1. Introduction
Duchenne muscular dystrophy (DMD) is an X-linked neuromuscular disorder characterized by progressive muscular degeneration. DMD affects about 1 in 3500 male births worldwide [
1]. Currently, dystrophin is considered one of the most important genes involved in DMD. This protein is a component of the dystrophin-associated protein complex (DAPC) [
1,
2]. Mutations in the dystrophin gene on the X chromosome cause DMD; they damage the DAPC, causing creatine kinase to pass through muscle cell membranes into the blood, in turn, leading to elevated serum creatine kinase levels [
2].
About 5–15% of DMD cases are caused by nonsense mutations, resulting in premature termination codons (PTCs) in the place of a sense codon. This leads to the production of a truncated dystrophin protein [
2,
3]. Consequently, several therapeutic approaches for treating DMD have focused on suppressing PTCs. One therapeutic approach in particular—the use of PTC-suppressing compounds (PTCSCs)—relies on the fact that translation termination is not 100% efficient [
3]. It depends on competition between the human eFR1 release factor, which recognizes the stop codon, and near-cognate aminoacyl-tRNAs, which carry anticodons with an altered third nucleotide base [
3]. Binding of near-cognate aminoacyl-tRNAs to the stop codon causes an amino acid to be incorporated, resulting in the continuous production of protein, as there is no longer a stop codon to be recognized [
3]. Thus, PTCSCs suppress PTCs by inducing translational readthrough, a mechanism that leads to the production of full-length protein by stimulating the binding of near-cognate tRNAs, to the stop codon [
3].
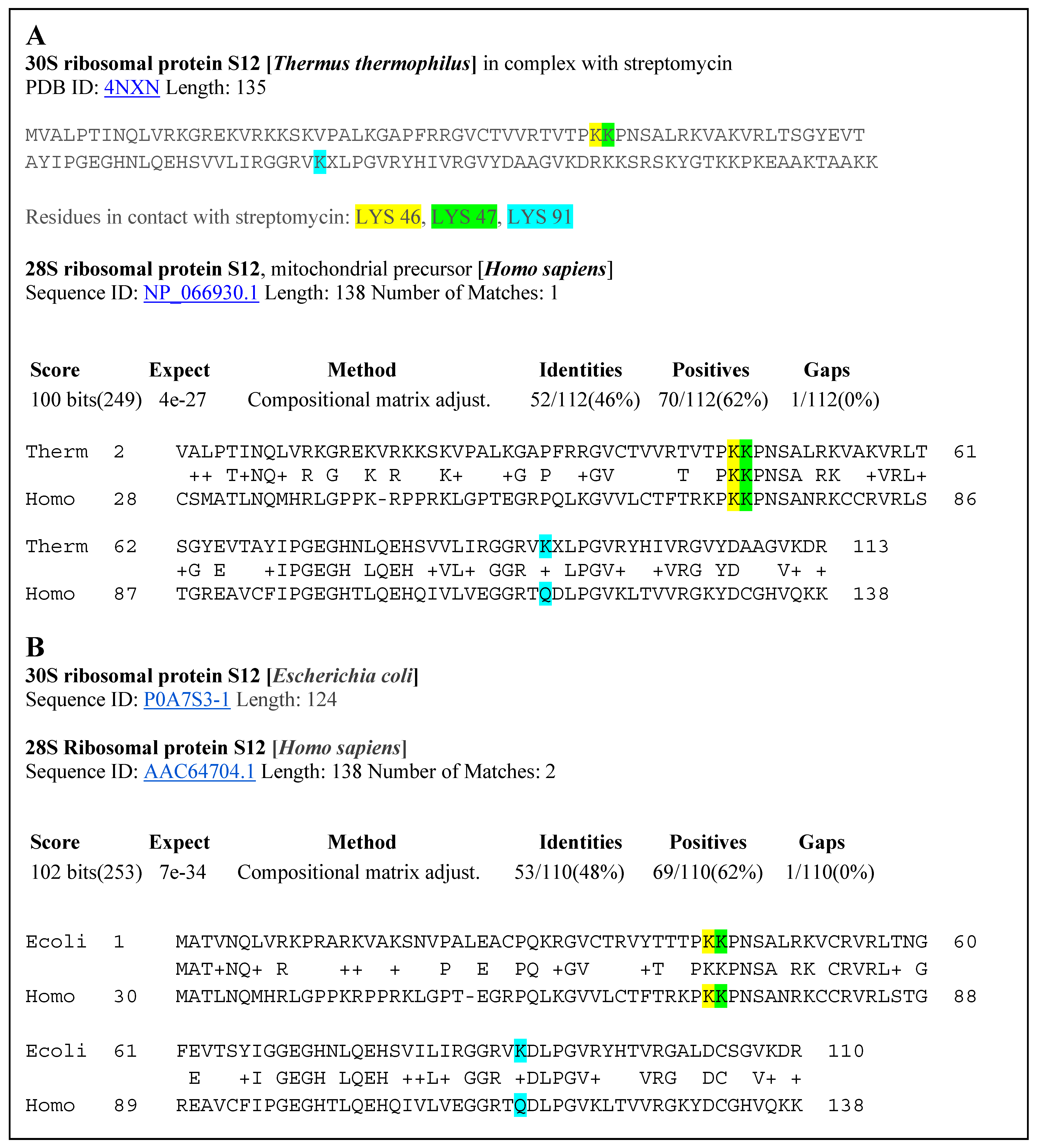
PTCSCs are hypothesized to bind to the 30S ribosomal subunit and interact with the 16S rRNA and S12 ribosomal protein [
4]. The 30S ribosomal subunit plays an important role in discriminating against near cognate aminoacyl tRNAs, ensuring translation accuracy. Binding of PTC-suppressing compounds to the 30S ribosomal subunit inhibits this process, resulting in codon misreading and altered translation of mRNA [
4]. Our hypothesis is that some ribosomal proteins have a high degree of homological correspondence to the analogous human ribosomal proteins, explaining the efficacy of these antibiotics for suppression of PTCs in humans.
One class of PTCSCs is aminoglycoside antibiotics. Aminoglycosides, such as gentamicin and paromomycin, have been found to stimulate readthrough and suppress PTCs. However, the use of these drugs is limited, as they may induce ototoxicity and nephrotoxicity, manifesting in side effects such as hearing loss and diminished renal function [
