
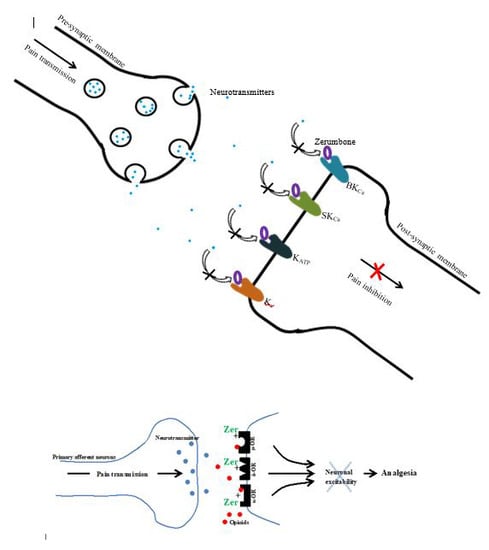
Zerumbone-Induced Analgesia Modulated via Potassium Channels and Opioid Receptors in Chronic Constriction Injury-Induced Neuropathic Pain

 ,
,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Involvement of Voltage-Dependent K+ Channels in Zerumbone’s Antiallodynic and Antihyperalgesic Effects
2.2. Involvement of ATP-Sensitive K+ Channels in Zerumbone’s Antiallodynic and Antihyperalgesic Effects
2.3. Involvement of Small- and Large-Conductance Ca2+-Activated K+ Channels in Zerumbone-Induced Antiallodynia and Antihyperalgesia
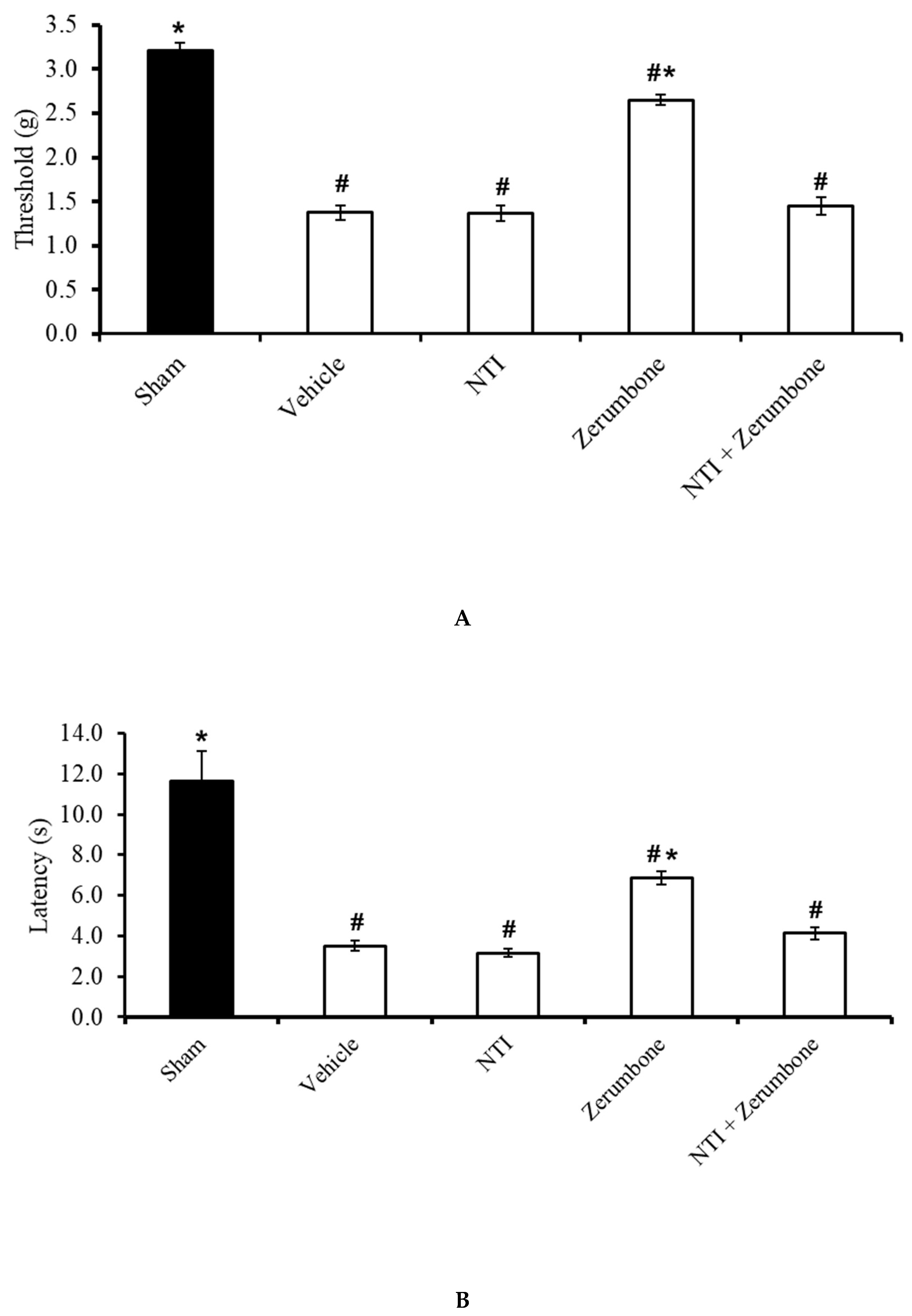
2.4. Involvement of Non-Selective Opioid Receptors
2.5. Involvement of Selective µ-Opioid Receptors
2.6. Involvement of Selective δ-Opioid Receptors
2.7. Involvement of Selective κ-Opioid Receptors
2.8. Rota Rod Assay
3. Discussion
4. Materials and Methods
4.1. Preparation of Zerumbone
4.2. Drugs and Chemicals
4.3. Animals
4.4. Induction of Neuropathic Pain
4.5. Experimental Design
4.6. Nociceptive Assays
4.6.1. VON Frey’s Filament Test
4.6.2. Hargreaves Test
4.7. Rota Rod Test
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chung, K.; Lee, B.H.; Yoon, Y.W.; Chung, J.M. Sympathetic sprouting in the dorsal root ganglia of the injured peripheral nerve in a rat neuropathic pain model. J. Comp. Neurol. 1996, 376, 241–252. [Google Scholar] [CrossRef]
- Ren, K.; Dubner, R. Inflammatory Models of Pain and Hyperalgesia. ILAR J. 1999, 40, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.N.; Meyer, R.A. Mechanisms of neuropathic pain. Neuron 2006, 52, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Fang, N.; Liu, M.; Cai, J.; Wan, Y.; Han, J.-S.; Xing, G.-G. Suppression of KCNQ/M (Kv7) potassium channels in dorsal root ganglion neurons contributes to the development of bone cancer pain in a rat model. Pain 2013, 154, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Deerinck, T.; Ellisman, M.; Schwarz, T. Differential distribution of closely related potassium channels in rat Schwann cells. J. Neurosci. 1995, 15, 3761–3774. [Google Scholar] [CrossRef]
- Arroyo, E.J.; Scherer, S.S. On the molecular architecture of myelinated fibers. Histochem. Cell Boil. 2000, 113, 1–18. [Google Scholar] [CrossRef]
- Kim, D.S.; Choi, J.O.; Rim, H.D.; Cho, H.J. Downregulation of voltage-gated potassium channel α gene expression in dorsal root ganglia following chronic constriction injury of the rat sciatic nerve. Mol. Brain Res. 2002, 105, 146–152. [Google Scholar] [CrossRef]
- Sarantopoulos, C.; McCallum, J.B.; Rigaud, M.; Fuchs, A.; Kwok, W.-M.; Hogan, Q.H. Opposing effects of spinal nerve ligation on calcium-activated potassium currents in axotomized and adjacent mammalian primary afferent neurons. Brain Res. 2007, 1132, 84–99. [Google Scholar] [CrossRef]
