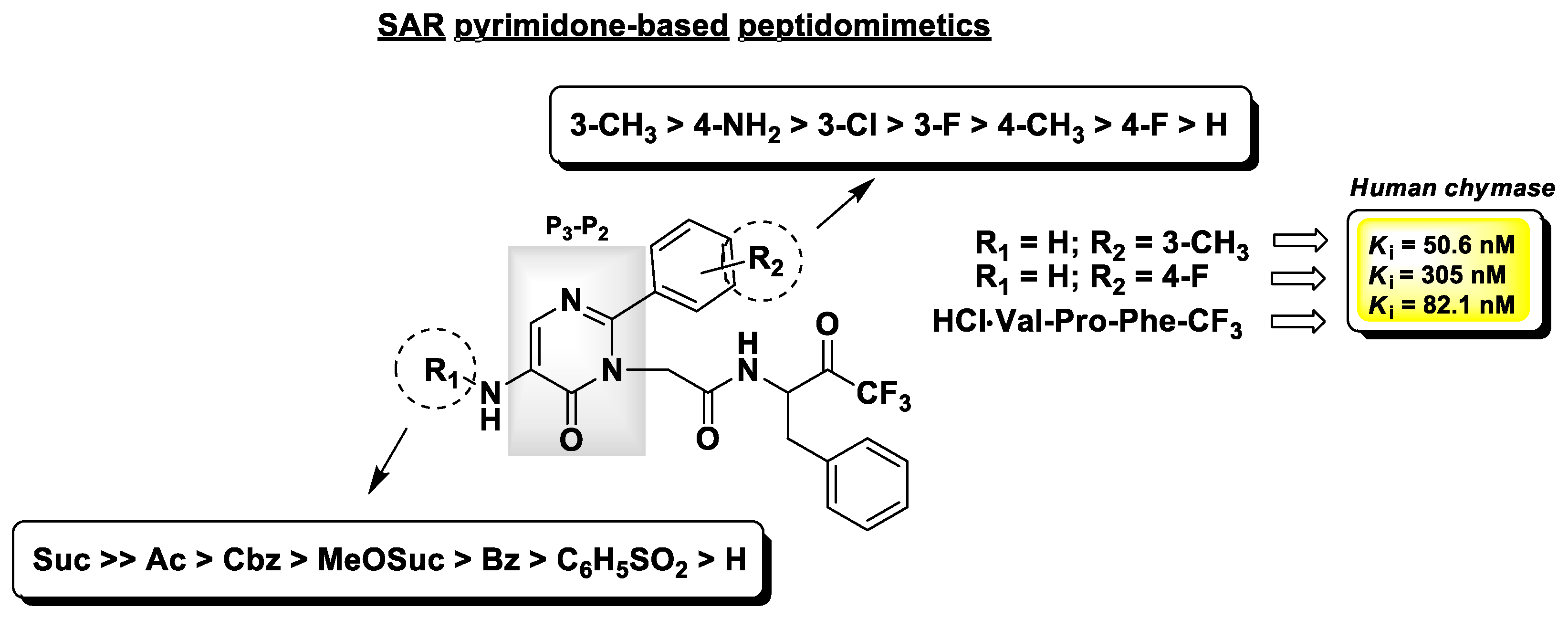
4.2. Chymase Inhibitors
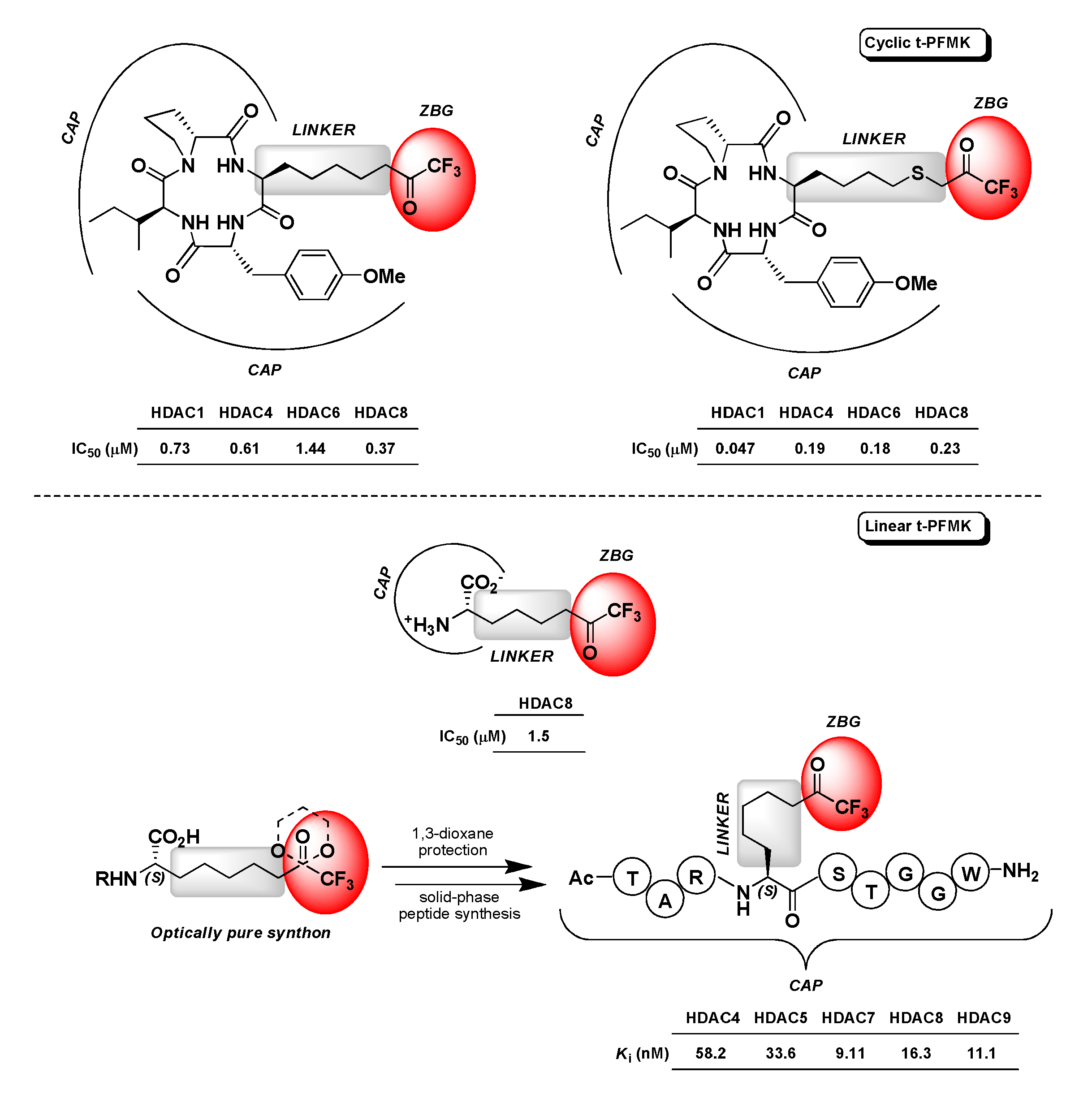
4.3. Histone Deacetylase Inhibitors
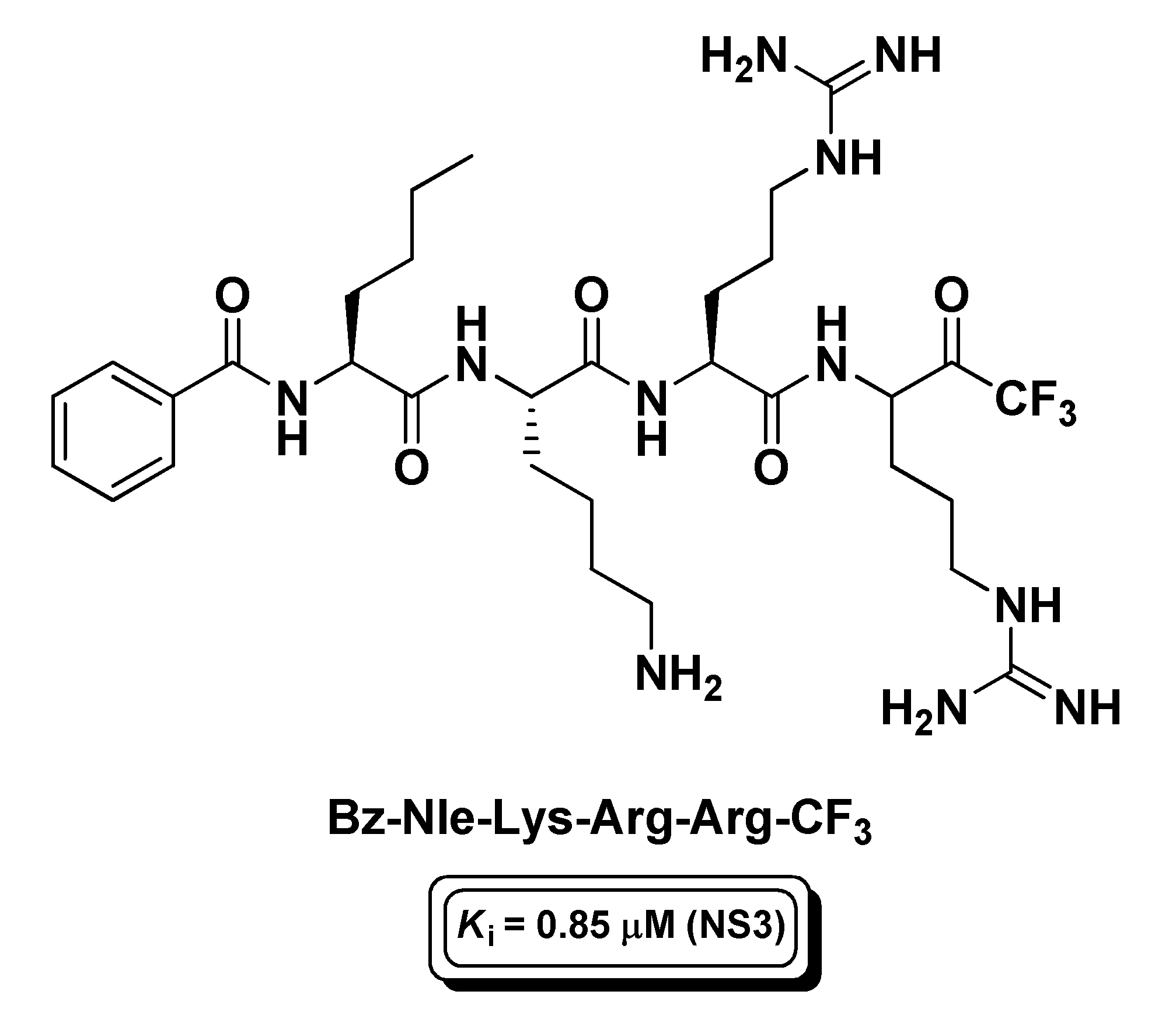
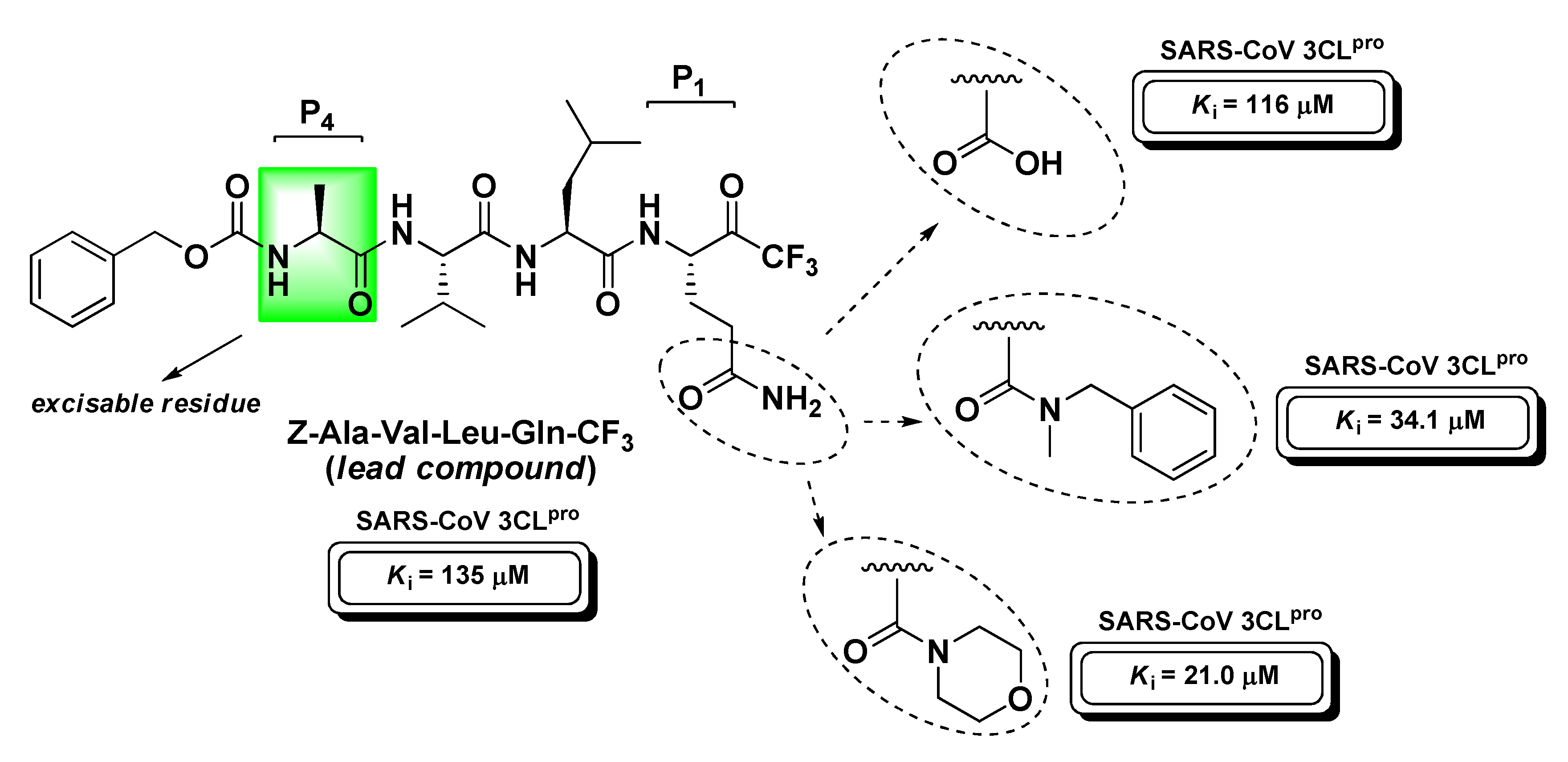
4.4. Viral Proteases Inhibitors
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 3CLpro | (Chymotrypsin-like protease) |
| Ac-PLVE-FMK | (Acetyl-Pro-Leu-Val-Glu(OMe)-CH2F) |
| Ac-PLVQ | (Acetyl-(Pro-Leu-Val-Gln)) |
| Ac-VLPE-FMK | (Acetyl-Val-Leu-Pro-Glu(OMe)-CH2F) |
| AMPA | (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) |
| Boc-Asp(OMe)-FMK | (Boc-Asp(OMe)-fluoromethyl ketone) |
| Casps | (Caspases) |
| Cat-B | (Cathepsin B) |
| Cat-C | (Cathepsin C) |
| Cats | (Cathepsins) |
| Cat-X | (Cathepsin X) |
| COVID-19 | (Coronavirus disease 2019) |
| d-PFMKs | (Peptidyl di-fluoromethyl ketones) |
| DUBs | (Deubiquitinating enzymes) |
| EP1113 | (Z-Val-(2-aminobenzoyl)-Asp-CH2F) |
| FP-2 | (Falcipain-2) |
| HAV | (Hepatitis A virus) |
| HDAC | (Histone deacetylase) |
| HLE | (Human leukocyte elastase) |
| m-FMK | (mono-Fluoromethyl ketone) |
| m-PFMKs | (Peptidyl mono-fluoromethyl ketones) |
| MPro | (SARS-CoV main protease) |
| Mu-Phe-HPhe-CH2F | (N-Morpholineurea-phenylalanyl-homophenylalanylfluoromethyl ketone) |
| NGLY1 | (Peptide N-glycanase) |
| N-glycanase | (Peptide N-glycanase) |
| NK cells | (Natural Killer cells) |
| NMDA | (N-methyl-d-aspartic acid) |
| PFMKs | (Peptidyl fluoromethyl ketones) |
| PLpro | (Papain-like protease) |
| SARS-CoV | (Severe acute respiratory syndrome coronavirus) |
| SARS-CoV-2 | (Severe acute respiratory syndrome coronavirus 2) |
| SENPs | (Sentrin/SUMO-specific proteases) |
| SUMOs | (Small ubiquitin-like modifiers) |
| TCP | (P. falciparum trophozoite cysteine proteinase) |
| THIQ-Leu-Phe-CH2F | (Tetrahydroisoquinoline Leu-Phe-CH2F) |
| TNFα | (Tumor necrosis factor) |
| t-PFMKs | (Peptidyl tri-fluoromethyl ketones) |
| ZBG | (Zinc binding group) |
| Z-DEVD-fmk | (Z-Asp(OMe)-Glu(OMe)-Val-Asp(OMe)-CH2F) |
| Z-DIPD-fmk | (Z-Asp(OMe)-Ile-Pro-Asp(OMe)-CH2F) |
| Z-IETD-fmk | (Z-Ile-Glu(OMe)-Thr-Asp(OMe)-CH2F) |
| Z-LEHD-fmk | (Z-Leu-Glu(OMe)-His-Asp(OMe)-CH2F) |
| Z-VAD-FMK | (Z-Valyl-alanyl-aspartyl-[O-methyl]-fluoromethyl ketone) |
| Z-VAD-fmk | (Z-Val-Ala-Asp-CH2F) |
| Z-YVAD-fmk | (Z-Tyr-Val-Ala-Asp(OMe)-CH2F) |
| β-CoVs | (β-Coronaviruses) |
References
- Barnes-Seeman, D.; Beck, J.; Springer, C. Fluorinated compounds in medicinal chemistry: Recent applications, synthetic advances and matched-pair analyses. Curr. Top. Med. Chem. 2014, 14, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Eichhold, T.H.; Hookfin, E.B.; Taiwo, Y.O.; De, B.; Wehmeyer, K.R. Isolation and quantification of fluoroacetate in rat tissues, following dosing of Z-Phe-Ala-CH2-F, a peptidyl fluoromethyl ketone protease inhibitor. J. Pharm. Biomed. Anal. 1997, 16, 459–467. [Google Scholar] [CrossRef]
- Kato, D.; Boatright, K.M.; Berger, A.B.; Nazif, T.; Blum, G.; Ryan, C.; Chehade, K.A.H.; Salvesen, G.S.; Bogyo, M. Activity-based probes that target diverse cysteine protease families. Nat. Chem. Biol. 2005, 1, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.C.; Asgian, J.L.; Ekici, O.D.; James, K.E. Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem. Rev. 2002, 102, 4639–4750. [Google Scholar] [CrossRef] [PubMed]
- Rayo, J.; Munoz, L.; Rosell, G.; Hammock, B.D.; Guerrero, A.; Luque, F.J.; Pouplana, R. Reactivity versus steric effects in fluorinated ketones as esterase inhibitors: A quantum mechanical and molecular dynamics study. J. Mol. Model. 2010, 16, 1753–1764. [Google Scholar] [CrossRef]
- Ngo, P.D.; Mansoorabadi, S.O.; Frey, P.A. Serine Protease Catalysis: A Computational Study of Tetrahedral Intermediates and Inhibitory Adducts. J. Phys. Chem. B 2016, 120, 7353–7359. [Google Scholar] [CrossRef]
- Rasnick, D. Synthesis of peptide fluoromethyl ketones and the inhibition of human cathepsin B. Anal. Biochem. 1985, 149, 461–465. [Google Scholar] [CrossRef]
- Jakos, T.; Pislar, A.; Jewett, A.; Kos, J. Cysteine Cathepsins in Tumor-Associated Immune Cells. Front. Immunol. 2019, 10, 2037. [Google Scholar] [CrossRef]
- Van Noorden, C.J.; Smith, R.E.; Rasnick, D. Cysteine proteinase activity in arthritic rat knee joints and the effects of a selective systemic inhibitor, Z-Phe-AlaCH2F. J. Rheumatol. 1988, 15, 1525–1535. [Google Scholar]
- Esser, R.E.; Watts, L.M.; Angelo, R.A.; Thornburg, L.P.; Prior, J.J.; Palmer, J.T. The effects of fluoromethyl ketone inhibitors of cathepsin B on adjuvant induced arthritis. J. Rheumatol. 1993, 20, 1176–1183. [Google Scholar]