5]. Their efficacy is also influenced by other factors, such as stop codon context and sequence specificity. Previous studies have found that readthrough levels induced by aminoglycosides have the order UGA > UAG >> UAA, and a +4 cytosine immediately following the PTC produced the most readthrough, due to the prevention of eRF1 recognition of the PTC [
5]. Aminoglycoside derivatives meant to reduce toxicity and improve therapeutic potential were developed by modifying the chemical structures of aminoglycosides [
6]. As examples of such drug-candidates, we can list NB54 and NB30, which are paromomycin derivatives, along with NB74 and NB84, which are geneticin (G418) derivatives [
6,
7].
Non-aminoglycosidic alternatives have also been identified through screens of compounds [
8]. One such drug-candidate is Ataluren, or PTC124, a PTC-suppressing oxadiazole compound of lower toxicity. Ataluren has been proposed to stimulate readthrough by binding to the h44 decoding center of the 16S rRNA in the 30S ribosomal subunit [
9]. While this drug has been found to have significantly lower toxicity, conflicting results have been found in regard to its efficacy [
10,
11]. Another PTC-suppressing compound, negamycin, exhibited lower toxicity than aminoglycosides [
11]. Negamycin follows a similar mechanism of action to aminoglycosides, binding to the ribosomal A site. [
11]. Furthermore, macrolides, antibiotics used to treat bacterial infections, have been tested as readthrough-inducing compounds [
12]. This family of compounds includes tylosin, josamycin, spiramycin, and azithromycin [
12,
13]. Macrolides were able to induce readthrough of nonsense mutations, indicating that they could be potential PTC-suppressing agents [
14]. Additional non-aminoglycosides that have been investigated include clitocine, escin, RTC #13, RTC #14, GJ071, GJ072, and amlexanox [
15,
16,
17,
18,
19,
20,
21]. Clitocine is an adenosine nucleotide analog [
15]. In contrast to aminoglycosides, clitocine is incorporated in replacement of the adenine in a stop codon, inducing readthrough [
15]. However, this compound is also affected by stop codon context, with the order of clitocine-induced readthrough levels for various PTCs being UAA >> UGA > UAG [
15]. Escin is an herbal anti-inflammatory drug that was able to induce read-through in a cystic fibrosis patient [
16]. This compound may be a potential PTC-suppressing compound used to treat DMD as well [
16]. High-throughput screening identified potential readthrough-inducing compounds RTC #13 and RTC #14 [
17,
18]. Studies have shown that RTC #13 was able to partially restore dystrophin levels in the muscles of mdx mice [
18]. More recently, GJ071 and GJ072 showed a similar read-through-inducing efficiency as PTC124, RTC #13, and RTC #14, as well as lower toxicity [
19].