- Busserolles, J.; Tsantoulas, C.; Eschalier, A.; García, J.A.L. Potassium channels in neuropathic pain. Pain 2016, 157, S7–S14. [Google Scholar] [CrossRef]
- Moulin, D.E.; Clark, A.J.; Gilron, I.; A Ware, M.; Watson, C.P.N.; Sessle, B.J.; Coderre, T.; Morley-Forster, P.K.; Stinson, J.; Boulanger, A.; et al. Pharmacological management of chronic neuropathic pain—Consensus statement and guidelines from the Canadian Pain Society. Pain Res. Manag. 2007, 12, 13–21. [Google Scholar] [CrossRef]
- Dworkin, R.H.; O’Connor, A.B.; Audette, J.; Baron, R.; Gourlay, G.K.; Haanpää, M.L.; Kent, J.L.; Krane, E.J.; Lebel, A.A.; Levy, R.M.; et al. Recommendations for the Pharmacological Management of Neuropathic Pain: An Overview and Literature Update. Mayo Clin. Proc. 2010, 85, S3–S14. [Google Scholar] [CrossRef] [PubMed]
- Raja, S.N.; Haythornthwaite, J.A.; Pappagallo, M.; Clark, M.R.; Travison, T.G.; Sabeen, S.; Royall, R.M.; Max, M.B. Opioids versus antidepressants in postherpetic neuralgia: A randomized, placebo-controlled trial. Neurology 2002, 59, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Moulin, D.E.; Palma, D.; Watling, C.; Schulz, V. Methadone in the Management of Intractable Neuropathic Noncancer Pain. Can. J. Neurol. Sci. 2005, 32, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Benyamin, R.; Trescot, A.M.; Datta, S.; Buenaventura, R.; Adlaka, R.; Sehgal, N.; E Glaser, S.; Vallejo, R. Opioid complications and side effects. Pain Phys. 2008, 11, 105–120. [Google Scholar]
- Dumas, E.O.; Pollack, G.M. Opioid Tolerance Development: A Pharmacokinetic/Pharmacodynamic Perspective. AAPS J. 2008, 10, 537–551. [Google Scholar] [CrossRef]
- Kosten, T.R.; George, T.P. The Neurobiology of Opioid Dependence: Implications for Treatment. Sci. Pr. Perspect. 2002, 1, 13–20. [Google Scholar] [CrossRef]
- Baby, S.; Dan, M.; Thaha, A.R.M.; Johnson, A.J.; Kurup, R.; Balakrishnapillai, P.; Lim, C.K. High content of zerumbone in volatile oils ofZingiber zerumbetfrom southern India and Malaysia. Flavour Fragr. J. 2009, 24, 301–308. [Google Scholar] [CrossRef]
- Koga, A.Y.; Beltrame, F.L.; Pereira, A.V. Several aspects of Zingiber zerumbet: A review. Rev. Bras. de Farm. 2016, 26, 385–391. [Google Scholar] [CrossRef]
- Butt, M.S.; Sultan, M.T. Ginger and its Health Claims: Molecular Aspects. Crit. Rev. Food Sci. Nutr. 2011, 51, 383–393. [Google Scholar] [CrossRef]
- Sahebkar, A. Potential efficacy of ginger as a natural supplement for nonalcoholic fatty liver disease. World J. Gastroenterol. 2011, 17, 271–272. [Google Scholar] [CrossRef]