- Tan, G.J.; Peng, Z.K.; Lu, J.P.; Tang, F.Q. Cathepsins mediate tumor metastasis. World J. Biol. Chem. 2013, 4, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Yagel, S.; Warner, A.H.; Nellans, H.N.; Lala, P.K.; Waghorne, C.; Denhardt, D.T. Suppression by cathepsin L inhibitors of the invasion of amnion membranes by murine cancer cells. Cancer Res. 1989, 49, 3553–3557. [Google Scholar] [PubMed]
- Rauber, P.; Angliker, H.; Walker, B.; Shaw, E. The Synthesis of Peptidylfluoromethanes and Their Properties as Inhibitors of Serine Proteinases and Cysteine Proteinases. Biochem. J. 1986, 239, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Angliker, H.; Wikstrom, P.; Rauber, P.; Shaw, E. The Synthesis of Lysylfluoromethanes and Their Properties as Inhibitors of Trypsin, Plasmin and Cathepsin-B. Biochem. J. 1987, 241, 871–875. [Google Scholar] [CrossRef] [PubMed]
- Angliker, H.; Wikstrom, P.; Rauber, P.; Stone, S.; Shaw, E. Synthesis and properties of peptidyl derivatives of arginylfluoromethanes. Biochem. J. 1988, 256, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.K.; Martin, L.A.; Watts, L.M.; Palmer, J.; Thornburg, L.; Prior, J.; Esser, R.E. Peptidyl fluoromethyl ketones as inhibitors of cathepsin B. Biochem. Pharm. 1992, 44, 1201–1207. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Kakegawa, H.; Narita, Y.; Hachiya, Y.; Hayakawa, T.; Kos, J.; Turk, V.; Katunuma, N. Significance of cathepsin B accumulation in synovial fluid of rheumatoid arthritis. Biochem. Biophys. Res. Commun. 2001, 283, 334–339. [Google Scholar] [CrossRef]
- Lawrence, C.P.; Kadioglu, A.; Yang, A.L.; Coward, W.R.; Chow, S.C. The cathepsin B inhibitor, z-FA-FMK, inhibits human T cell proliferation in vitro and modulates host response to pneumococcal infection in vivo. J. Immunol. 2006, 177, 3827–3836. [Google Scholar] [CrossRef]
- Kam, C.M.; Gotz, M.G.; Koot, G.; McGuire, M.; Thiele, D.; Hudig, D.; Powers, J.C. Design and evaluation of inhibitors for dipeptidyl peptidase I (Cathepsin C). Arch. Biochem. Biophys. 2004, 427, 123–134. [Google Scholar] [CrossRef]
- Turk, D.; Janjic, V.; Stern, I.; Podobnik, M.; Lamba, D.; Dahl, S.W.; Lauritzen, C.; Pedersen, J.; Turk, V.; Turk, B. Structure of human dipeptidyl peptidase I (cathepsin C): Exclusion domain added to an endopeptidase framework creates the machine for activation of granular serine proteases. EMBO J. 2001, 20, 6570–6582. [Google Scholar] [CrossRef]
- Rudzinska, M.; Parodi, A.; Maslova, V.D.; Efremov, Y.M.; Gorokhovets, N.V.; Makarov, V.A.; Popkov, V.A.; Golovin, A.V.; Zernii, E.Y.; Zamyatnin, A.A. Cysteine Cathepsins Inhibition Affects Their Expression and Human Renal Cancer Cell Phenotype. Cancers 2020, 12, 1310. [Google Scholar] [CrossRef] [PubMed]
- Gorokhovets, N.V.; Makarov, V.A.; Petushkova, A.I.; Prokopets, O.S.; Rubtsov, M.A.; Savvateeva, L.V.; Zernii, E.Y.; Zamyatnin, A.A., Jr. Rational Design of Recombinant Papain-Like Cysteine Protease: Optimal Domain Structure and Expression Conditions for Wheat-Derived Enzyme Triticain-alpha. Int. J. Mol. Sci. 2017, 18, 1395. [Google Scholar] [CrossRef] [PubMed]
- Cocchiaro, P.; de Pasquale, V.; Della Morte, R.; Tafuri, S.; Avallone, L.; Pizard, A.; Moles, A.; Pavone, L.M. The Multifaceted Role of the Lysosomal Protease Cathepsins in Kidney Disease. Front. Cell Dev. Biol. 2017, 5, 114. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 326 Pt 1, 1–16. [Google Scholar] [CrossRef]
- MacKenzie, S.H.; Schipper, J.L.; Clark, A.C. The potential for caspases in drug discovery. Curr. Opin. Drug Discov. Dev. 2010, 13, 568–576. [Google Scholar]
- Cheng, Y.; Deshmukh, M.; D’costa, A.; Demaro, J.A.; Gidday, J.M.; Shah, A.; Sun, Y.; Jacquin, M.F.; Johnson, E.M.; Holtzman, D.M. Caspase inhibitor affords neuroprotection with delayed administration in a rat model of neonatal hypoxic-ischemic brain injury. J. Clin. Investig. 1998, 101, 1992–1999. [Google Scholar] [CrossRef]
- Allen, J.W.; Knoblach, S.M.; Faden, A.I. Combined mechanical trauma and metabolic impairment in vitro induces NMDA receptor-dependent neuronal cell death and caspase-3-dependent apoptosis. FASEB J. 1999, 13, 1875–1882. [Google Scholar] [CrossRef]
- Werth, J.L.; Deshmukh, M.; Cocabo, J.; Johnson, E.M., Jr.; Rothman, S.M. Reversible physiological alterations in sympathetic neurons deprived of NGF but protected from apoptosis by caspase inhibition or Bax deletion. Exp. Neurol. 2000, 161, 203–211. [Google Scholar] [CrossRef]
- Chan, Y.M.; Wu, W.; Yip, H.K.; So, K.F.; Oppenheim, R.W. Caspase inhibitors promote the survival of avulsed spinal motoneurons in neonatal rats. Neuroreport 2001, 12, 541–545. [Google Scholar] [CrossRef]