Computational approaches for identification of PTCSCs help to expedite the reduction of large molecular databases to the sets of molecules which are most probable to exhibit an optimized blend of features to be tested in vitro; inhibitors which are efficacious and low in toxicity. Only one paper describing the deployment or development of such screens for DMD was found [
22], despite the potential development of such screens and their widespread use in the search for drugs to treat other diseases. Yusuke et al. developed an in-silico tool that can design nucleotide analogs which recognize, bind, and block transcription or splice sites of pre-mRNAs, morpholino sequences, for exon skipping [
22]. Remarkably, they report that most of their computationally derived morpholinos are more efficient at promoting Exon 51 skipping in vitro than Eteplirsen [
22], the current FDA approved treatment of this type for this disease.
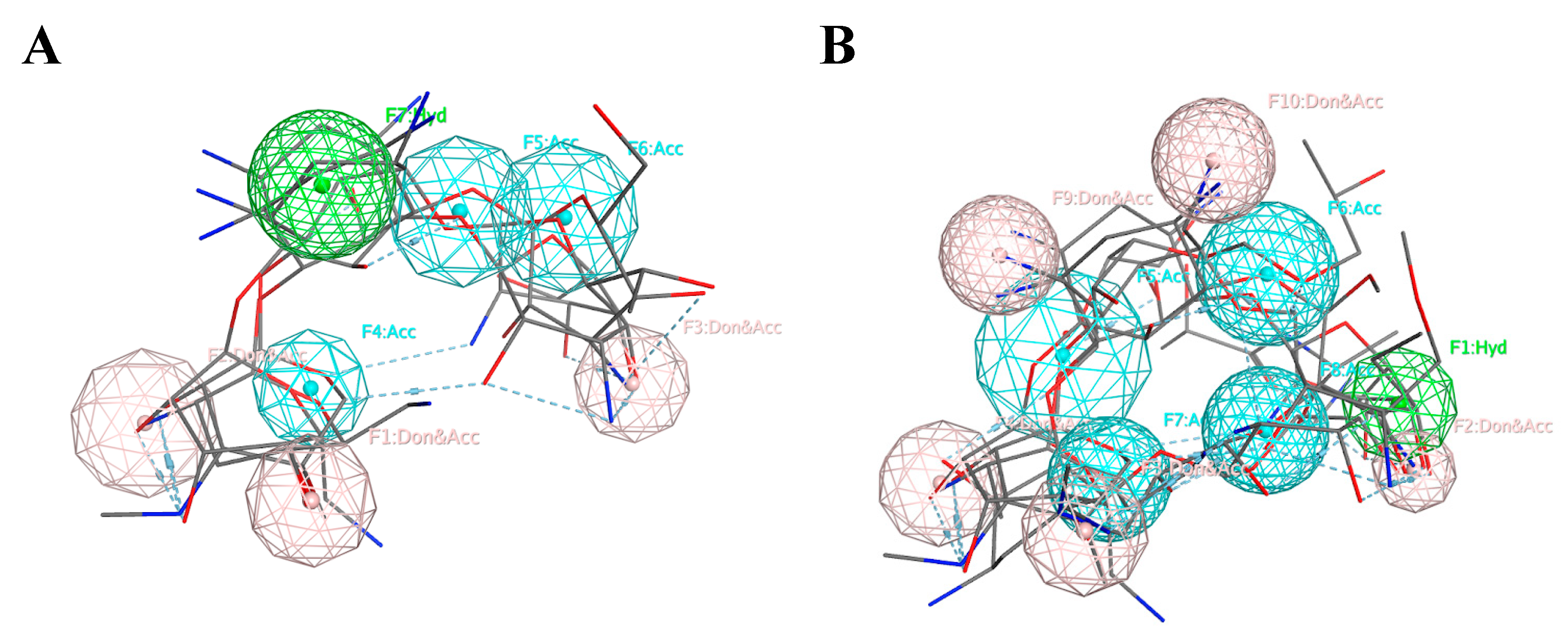
Computational drug design employs different methods. Pharmacophore-based methods have proven to be some of the most effective for computational drug design approaches. Researchers create a set of functional centers based on either ligand or receptor and use it for selection from conformational databases of compounds. Another popular, modern method uses quantitative descriptions of already existing agonists or antagonists of the specific proteins, called quantitative structure-activity relationship (QSAR) descriptors, to create a machine learning (ML) model that would be used for the elucidation of new compounds that share this activity on the target protein.
Here, we point out some successful stories of using pharmacophore-based and ML-based searches for new drug design.
In one study, an inhibitor of the promising drug target acid sphingomyelinase (ASM) was selected using a pharmacophore model created on the basis of known ASM inhibitors including α-mangostin [
23]. A database search using this pharmacophore model revealed 23 potential inhibitors, 10 of which were found to be effective in experimental studies.
A machine learning system for elucidation of potential drug candidates for the 5-HT
2B receptor (5-hydroxytryptamine receptor 2B) inhibition was developed using known inhibitors with Ki < 500 nM and inactive molecules with Ki > 1000 nM. The author of this study used a NSFP fingerprint-based ML method and virtual docking of the obtained compounds to the binding sites. Nine potential inhibitors were selected, five of which showed binding to the 5-HT