- Sultana, S.; Ripa, F.; Hamid, K. Comparative antioxidant activity study of some commonly used spices in Bangladesh. Pak. J. Boil. Sci. 2010, 13, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, M.; Perimal, E.; Zakaria, Z.; Mokhtar, F.; Akhtar, M.; Lajis, N.; Israf, D. Preliminary analysis of the antinociceptive activity of zerumbone. Fitoterapia 2009, 80, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Perimal, E.K.; Akhtar, M.N.; Mohamad, A.S.; Khalid, M.H.; Ming, O.H.; Khalid, S.; Tatt, L.M.; Kamaldin, M.N.; Zakaria, Z.A.; Israf, D.A.; et al. Zerumbone-Induced Antinociception: Involvement of the l-Arginine-Nitric Oxide-cGMP -PKC-K+ATP Channel Pathways. Basic Clin. Pharmacol. Toxicol. 2010, 108, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, M.R.; Perimal, E.; Akhtar, M.; Mohamad, A.; Khalid, M.; Tasrip, N.; Mokhtar, F.; Zakaria, Z.; Lajis, N.; Israf, D. Anti-inflammatory effect of zerumbone on acute and chronic inflammation models in mice. Fitoterapia 2010, 81, 855–858. [Google Scholar] [CrossRef]
- Zulazmi, N.A.; Gopalsamy, B.; Farouk, A.A.O.; Sulaiman, M.R.; Bharatham, B.H.; Perimal, E.K. Antiallodynic and antihyperalgesic effects of zerumbone on a mouse model of chronic constriction injury-induced neuropathic pain. Fitoterapia 2015, 105, 215–221. [Google Scholar] [CrossRef]
- Zulazmi, N.A.; Gopalsamy, B.; Min, J.C.S.; Akira, A.; Sulaiman, M.R.; Bharatham, B.H.; Perimal, E.K. Zerumbone Alleviates Neuropathic Pain through the Involvement of l-Arginine-Nitric Oxide-cGMP-K+ ATP Channel Pathways in Chronic Constriction Injury in Mice Model. Molecules 2017, 22, 555. [Google Scholar] [CrossRef] [PubMed]
- Chia, J.S.M.; Farouk, A.A.O.; Mohamad, A.S.; Sulaiman, M.R.; Perimal, E.K. Zerumbone alleviates chronic constriction injury-induced allodynia and hyperalgesia through serotonin 5-HT receptors. Biomed. Pharm. 2016, 83, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Gopalsamy, B.; Farouk, A.A.O.; Tengku Mohamad, T.A.S.; Sulaiman, M.R.; Perimal, E.K. Antiallodynic and antihyperalgesic activities of zerumbone via the suppression of IL-1beta, IL-6, and TNF-alpha in a mouse model of neuropathic pain. J Pain Res. 2017, 10, 2605–2619. [Google Scholar] [CrossRef] [PubMed]
- Kitayama, T.; Okamoto, T.; Hill, R.K.; Kawai, Y.; Takahashi, S.; Yonemori, S.; Yamamoto, Y.; Ohe, K.; Uemura, S.; Sawada, S. Chemistry of Zerumbone. 1. Simplified Isolation, Conjugate Addition Reactions, and a Unique Ring Contracting Transannular Reaction of Its Dibromide. J. Org. Chem. 1999, 64, 2667–2672. [Google Scholar] [CrossRef]