- Brown, T.L.; Patil, S.; Basnett, R.K.; Howe, P.H. Caspase inhibitor BD-fmk distinguishes transforming growth factor beta-induced apoptosis from growth inhibition. Cell Growth Differ. 1998, 9, 869–875. [Google Scholar] [PubMed]
- Garcia-Calvo, M.; Peterson, E.P.; Leiting, B.; Ruel, R.; Nicholson, D.W.; Thornberry, N.A. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J. Biol. Chem. 1998, 273, 32608–32613. [Google Scholar] [CrossRef] [PubMed]
- Cowburn, A.S.; White, J.F.; Deighton, J.; Walmsley, S.R.; Chilvers, E.R. z-VAD-fmk augmentation of TNF alpha-stimulated neutrophil apoptosis is compound specific and does not involve the generation of reactive oxygen species. Blood 2005, 105, 2970–2972. [Google Scholar] [CrossRef]
- Clark, R.S.; Nathaniel, P.D.; Zhang, X.; Dixon, C.E.; Alber, S.M.; Watkins, S.C.; Melick, J.A.; Kochanek, P.M.; Graham, S.H. boc-Aspartyl(OMe)-fluoromethylketone attenuates mitochondrial release of cytochrome c and delays brain tissue loss after traumatic brain injury in rats. J. Cereb. Blood Flow Metab. 2007, 27, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, J.-C.; Zhou, Z.-L.; Yang, W.; Guastella, J.; Drewe, J.; Cai, S.X. Dipeptidyl aspartyl fluoromethylketones as potent caspase-3 inhibitors: SAR of the P2 amino acid. Bioorg. Med. Chem. Lett. 2004, 14, 1269–1272. [Google Scholar] [CrossRef]
- Yang, W.; Guastella, J.; Huang, J.C.; Wang, Y.; Zhang, L.; Xue, D.; Tran, M.; Woodward, R.; Kasibhatla, S.; Tseng, B.; et al. MX1013, a dipeptide caspase inhibitor with potent in vivo antiapoptotic activity. Br. J. Pharm. 2003, 140, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Mittl, P.R.; Di Marco, S.; Krebs, J.F.; Bai, X.; Karanewsky, D.S.; Priestle, J.P.; Tomaselli, K.J.; Grutter, M.G. Structure of recombinant human CPP32 in complex with the tetrapeptide acetyl-Asp-Val-Ala-Asp fluoromethyl ketone. J. Biol. Chem. 1997, 272, 6539–6547. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Guan, L.; Jia, S.; Tseng, B.; Drewe, J.; Cai, S.X. Dipeptidyl aspartyl fluoromethylketones as potent caspase inhibitors: Peptidomimetic replacement of the P2 α-amino acid by a α-hydroxy acid. Bioorg. Med. Chem. Lett. 2005, 15, 1379–1383. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jia, S.; Tseng, B.; Drewe, J.; Cai, S.X. Dipeptidyl aspartyl fluoromethylketones as potent caspase inhibitors: Peptidomimetic replacement of the P2 amino acid by 2-aminoaryl acids and other non-natural amino acids. Bioorg. Med. Chem. Lett. 2007, 17, 6178–6182. [Google Scholar] [CrossRef]
- Thornberry, N.A. Caspases: Key mediators of apoptosis. Chem. Biol. 1998, 5, R97–R103. [Google Scholar] [CrossRef]
- Rodriguez, I.; Matsuura, K.; Ody, C.; Nagata, S.; Vassalli, P. Systemic injection of a tripeptide inhibits the intracellular activation of CPP32-like proteases in vivo and fully protects mice against Fas-mediated fulminant liver destruction and death. J. Exp. Med. 1996, 184, 2067–2072. [Google Scholar] [CrossRef]
- Chandler, J.M.; Cohen, G.M.; MacFarlane, M. Different subcellular distribution of caspase-3 and caspase-7 following Fas-induced apoptosis in mouse liver. J. Biol. Chem. 1998, 273, 10815–10818. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Friedlander, R.M.; Gagliardini, V.; Ayata, C.; Fink, K.; Huang, Z.; Shimizu-Sasamata, M.; Yuan, J.; Moskowitz, M.A. Inhibition of interleukin 1β converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc. Natl. Acad. Sci. USA 1997, 94, 2007–2012. [Google Scholar] [CrossRef] [PubMed]
- Endres, M.; Namura, S.; Shimizu-Sasamata, M.; Waeber, C.; Zhang, L.; Gomez-Isla, T.; Hyman, B.T.; Moskowitz, M.A. Attenuation of delayed neuronal death after mild focal ischemia in mice by inhibition of the caspase family. J. Cereb. Blood Flow Metab. 1998, 18, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Wiessner, C.; Sauer, D.; Alaimo, D.; Allegrini, P.R. Protective effect of a caspase inhibitor in models for cerebral ischemia in vitro and in vivo. Cell Mol. Biol. (Noisy-Le-Grand) 2000, 46, 53–62. [Google Scholar]
- Li, H.; Colbourne, F.; Sun, P.; Zhao, Z.; Buchan, A.M.; Iadecola, C.J.S. Caspase inhibitors reduce neuronal injury after focal but not global cerebral ischemia in rats. Stroke 2000, 31, 176–180. [Google Scholar] [CrossRef]
- Yaoita, H.; Ogawa, K.; Maehara, K.; Maruyama, Y. Attenuation of ischemia/reperfusion injury in rats by a caspase inhibitor. Circulation 1998, 97, 276–281. [Google Scholar] [CrossRef]