2B receptor and one with Ki = 0.3 mM [
24]. In the same study, Bruns used various machine learning algorithms, including self organizing maps (SOM), convolution neural networks (CNN), and recurring neural networks (RNN), for development of novel inhibitors of the CXC chemokine receptor 4 (CXCR4) based on known inhibitors and non-inhibitor compounds. With the most effective SOM ML system, he elucidated a new inhibitor with an EC
50 (half maximal effective concentration) < 10μM.
In another study, Li and colleagues [
25] conducted classical ML-based elucidation of new inhibitors of topoisomerase I. They prepared an input set containing 481 inhibitors and 480 non-inhibitor compounds. They used 189 molecular descriptors, k-nearest neighbors (KNN), radio frequency (RF), and support vector machines (SVM) ML methods to develop their models and conducted further virtual screening of the Maybridge database. Following selection, molecular docking was conducted with AutoDock Vina software [
26]. The authors elucidated several compounds with docking energies better than −10 kcal/mol and similar scaffolds to known topoisomerase I inhibitors.
In our recent study [
27], we attempted to compare the results of pharmacophore-based and ML-based drug design, and confirmed that results of pharmacophore-based and deep learning (DL)-based drug selection were similar in a significant part of the predicted drug candidates.
In the current study, we also attempted to concentrate on the pharmacophore-based, DL, and ML-based techniques for predictions of the same compounds, which we can consider the more robust predictions for further testing.
Here, we deployed pharmacophore-modeling along with ML and DL based approaches to allow for identification of novel drug candidates with PTC-suppressing ability for the treatment of DMD. A pharmacophore model was developed based on common pharmacophore features of existing PTC-suppressing compounds and used to screen for compounds with structural correspondence to these pharmacophore features. To validate pharmacophore-based results, ML and DL models were developed for prediction of PTC-suppressing compounds using QSAR descriptors. This study develops an ensemble of models to predict new compounds with PTC-suppressing ability and lays the foundation for future investigation of the identified PTC-suppressing drug candidates for treatment of DMD.