- Maimone, T.J.; Baran, P.S. Modern synthetic efforts toward biologically active terpenes. Nat. Chem. Biol. 2007, 3, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Jantan, I.; Arshad, L.; Bukhari, S.N.A. Exploring the immunomodulatory and anticancer properties of zerumbone. Food Funct. 2017, 8, 3410–3431. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Youn, K.; Ji, Y.; Lee, S.; Lim, G.; Lee, J.; Ho, C.-T.; Leem, S.-H.; Jun, M. Biological and Computational Studies for Dual Cholinesterases Inhibitory Effect of Zerumbone. Nutrients 2020, 12, 1215. [Google Scholar] [CrossRef] [PubMed]
- Fatima, A.; Abdul, B.; Abdullah, R.; Karjiban, R.; Lee, V. Docking studies reveal zerumbone targets β-catenin of the Wnt-β-catenin pathway in breast cancer. J. Serbian Chem. Soc. 2018, 83, 575–591. [Google Scholar] [CrossRef]
- Eid, E.E.M.; Azam, F.; Hassan, M.; Taban, I.M.; Halim, M.A. Zerumbone binding to estrogen receptors: An in-silico investigation. J. Recept. Signal Transduct. 2018, 38, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Murakami, A.; Takahashi, D.; Kinoshita, T.; Koshimizu, K.; Kim, H.W.; Yoshihiro, A.; Nakamura, Y.; Jiwajinda, S.; Terao, J.; Ohigashi, H. Zerumbone, a Southeast Asian ginger sesquiterpene, markedly suppresses free radical generation, proinflammatory protein production, and cancer cell proliferation accompanied by apoptosis: The alpha,beta-unsaturated carbonyl group is a prerequisite. Carcinogenesis 2002, 23, 795–802. [Google Scholar] [CrossRef]
- Singh, S.P.; Nongalleima, K.; Singh, N.I.; Doley, P.; Singh, C.B.; Singh, T.R.; Sahoo, D. Zerumbone reduces proliferation of HCT116 colon cancer cells by inhibition of TNF-alpha. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef]
- Chen, L.; Chen, W.; Qian, X.; Fang, Y.; Zhu, N. Liquiritigenin alleviates mechanical and cold hyperalgesia in a rat neuropathic pain model. Sci. Rep. 2014, 4, 5676. [Google Scholar] [CrossRef]
- Deacon, R. Measuring motor coordination in mice. J. Vis. Exp. 2013, 75, e2609. [Google Scholar] [CrossRef]
- Rasband, M.N.; Trimmer, J.S.; Schwarz, T.L.; Levinson, S.R.; Ellisman, M.H.; Schachner, M.; Shrager, P. Potassium Channel Distribution, Clustering, and Function in Remyelinating Rat Axons. J. Neurosci. 1998, 18, 36–47. [Google Scholar] [CrossRef]
- McKeown, L.; Swanton, L.; Robinson, P.; Jones, O.T. Surface expression and distribution of voltage-gated potassium channels in neurons (Review). Mol. Membr. Boil. 2008, 25, 332–343. [Google Scholar] [CrossRef]
- Taglialatela, M.; Vandongen, A.M.; A Drewe, J.; Joho, R.H.; Brown, A.M.; E Kirsch, G. Patterns of internal and external tetraethylammonium block in four homologous K+ channels. Mol. Pharmacol. 1991, 40, 299–307. [Google Scholar]
- Del Camino, D.; Holmgren, M.; Liu, Y.; Yellen, G. Blocker protection in the pore of a voltage-gated K+ channel and its structural implications. Nature 2000, 403, 321–325. [Google Scholar] [CrossRef]