- Huang, J.Q.; Radinovic, S.; Rezaiefar, P.; Black, S.C. In vivo myocardial infarct size reduction by a caspase inhibitor administered after the onset of ischemia. Eur. J. Pharm. 2000, 402, 139–142. [Google Scholar] [CrossRef]
- Mocanu, M.M.; Baxter, G.F.; Yellon, D.M. Caspase inhibition and limitation of myocardial infarct size: Protection against lethal reperfusion injury. Br. J. Pharm. 2000, 130, 197–200. [Google Scholar] [CrossRef]
- Farber, A.; Connors, J.P.; Friedlander, R.M.; Wagner, R.J.; Powell, R.J.; Cronenwett, J.L. A specific inhibitor of apoptosis decreases tissue injury after intestinal ischemia-reperfusion in mice. J. Vasc. Surg. 1999, 30, 752–760. [Google Scholar] [CrossRef]
- Iwata, A.; Harlan, J.M.; Vedder, N.B.; Winn, R.K. The caspase inhibitor z-VAD is more effective than CD18 adhesion blockade in reducing muscle ischemia-reperfusion injury: Implication for clinical trials. Blood 2002, 100, 2077–2080. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Daemen, M.A.; van’t Veer, C.; Denecker, G.; Heemskerk, V.H.; Wolfs, T.G.; Clauss, M.; Vandenabeele, P.; Buurman, W.A. Inhibition of apoptosis induced by ischemia-reperfusion prevents inflammation. J. Clin. Investig. 1999, 104, 541–549. [Google Scholar] [CrossRef]
- Revesz, L.; Briswalter, C.; Heng, R.; Leutwiler, A.; Mueller, R.; Wuethrich, H.-J.J.T.L. Synthesis of P1 aspartate-based peptide acyloxymethyl and fluoromethyl ketones as inhibitors of interleukin-1β-converting enzyme. Tetrahedron Lett. 1994, 35, 9693–9696. [Google Scholar] [CrossRef]
- Rozman-Pungercar, J.; Kopitar-Jerala, N.; Bogyo, M.; Turk, D.; Vasiljeva, O.; Stefe, I.; Vandenabeele, P.; Bromme, D.; Puizdar, V.; Fonovic, M.; et al. Inhibition of papain-like cysteine proteases and legumain by caspase-specific inhibitors: When reaction mechanism is more important than specificity. Cell Death Differ. 2003, 10, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.S.; Novak, R.; Herzog, K.H.; Bodner, S.M.; Cleveland, J.L.; Tuomanen, E.I. Neuroprotection by a caspase inhibitor in acute bacterial meningitis. Nat. Med. 1999, 5, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Adrain, C.; Creagh, E.M.; Martin, S.J.J.T.E.J. Apoptosis-associated release of Smac/DIABLO from mitochondria requires active caspases and is blocked by Bcl-2. EMBO J. 2001, 20, 6627–6636. [Google Scholar] [CrossRef] [PubMed]
- Caserta, T.M.; Smith, A.N.; Gultice, A.D.; Reedy, M.A.; Brown, T.L. Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis 2003, 8, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Qiu, J.; Hirt, L.; Dalkara, T.; Moskowitz, M.A. Synergistic protective effect of caspase inhibitors and bFGF against brain injury induced by transient focal ischaemia. Br. J. Pharm. 2001, 133, 345–350. [Google Scholar] [CrossRef]
- Iwata, A.; Nishio, K.; Winn, R.K.; Chi, E.Y.; Henderson, W.R., Jr.; Harlan, J.M. A broad-spectrum caspase inhibitor attenuates allergic airway inflammation in murine asthma model. J. Immunol. 2003, 170, 3386–3391. [Google Scholar] [CrossRef]
- Okuda, Y.; Sakoda, S.; Fujimura, H.; Yanagihara, T. The effect of apoptosis inhibitors on experimental autoimmune encephalomyelitis: Apoptosis as a regulatory factor. Biochem. Biophys. Res. Commun. 2000, 267, 826–830. [Google Scholar] [CrossRef]
- Kopalli, S.R.; Kang, T.B.; Koppula, S. Necroptosis inhibitors as therapeutic targets in inflammation mediated disorders—A review of the current literature and patents. Expert Opin. Ther. Pat. 2016, 26, 1239–1256. [Google Scholar] [CrossRef]
- Shaalan, A.; Carpenter, G.; Proctor, G. Caspases are key regulators of inflammatory and innate immune responses mediated by TLR3 in vivo. Mol. Immunol. 2018, 94, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yao, X.; Zhu, Y.; Zhang, H.; Wang, H.; Ma, Q.; Yan, F.; Yang, Y.; Zhang, J.; Shi, H.; et al. The Caspase Inhibitor Z-VAD-FMK Alleviates Endotoxic Shock via Inducing Macrophages Necroptosis and Promoting MDSCs-Mediated Inhibition of Macrophages Activation. Front. Immunol. 2019, 10, 1824. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Shi, L.; Zou, X.; Zheng, X.; Zhang, F.; Ding, X.; Zhu, H.; Shen, Y. Caspase inhibitor zVAD-fmk protects against acute pancreatitis-associated lung injury via inhibiting inflammation and apoptosis. Pancreatology 2016, 16, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Pero, M.E.; Zullo, G.; Esposito, L.; Iannuzzi, A.; Lombardi, P.; De Canditiis, C.; Neglia, G.; Gasparrini, B. Inhibition of apoptosis by caspase inhibitor Z-VAD-FMK improves cryotolerance of in vitro derived bovine embryos. Theriogenology 2018, 108, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Rudel, T. Caspase inhibitors in prevention of apoptosis. Herz 1999, 24, 236–241. [Google Scholar] [CrossRef]