3. Discussion
Many compounds that have been developed to treat DMD by stimulating translational readthrough are limited, due to their low efficacy or high toxicity. Aminoglycosides, such as gentamicin and paromomycin, have been found to induce ototoxicity and nephrotoxicity when administered to patients [
37]. The use of non-aminoglycosides to treat DMD is still being investigated; however, the efficacy of these compounds varies, due to the stop codon context and sequence specificity.
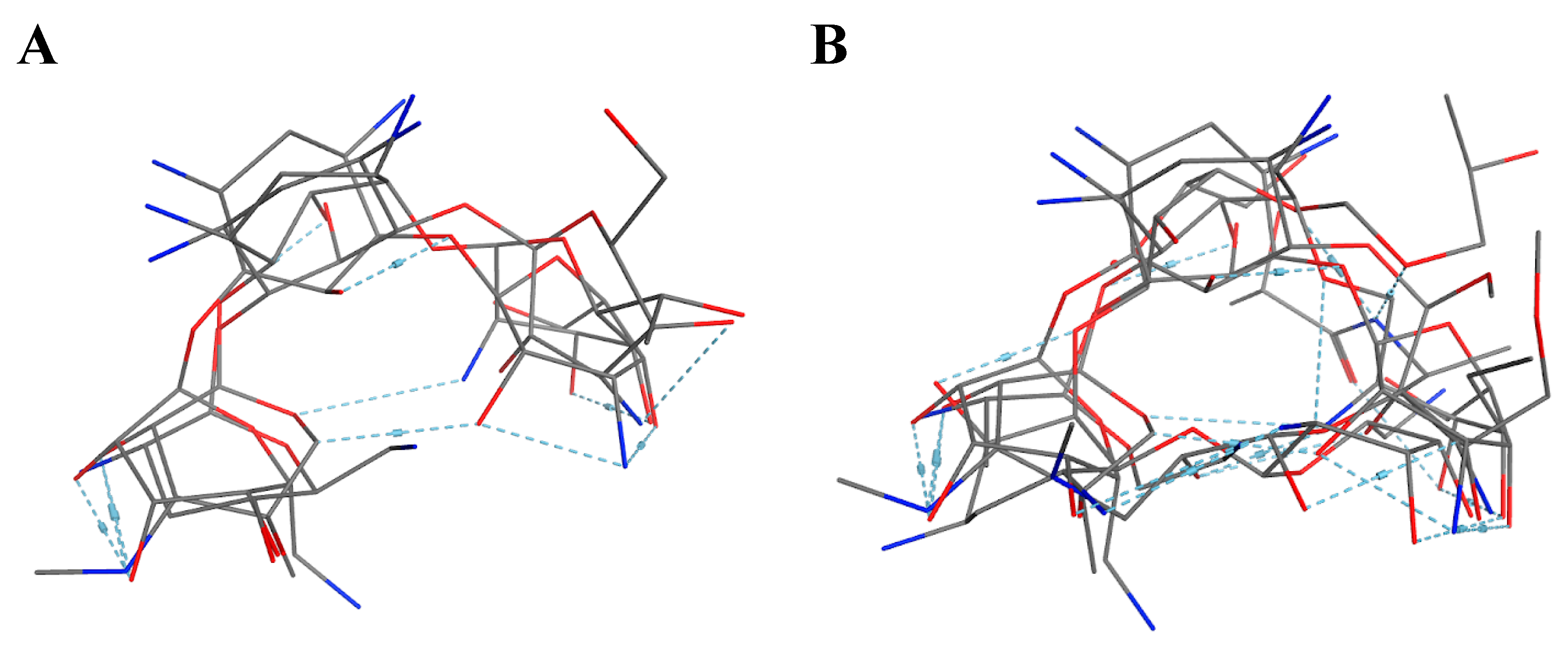
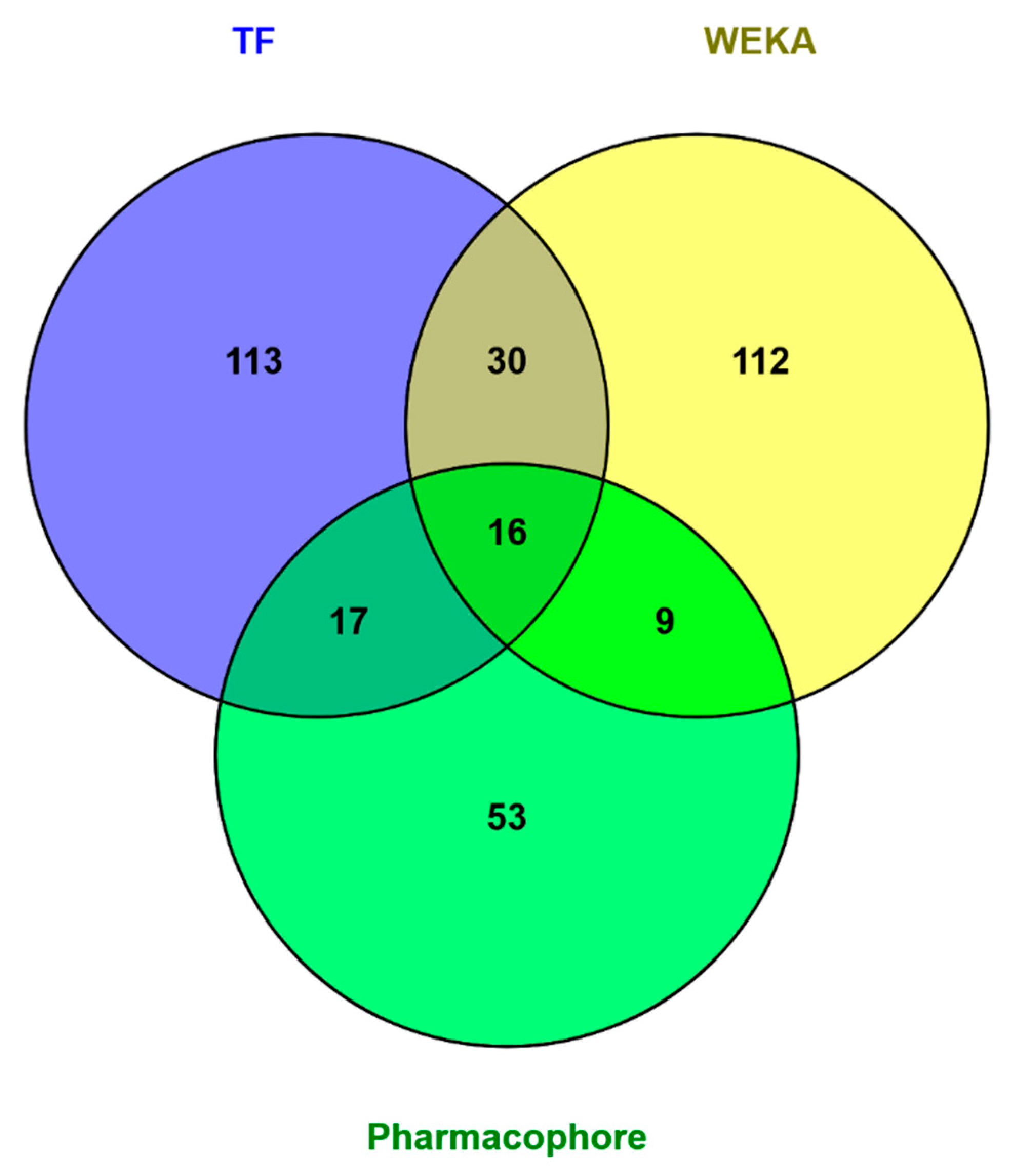
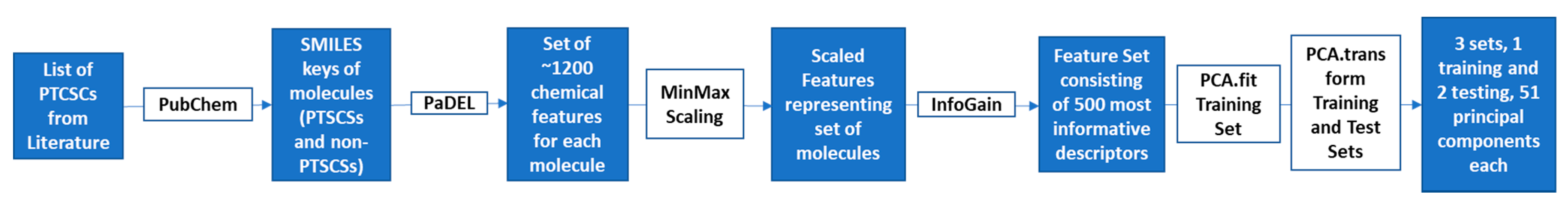
We implemented pharmacophore-based, ML, and DL approaches to address this issue, with the aim of identifying potent compounds, including FDA-approved compounds, with PTC-suppressing capability. A literature search was conducted to identify known PTC-suppressing compounds. We then clustered the chosen compounds by molecular fingerprints. Compounds within each cluster were structurally aligned. Using these alignments, we developed two pharmacophore models. These models were used to conduct a search on the NCI database and DrugBank database of FDA-approved