- Wuttke, T.V.; Seebohm, G.; Bail, S.; Maljevic, S.; Lerche, H. The New Anticonvulsant Retigabine Favors Voltage-Dependent Opening of the Kv7.2 (KCNQ2) Channel by Binding to Its Activation Gate. Mol. Pharmacol. 2005, 67, 1009–1017. [Google Scholar] [CrossRef]
- Lü, Q.; Peevey, J.; Jow, F.; Monaghan, M.M.; Mendoza, G.; Zhang, H.; Wu, J.; Kim, C.Y.; Bicksler, J.; Greenblatt, L.; et al. Disruption of Kv1.1 N-type inactivation by novel small molecule inhibitors (disinactivators). Bioorganic Med. Chem. 2008, 16, 3067–3075. [Google Scholar] [CrossRef]
- Rose, K.; Ooi, L.; Dalle, C.; Robertson, B.; Wood, I.C.; Gamper, N. Transcriptional repression of the M channel subunit Kv7.2 in chronic nerve injury. Pain 2011, 152, 742–754. [Google Scholar] [CrossRef]
- Liu, B.; Linley, J.E.; Du, X.; Zhang, X.; Ooi, L.; Zhang, H.; Gamper, N. The acute nociceptive signals induced by bradykinin in rat sensory neurons are mediated by inhibition of M-type K+ channels and activation of Ca2+-activated Cl- channels. J. Clin. Investig. 2010, 120, 1240–1252. [Google Scholar] [CrossRef]
- Passmore, G.M.; Selyanko, A.A.; Mistry, M.; Al-Qatari, M.; Marsh, S.J.; Matthews, E.A.; Dickenson, A.H.; Brown, T.A.; Burbidge, S.A.; Main, M.; et al. KCNQ/M Currents in Sensory Neurons: Significance for Pain Therapy. J. Neurosci. 2003, 23, 7227–7236. [Google Scholar] [CrossRef]
- Wulff, H.; Castle, N.A.; Pardo, L.A. Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov. 2009, 8, 982–1001. [Google Scholar] [CrossRef]
- Gribble, F.M.; Reimann, F. Sulphonylurea action revisited: The post-cloning era. Diabetologia 2003, 46, 875–891. [Google Scholar] [CrossRef]
- Ocaña, M.; Baeyens, J.M. Differential effects of K+ channel blockers on antinociception induced by alpha 2-adrenoceptor, GABAB and kappa-opioid receptor agonists. Br. J. Pharmacol. 1993, 110, 1049–1054. [Google Scholar]
- Robles, L.-I.; Barrios, M.; Del Pozo, E.; Dordal, A.; Baeyens, J. Effects of K+ channel blockers and openers on antinociception induced by agonists of 5-HT1A receptors. Eur. J. Pharmacol. 1996, 295, 181–188. [Google Scholar] [CrossRef]
- Ocaña, M.; Del Pozo, E.; Barrios, M.; Robles, L.I.; Baeyens, J. An ATP-dependent potassium channel blocker antagonizes morphine analgesia. Eur. J. Pharmacol. 1990, 186, 377–378. [Google Scholar] [CrossRef]
- Zushida, K.; Onodera, K.; Kamei, J. Effect of diabetes on pinacidil-induced antinociception in mice. Eur. J. Pharmacol. 2002, 453, 209–215. [Google Scholar] [CrossRef]
- Campbell, V.C.; Welch, S.P. The role of minoxidil on endogenous opioid peptides in the spinal cord: A putative co-agonist relationship between K-ATP openers and opioids. Eur. J. Pharmacol. 2001, 417, 91–98. [Google Scholar] [CrossRef]