- Chen, J.; Nagayama, T.; Jin, K.L.; Stetler, R.A.; Zhu, R.L.; Graham, S.H.; Simon, R.P. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J. Neurosci. 1998, 18, 4914–4928. [Google Scholar] [CrossRef]
- Zhan, R.Z.; Wu, C.R.; Fujihara, H.; Taga, K.; Qi, S.H.; Naito, M.; Shimoji, K. Both caspase-dependent and caspase-independent pathways may be involved in hippocampal CA1 neuronal death because of loss of cytochrome c from mitochondria in a rat forebrain ischemia model. J. Cerebr. Blood Flow Met. 2001, 21, 529–540. [Google Scholar] [CrossRef]
- Zhao, H.; Yenari, M.A.; Cheng, D.; Sapolsky, R.M.; Steinberg, G.K. Biphasic cytochrome c release after transient global ischemia and its inhibition by hypothermia. J. Cereb. Blood Flow Metab. 2005, 25, 1119–1129. [Google Scholar] [CrossRef]
- Teschendorf, P.; Vogel, P.; Wippel, A.; Krumnikl, J.J.; Spohr, F.; Bottiger, B.W.; Popp, E. The effect of intracerebroventricular application of the caspase-3 inhibitor zDEVD-FMK on neurological outcome and neuronal cell death after global cerebral ischaemia due to cardiac arrest in rats. Resuscitation 2008, 78, 85–91. [Google Scholar] [CrossRef]
- Nersesyan, H.; Hyder, F.; Rothman, D.L.; Blumenfeld, H. Dynamic fMRI and EEG recordings during spike-wave seizures and generalized tonic-clonic seizures in WAG/Rij rats. J. Cereb. Blood Flow Metab. 2004, 24, 589–599. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.J.; Rubin, L.L.; Philpott, K.L. Involvement of caspases in sympathetic neuron apoptosis. J. Cell Sci. 1997, 110 Pt 18, 2165–2173. [Google Scholar]
- Kanthasamy, A.G.; Anantharam, V.; Zhang, D.; Latchoumycandane, C.; Jin, H.; Kaul, S.; Kanthasamy, A. A novel peptide inhibitor targeted to caspase-3 cleavage site of a proapoptotic kinase protein kinase C delta (PKCδ) protects against dopaminergic neuronal degeneration in Parkinson’s disease models. Free Radic. Biol. Med. 2006, 41, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Kaul, S.; Kanthasamy, A.; Kitazawa, M.; Anantharam, V.; Kanthasamy, A.G. Caspase-3 dependent proteolytic activation of protein kinase C delta mediates and regulates 1-methyl-4-phenylpyridinium (MPP+)-induced apoptotic cell death in dopaminergic cells: Relevance to oxidative stress in dopaminergic degeneration. Eur. J. Neurosci. 2003, 18, 1387–1401. [Google Scholar] [CrossRef] [PubMed]
- Ono, Y.; Sorimachi, H. Calpains—An elaborate proteolytic system. Biochim. Biophys. Acta 2012, 1824, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Kawai, M.; Quincy, D.A.; Lane, B.; Mollison, K.W.; Or, Y.S.; Luly, J.R.; Carter, G.W. Structure-function studies in a series of carboxyl-terminal octapeptide analogues of anaphylatoxin C5a. J. Med. Chem. 1992, 35, 220–223. [Google Scholar] [CrossRef]
- Chatterjee, S.; Ator, M.A.; Bozyczko-Coyne, D.; Josef, K.; Wells, G.; Tripathy, R.; Iqbal, M.; Bihovsky, R.; Senadhi, S.E.; Mallya, S.; et al. Synthesis and biological activity of a series of potent fluoromethyl ketone inhibitors of recombinant human calpain I. J. Med. Chem. 1997, 40, 3820–3828. [Google Scholar] [CrossRef]
- Chatterjee, S.; Josef, K.; Wells, G.; Iqbal, M.; Bihovsky, R.; Mallamo, J.P.; Ator, M.A.; BozyczkoCoyne, D.; Mallya, S.; Senadhi, S.; et al. Potent fluoromethyl ketone inhibitors of recombinant human calpain I+. Bioorg. Med. Chem. Lett. 1996, 6, 1237–1240. [Google Scholar] [CrossRef]
- Sasaki, T.; Kikuchi, T.; Yumoto, N.; Yoshimura, N.; Murachi, T. Comparative specificity and kinetic studies on porcine calpain I and calpain II with naturally occurring peptides and synthetic fluorogenic substrates. J. Biol. Chem. 1984, 259, 12489–12494. [Google Scholar]
- Suzuki, T.; Park, H.; Lennarz, W.J. Cytoplasmic peptide:N-glycanase (PNGase) in eukaryotic cells: Occurrence, primary structure, and potential functions. FASEB J. 2002, 16, 635–641. [Google Scholar] [CrossRef]
- Misaghi, S.; Korbel, G.A.; Kessler, B.; Spooner, E.; Ploegh, H.L. z-VAD-fmk inhibits peptide:N-glycanase and may result in ER stress. Cell Death Differ. 2006, 13, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Misaghi, S.; Pacold, M.E.; Blom, D.; Ploegh, H.L.; Korbel, G.A. Using a small molecule inhibitor of peptide: N-glycanase to probe its role in glycoprotein turnover. Chem. Biol. 2004, 11, 1677–1687. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Hara, I.; Nakano, M.; Zhao, G.; Lennarz, W.J.; Schindelin, H.; Taniguchi, N.; Totani, K.; Matsuo, I.; Ito, Y. Site-specific labeling of cytoplasmic peptide:N-glycanase by N,N′-diacetylchitobiose-related compounds. J. Biol. Chem. 2006, 281, 22152–22160. [Google Scholar] [CrossRef] [PubMed]