drugs. We selected 16 FDA-approved drugs as a consensus of all three approaches. Similar studies may be done in the future to further identify PTC-suppressing compounds for the treatment of DMD using larger, more expansive databases, such as ChEMBL.
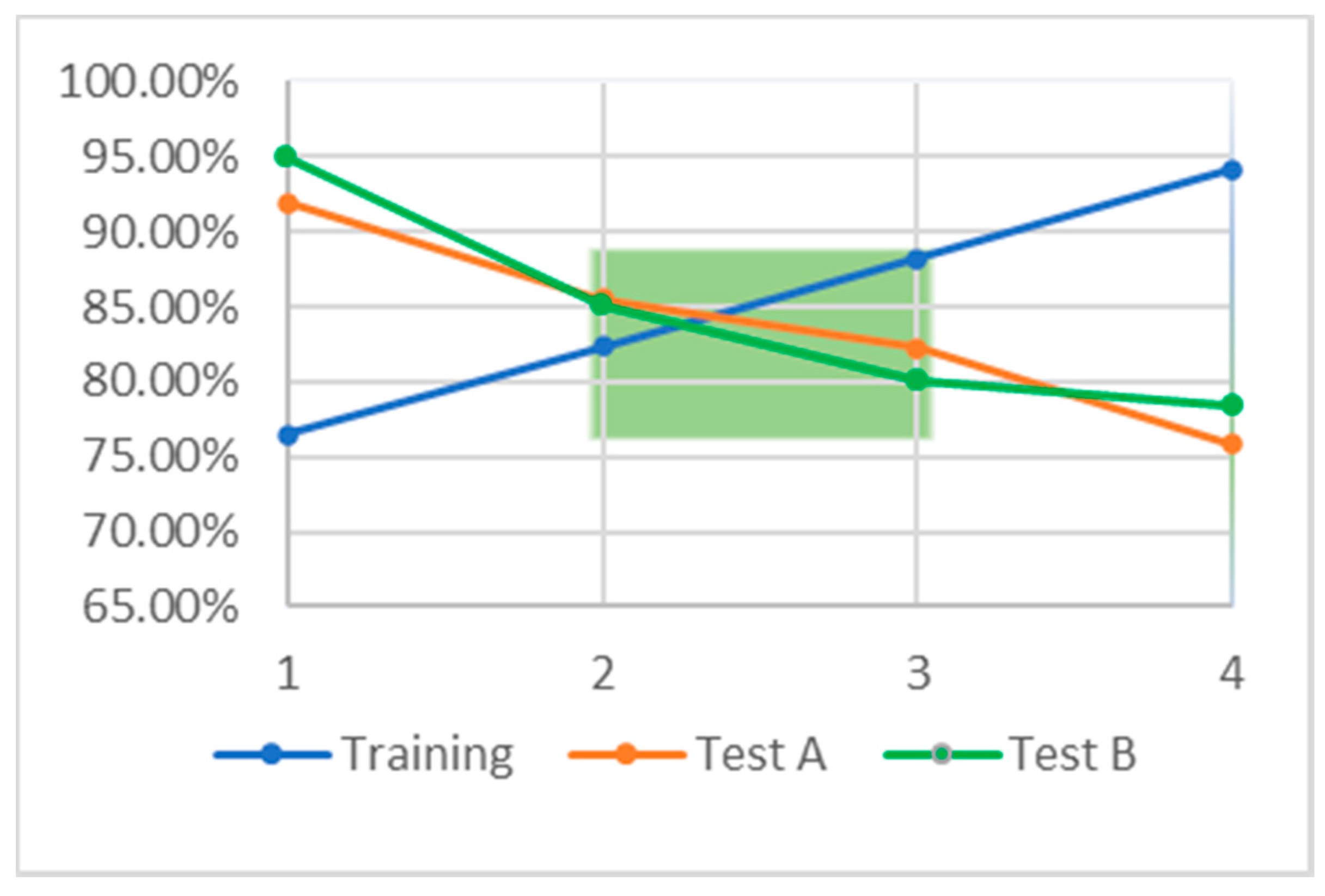
Based on our QSAR models, we observed approximately linear relationships between the biological activities and structural attributes of each of the compounds. Our QSAR models show that the inhibitory activity against the 30S ribosome of drugs found by the consensus of pharmacophore-based search, ML, and DL methods have a close to linear relationship with the descriptor of molecular refractivity. Moreover, we confirmed that the bacterial ribosomal proteins interacting with antibiotics have a significant homology with the human ribosomal proteins. This suggests an answer to the question of why these antibiotics cause PTC suppression in human patients.
We applied all three models to the database of FDA approved drugs and obtained interesting results. All three models, WEKA, TensorFlow, and Pharmacophore, agreed on 16 molecules. These FDA approved drugs are prime candidates for