- Sun, H.-S.; Feng, Z.-P. Neuroprotective role of ATP-sensitive potassium channels in cerebral ischemia. Acta Pharmacol. Sin. 2012, 34, 24–32. [Google Scholar] [CrossRef]
- Wulf-Johansson, H.; Hay-Schmidt, A.; Poulsen, A.N.; Klaerke, D.A.; Olesen, J.; Jansen, I. Expression of BKCa channels and the modulatory β-subunits in the rat and porcine trigeminal ganglion. Brain Res. 2009, 1292, 1–13. [Google Scholar] [CrossRef]
- Furukawa, N.; Takasusuki, T.; Fukushima, T.; Hori, Y. Presynaptic large-conductance calcium-activated potassium channels control synaptic transmission in the superficial dorsal horn of the mouse. Neurosci. Lett. 2008, 444, 79–82. [Google Scholar] [CrossRef]
- Chen, S.-R.; Cai, Y.-Q.; Pan, H.-L. Plasticity and emerging role of BKCa channels in nociceptive control in neuropathic pain. J. Neurochem. 2009, 110, 352–362. [Google Scholar] [CrossRef]
- Liu, C.-Y.; Lu, Z.-Y.; Li, N.; Yu, L.-H.; Zhao, Y.-F.; Ma, B. The role of large-conductance, calcium-activated potassium channels in a rat model of trigeminal neuropathic pain. Cephalalgia 2014, 35, 16–35. [Google Scholar] [CrossRef]
- Horrigan, F.T.; Cui, J.; Aldrich, R.W. Allosteric Voltage Gating of Potassium Channels I. J. Gen. Physiol. 1999, 114, 277–304. [Google Scholar] [CrossRef]
- Hill, M.A.; Yang, Y.; Ella, S.R.; Davis, M.J.; Braun, A.P. Large conductance, Ca2+-activated K+ channels (BKCa) and arteriolar myogenic signaling. FEBS Lett. 2010, 584, 2033–2042. [Google Scholar] [CrossRef]
- Cui, J.; Yang, H.; Lee, U.S. Molecular mechanisms of BK channel activation. Cell. Mol. Life Sci. 2008, 66, 852–875. [Google Scholar] [CrossRef]
- Ocaña, M.; Cendán, C.M.; Cobos, E.J.; Entrena, J.M.; Baeyens, J.M. Potassium channels and pain: Present realities and future opportunities. Eur. J. Pharmacol. 2004, 500, 203–219. [Google Scholar] [CrossRef]
- Cao, Y.; Dreixler, J.C.; Roizen, J.D.; Roberts, M.T.; Houamed, K.M. Modulation of recombinant small-conductance Ca(2+)-activated K(+) channels by the muscle relaxant chlorzoxazone and structurally related compounds. J. Pharmacol. Exp. Ther. 2001, 296, 683–689. [Google Scholar]
- Thompson, J.M.; Ji, G.; Neugebauer, V. Small-conductance calcium-activated potassium (SK) channels in the amygdala mediate pain-inhibiting effects of clinically available riluzole in a rat model of arthritis pain. Mol. Pain 2015, 11, 51. [Google Scholar] [CrossRef]
- Granados-Soto, V.; Argüelles, C.F.; Ortiz, M.I. The peripheral antinociceptive effect of resveratrol is associated with activation of potassium channels. Neuropharmacology 2002, 43, 917–923. [Google Scholar] [CrossRef]
- Kapitzke, D.; Vetter, I.; Cabot, P.J. Endogenous opioid analgesia in peripheral tissues and the clinical implications for pain control. Ther. Clin. Risk Manag. 2005, 1, 279–297. [Google Scholar]