- Witte, M.D.; Descals, C.V.; de Lavoir, S.V.; Florea, B.I.; van der Marel, G.A.; Overkleeft, H.S. Bodipy-VAD-Fmk, a useful tool to study yeast peptide N-glycanase activity. Org. Biomol. Chem. 2007, 5, 3690–3697. [Google Scholar] [CrossRef]
- Geoffroy, M.C.; Hay, R.T. An additional role for SUMO in ubiquitin-mediated proteolysis. Nat. Rev. Mol. Cell Biol. 2009, 10, 564–568. [Google Scholar] [CrossRef]
- Kunz, K.; Piller, T.; Muller, S. SUMO-specific proteases and isopeptidases of the SENP family at a glance. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef]
- Dobrotă, C.; Fasci, D.; Hădade, N.D.; Roiban, G.D.; Pop, C.; Meier, V.M.; Dumitru, I.; Matache, M.; Salvesen, G.S.; Funeriu, D.P.J.C. Glycine Fluoromethylketones as SENP-Specific Activity Based Probes. ChemBioChem 2012, 13, 80–84. [Google Scholar] [CrossRef]
- Shen, L.N.; Dong, C.; Liu, H.; Naismith, J.H.; Hay, R.T.J.B.J. The structure of SENP1–SUMO-2 complex suggests a structural basis for discrimination between SUMO paralogues during processing. Biochem. J. 2006, 397, 279–288. [Google Scholar] [CrossRef]
- Xu, Z.; Chau, S.F.; Lam, K.H.; Chan, H.Y.; Ng, T.B.; Au, S.W.J.B.J. Crystal structure of the SENP1 mutant C603S–SUMO complex reveals the hydrolytic mechanism of SUMO-specific protease. Biochem. J. 2006, 398, 345–352. [Google Scholar] [CrossRef]
- Rosenthal, P.J.; Wollish, W.S.; Palmer, J.T.; Rasnick, D. Antimalarial effects of peptide inhibitors of a Plasmodium falciparum cysteine proteinase. J. Clin. Investig. 1991, 88, 1467–1472. [Google Scholar] [CrossRef]
- Ettari, R.; Bova, F.; Zappalà, M.; Grasso, S.; Micale, N.J.M.r.r. Falcipain-2 inhibitors. Med. Res. Rev. 2010, 30, 136–167. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, P.J.; Lee, G.K.; Smith, R.E. Inhibition of a Plasmodium vinckei cysteine proteinase cures murine malaria. J. Clin. Investig. 1993, 91, 1052–1056. [Google Scholar] [CrossRef]
- Sajid, M.; Robertson, S.A.; Brinen, L.S.; McKerrow, J.H. Cruzain. In Cysteine Proteases of Pathogenic Organisms; Springer: Boston, MA, USA, 2011; pp. 100–115. [Google Scholar]
- Harth, G.; Andrews, N.; Mills, A.A.; Engel, J.C.; Smith, R.; McKerrow, J.H. Peptide-fluoromethyl ketones arrest intracellular replication and intercellular transmission of Trypanosoma cruzi. Mol. Biochem. Parasitol. 1993, 58, 17–24. [Google Scholar] [CrossRef]
- Gillmor, S.A.; Craik, C.S.; Fletterick, R.J. Structural determinants of specificity in the cysteine protease cruzain. Protein Sci. 1997, 6, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.S.; Guan, Y.; Yuen, K.Y. Severe acute respiratory syndrome. Nat. Med. 2004, 10, S88–S97. [Google Scholar] [CrossRef] [PubMed]
- Hegyi, A.; Ziebuhr, J. Conservation of substrate specificities among coronavirus main proteases. J. Gen. Virol. 2002, 83, 595–599. [Google Scholar] [CrossRef]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef]
- Jiang, S.; Du, L.; Shi, Z. An emerging coronavirus causing pneumonia outbreak in Wuhan, China: Calling for developing therapeutic and prophylactic strategies. Emerg. Microbes Infect. 2020, 9, 275–277. [Google Scholar] [CrossRef]
- Lu, S. Timely development of vaccines against SARS-CoV-2. Emerg. Microbes Infect. 2020, 9, 542–544. [Google Scholar] [CrossRef]
- Zhang, H.Z.; Zhang, H.; Kemnitzer, W.; Tseng, B.; Cinatl, J.; Michaelis, M.; Doerr, H.W.; Cai, S.X. Design and synthesis of dipeptidyl glutaminyl fluoromethyl ketones as potent severe acute respiratory syndrome coronovirus (SARS-CoV) inhibitors. J. Med. Chem. 2006, 49, 1198–1201. [Google Scholar] [CrossRef]
- Morris, T.S.; Frormann, S.; Shechosky, S.; Lowe, C.; Lall, M.S.; Gauss-Muller, V.; Purcell, R.H.; Emerson, S.U.; Vederas, J.C.; Malcolm, B.A. In vitro and ex vivo inhibition of hepatitis A virus 3C proteinase by a peptidyl monofluoromethyl ketone. Bioorg. Med. Chem. 1997, 5, 797–807. [Google Scholar] [CrossRef]
- Miele, M.; Citarella, A.; Micale, N.; Holzer, W.; Pace, V. Direct and Chemoselective Synthesis of Tertiary Difluoroketones via Weinreb Amide Homologation with a CHF2-Carbene Equivalent. Org. Lett. 2019, 21, 8261–8265. [Google Scholar] [CrossRef] [PubMed]
- Pattison, G. Methods for the Synthesis of α, α-Difluoroketones. Eur. J. Org. Chem. 2018, 2018, 3520–3540. [Google Scholar] [CrossRef]
- Imperiali, B.; Abeles, R.H. Inhibition of serine proteases by peptidyl fluoromethyl ketones. Biochemistry 1986, 25, 3760–3767. [Google Scholar] [CrossRef] [PubMed]