in vitro testing to start the process of repurposing them to treat DMD. Pharmacophore modeling is a better predictor of biological activity than are QSAR methods, because in QSAR, we are limited to compounds targeting the same molecule. The molecules contained in the overlap between the pharmacophore model and TensorFlow model predictions sets are slightly more promising than the WEKA ML models because the TensorFlow DL model performed better than the WEKA model on independent testing sets.
Molecules predicted to have activity by more than one method should, in general, be weighted significantly higher than those which are only labeled active by only one model. Ensemble modeling methods, those in which multiple models are developed to make a single prediction, have been demonstrated to be more powerful than prediction methods dependent on only one model. Using an ensemble modeling method to make predictions is analogous to getting multiple professional opinions on a subject matter before making a decision. It allows for the prediction to account for many, well informed opinions, selecting the options which make sense from multiple perspectives.
Our analysis presents several potent compounds with PTC-suppressing ability. Ten of the 16 compounds identified using pharmacophore, ML, and DL models target the 30S ribosomal subunit, the primary target of existing PTC-suppressing compounds, indicating that they are viable options for repurposing for DMD treatment. Of the other six compounds, diazolidinyl urea, steviolbioside, streptozocin, and rutin have similar mechanisms of action (inhibition of DNA/protein synthesis), and their targets have similar structures to the 30S ribosomal subunit. This indicates that these compounds may have potential PTC-suppressing ability as well. With regard to the structures of the compounds found from all three approaches, 10 of the 16 compounds contain a 2-deoxystreptamine (2 DOS), which has been identified as a key structural feature in novel aminoglycoside structures [
8]. In general, the structures of the compounds indicate PTC-suppressing ability. These compounds warrant further analysis of their pharmacokinetic properties and experimental validation of the development of these compounds for drug design.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


