- Breaux, E.J.; Bender, M.L. The binding of specific and non-specific aldehyde substrate analogs to alpha-chymotrypsin. FEBS Lett. 1975, 56, 81–84. [Google Scholar] [CrossRef]
- Powers, J.C.; Tuhy, P.M. Active-site specific inhibitors of elastase. Biochemistry 1973, 12, 4767–4774. [Google Scholar] [CrossRef] [PubMed]
- Sham, H.L.; Wideburg, N.E.; Spanton, S.G.; Kohlbrenner, W.E.; Betebenner, D.A.; Kempf, D.J.; Norbeck, D.W.; Plattner, J.J.; Erickson, J.W. Synthesis of (2s,5s,4r)-2,5-Diamino-3,3-Difluoro-1,6-Diphenylhydroxyhexane—The Core Unit of a Potent Hiv Proteinase-Inhibitor. J. Chem. Soc. Chem. Commun. 1991, 110–112. [Google Scholar] [CrossRef]
- Prabu-Jeyabalan, M.; Nalivaika, E.; Schiffer, C.A. Substrate shape determines specificity of recognition for HIV-1 protease: Analysis of crystal structures of six substrate complexes. Structure 2002, 10, 369–381. [Google Scholar] [CrossRef]
- Navia, M.A.; Fitzgerald, P.M.; McKeever, B.M.; Leu, C.T.; Heimbach, J.C.; Herber, W.K.; Sigal, I.S.; Darke, P.L.; Springer, J.P. Three-dimensional structure of aspartyl protease from human immunodeficiency virus HIV-1. Nature 1989, 337, 615–620. [Google Scholar] [CrossRef]
- Wlodawer, A.; Miller, M.; Jaskolski, M.; Sathyanarayana, B.K.; Baldwin, E.; Weber, I.T.; Selk, L.M.; Clawson, L.; Schneider, J.; Kent, S.B. Conserved folding in retroviral proteases: Crystal structure of a synthetic HIV-1 protease. Science 1989, 245, 616–621. [Google Scholar] [CrossRef]
- Haufe, G.; Leroux, F. Fluorine in Life Sciences: Pharmaceuticals, Medicinal Diagnostics, and Agrochemicals: Progress in Fluorine Science Series; Academic Press: Cambridge, MA, USA, 2018. [Google Scholar]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil Elastase, Proteinase 3, and Cathepsin G as Therapeutic Targets in Human Diseases. Pharm. Rev. 2010, 62, 726–759. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.C.; Stein, R.L.; Giles, R.E.; Krell, R.D. Biochemistry and pharmacology of ICI 200,880, a synthetic peptide inhibitor of human neutrophil elastase. Ann. N. Y. Acad. Sci. 1991, 624, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Mehta, J.L.; Nichols, W.W.; Nicolini, F.A.; Hendricks, J.; Donnelly, W.H.; Saldeen, T.G. Neutrophil elastase inhibitor ICI 200,880 protects against attenuation of coronary flow reserve and myocardial dysfunction following temporary coronary artery occlusion in the dog. Cardiovasc. Res. 1994, 28, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, L.H.; Radhakrishnan, R.; Rosenfield, R.E., Jr.; Meyer, E.F., Jr.; Trainor, D.A.; Stein, M. X-ray diffraction analysis of the inhibition of porcine pancreatic elastase by a peptidyl trifluoromethylketone. J. Mol. Biol. 1988, 201, 423–428. [Google Scholar] [CrossRef]
- Brown, F.J.; Andisik, D.W.; Bernstein, P.R.; Bryant, C.B.; Ceccarelli, C.; Damewood, J.R., Jr.; Edwards, P.D.; Earley, R.A.; Feeney, S.; Green, R.C.; et al. Design of orally active, non-peptidic inhibitors of human leukocyte elastase. J. Med. Chem. 1994, 37, 1259–1261. [Google Scholar] [CrossRef]
- Veale, C.A.; Bernstein, P.R.; Bryant, C.; Ceccarelli, C.; Damewood, J.R., Jr.; Earley, R.; Feeney, S.W.; Gomes, B.; Kosmider, B.J.; Steelman, G.B.; et al. Nonpeptidic inhibitors of human leukocyte elastase. 5. Design, synthesis, and X-ray crystallography of a series of orally active 5-aminopyrimidin-6-one-containing trifluoromethyl ketones. J. Med. Chem. 1995, 38, 98–108. [Google Scholar] [CrossRef]
- Veale, C.A.; Bernstein, P.R.; Bohnert, C.M.; Brown, F.J.; Bryant, C.; Damewood, J.R., Jr.; Earley, R.; Feeney, S.W.; Edwards, P.D.; Gomes, B.; et al. Orally active trifluoromethyl ketone inhibitors of human leukocyte elastase. J. Med. Chem. 1997, 40, 3173–3181. [Google Scholar] [CrossRef]
- Peet, N.P.; Burkhart, J.P.; Angelastro, M.R.; Giroux, E.L.; Mehdi, S.; Bey, P.; Kolb, M.; Neises, B.; Schirlin, D. Synthesis of Peptidyl Fluoromethyl Ketones and Peptidyl Alpha-Keto Esters as Inhibitors of Porcine Pancreatic Elastase, Human Neutrophil Elastase, and Rat and Human Neutrophil Cathepsin-G. J. Med. Chem. 1990, 33, 394–407. [Google Scholar] [CrossRef]
- Neil, D.; Rawlings, N.D.; Salvesen, G. Handbook of Proteolytic Enzymes; Academ Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Pereira, P.J.B.; Wang, Z.-M.; Rubin, H.; Huber, R.; Bode, W.; Schechter, N.M.; Strobl, S. The 2.2 Å crystal structure of human chymase in complex with succinyl-Ala-Ala-Pro-Phe-chloromethylketone: Structural explanation for its dipeptidyl carboxypeptidase specificity. J. Mol. Biol. 1999, 286, 163–173. [Google Scholar] [CrossRef]
- Akahoshi, F.; Ashimori, A.; Yoshimura, T.; Imada, T.; Nakajima, M.; Mitsutomi, N.; Kuwahara, S.; Ohtsuka, T.; Fukaya, C.; Miyazaki, M.J.B.; et al. Non-Peptidic inhibitors of human chymase. Synthesis, structure–activity relationships, and pharmacokinetic profiles of a series of 5-amino-6-oxo-1, 6-dihydropyrimidine-containing trifluoromethyl ketones. Bioorg. Med. Chem. 2001, 9, 301–315. [Google Scholar] [CrossRef]
- Madsen, A.S.; Kristensen, H.M.; Lanz, G.; Olsen, C.A.J.C. The effect of various zinc binding groups on inhibition of histone deacetylases 1–11. ChemMedChem 2014, 9, 614–626. [Google Scholar] [CrossRef] [PubMed]
- Milazzo, G.; Mercatelli, D.; Di Muzio, G.; Triboli, L.; De Rosa, P.; Perini, G.; Giorgi, F.M. Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability. Genes (Basel) 2020, 11, 556. [Google Scholar] [CrossRef] [PubMed]
- Rajak, H.; Singh, A.; Raghuwanshi, K.; Kumar, R.; Dewangan, P.K.; Veerasamy, R.; Sharma, P.C.; Dixit, A.; Mishra, P. A Structural Insight into Hydroxamic Acid Based Histone Deacetylase Inhibitors for the Presence of Anticancer Activity. Curr. Med. Chem. 2014, 21, 2642–2664. [Google Scholar] [CrossRef] [PubMed]
- Jose, B.; Oniki, Y.; Kato, T.; Nishino, N.; Sumida, Y.; Yoshida, M. Novel histone deacetylase inhibitors: Cyclic tetrapeptide with trifluoromethyl and pentafluoroethyl ketones. Bioorg. Med. Chem. Lett. 2004, 14, 5343–5346. [Google Scholar] [CrossRef]
- Ilies, M.; Dowling, D.P.; Lombardi, P.M.; Christianson, D.W. Synthesis of a new trifluoromethylketone analogue of l-arginine and contrasting inhibitory activity against human arginase I and histone deacetylase 8. Bioorg. Med. Chem. Lett. 2011, 21, 5854–5858. [Google Scholar] [CrossRef][Green Version]
- Moreno-Yruela, C.; Olsen, C.A. Synthesis of Trifluoromethyl Ketone Containing Amino Acid Building Blocks for the Preparation of Peptide-Based Histone Deacetylase (HDAC) Inhibitors. Synth. Stuttg. 2018, 50, 4037–4046. [Google Scholar] [CrossRef]
- Lin, K.H.; Ali, A.; Rusere, L.; Soumana, D.I.; Kurt Yilmaz, N.; Schiffer, C.A. Dengue Virus NS2B/NS3 Protease Inhibitors Exploiting the Prime Side. J. Virol. 2017, 91, e00045-17. [Google Scholar] [CrossRef]
- Yin, Z.; Patel, S.J.; Wang, W.L.; Wang, G.; Chan, W.L.; Rao, K.R.; Alam, J.; Jeyaraj, D.A.; Ngew, X.; Patel, V.; et al. Peptide inhibitors of Dengue virus NS3 protease. Part 1: Warhead. Bioorg. Med. Chem. Lett. 2006, 16, 36–39. [Google Scholar] [CrossRef]
- Gibbs, A.C.; Steele, R.; Liu, G.; Tounge, B.A.; Montelione, G.T. Inhibitor Bound Dengue NS2B-NS3pro Reveals Multiple Dynamic Binding Modes. Biochemistry 2018, 57, 1591–1602. [Google Scholar] [CrossRef]
- Sydnes, M.O.; Hayashi, Y.; Sharma, V.K.; Hamada, T.; Bacha, U.; Barrila, J.; Freire, E.; Kiso, Y. Synthesis of glutamic acid and glutamine peptides possessing a trifluoromethyl ketone group as SARS-CoV 3CL protease inhibitors. Tetrahedron 2006, 62, 8601–8609. [Google Scholar] [CrossRef]
- Shao, Y.M.; Yang, W.B.; Kuo, T.H.; Tsai, K.C.; Lin, C.H.; Yang, A.S.; Liang, P.H.; Wong, C.H. Design, synthesis, and evaluation of trifluoromethyl ketones as inhibitors of SARS-CoV 3CL protease. Bioorg. Med. Chem. 2008, 16, 4652–4660. [Google Scholar] [CrossRef] [PubMed]
- Bacha, U.; Barrila, J.; Gabelli, S.B.; Kiso, Y.; Mario Amzel, L.; Freire, E. Development of Broad-Spectrum Halomethyl Ketone Inhibitors Against Coronavirus Main Protease 3CLpro. Chem. Biol. Drug Des. 2008, 72, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Regnier, T.; Sarma, D.; Hidaka, K.; Bacha, U.; Freire, E.; Hayashi, Y.; Kiso, Y. New developments for the design, synthesis and biological evaluation of potent SARS-CoV 3CLpro inhibitors. Bioorg. Med. Chem. Lett. 2009, 19, 2722–2727. [Google Scholar] [CrossRef] [PubMed]



















© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Citarella, A.; Micale, N. Peptidyl Fluoromethyl Ketones and Their Applications in Medicinal Chemistry. Molecules 2020, 25, 4031. https://doi.org/10.3390/molecules25174031
Citarella A, Micale N. Peptidyl Fluoromethyl Ketones and Their Applications in Medicinal Chemistry. Molecules. 2020; 25(17):4031. https://doi.org/10.3390/molecules25174031
Chicago/Turabian StyleCitarella, Andrea, and Nicola Micale. 2020. "Peptidyl Fluoromethyl Ketones and Their Applications in Medicinal Chemistry" Molecules 25, no. 17: 4031. https://doi.org/10.3390/molecules25174031
APA StyleCitarella, A., & Micale, N. (2020). Peptidyl Fluoromethyl Ketones and Their Applications in Medicinal Chemistry. Molecules, 25(17), 4031. https://doi.org/10.3390/molecules25174031