- Chang, K.J.; Cuatrecasas, P. Multiple opiate receptors. Enkephalins and morphine bind to receptors of different specificity. J. Boil. Chem. 1979, 254, 2610–2618. [Google Scholar]
- Stoeber, M.; Jullié, D.; Li, J.; Chakraborty, S.; Majumdar, S.; Lambert, N.A.; Manglik, A.; Von Zastrow, M. Agonist-selective recruitment of engineered protein probes and of GRK2 by opioid receptors in living cells. eLife 2020, 9, e54208. [Google Scholar] [CrossRef]
- Al-Hasani, R.; Bruchas, M.R. Molecular Mechanisms of Opioid Receptor-dependent Signaling and Behavior. Anesthesiology 2011, 115, 1363–1381. [Google Scholar] [CrossRef]
- Ossipov, M.H.; Morimura, K.; Porreca, F. Descending pain modulation and chronification of pain. Curr. Opin. Support Palliat Care 2014, 8, 143–151. [Google Scholar]
- Pradhan, A.A.; Clarke, P.B. Comparison between delta-opioid receptor functional response and autoradiographic labeling in rat brain and spinal cord. J. Comp. Neurol. 2005, 481, 416–426. [Google Scholar] [CrossRef]
- Poulin, J.-F.; Chevalier, B.; Laforest, S.; Drolet, G. Enkephalinergic afferents of the centromedial amygdala in the rat. J. Comp. Neurol. 2006, 496, 859–876. [Google Scholar] [CrossRef]
- Le Merrer, J.; Becker, J.A.J.; Befort, K.; Kieffer, B.L. Reward processing by the opioid system in the brain. Physiol. Rev. 2009, 89, 1379–1412. [Google Scholar] [CrossRef]
- Basbaum, A.I.; Fields, H.L. Endogenous pain control mechanisms: Review and hypothesis. Ann. Neurol. 1978, 4, 451–462. [Google Scholar] [CrossRef]
- Basbaum, A.I.; Fields, H.L. Endogenous pain control systems: Brainstem spinal pathways and endorphin circuitry. Annu. Rev. Neurosci. 1984, 7, 309–338. [Google Scholar] [CrossRef]
- Drake, C.T.; Chavkin, C.; Milner, T.A. Opioid systems in the dentate gyrus. Prog. Brain Res. 2007, 163, 245–814. [Google Scholar] [CrossRef]
- Upton, N.; Sewell, R.D.; Spencer, P.S. Analgesic actions of mu- and kappa-opiate agonists in rats. Arch. Int. de Pharmacodyn. et de Ther. 1983, 262, 199–207. [Google Scholar]
- Tyers, M.B. A classification of opiate receptors that mediate antinociception in animals. Br. J. Pharmacol. 1980, 69, 503–512. [Google Scholar] [CrossRef]
- Goodman, A.J.; Le Bourdonnec, B.; Dolle, R.E. Mu Opioid Receptor Antagonists: Recent Developments. Chem. Med. Chem. 2007, 2, 1552–1570. [Google Scholar] [CrossRef]
- Gaveriaux-Ruff, C.; Karchewski, L.A.; Hever, X.; Matifas, A.; Kieffer, B.L. Inflammatory pain is enhanced in delta opioid receptor-knockout mice. Eur. J. Neurosci. 2008, 27, 2558–2567. [Google Scholar] [CrossRef] [PubMed]
- Nadal, X.; Banos, J.E.; Kieffer, B.L.; Maldonado, R. Neuropathic pain is enhanced in delta-opioid receptor knockout mice. Eur. J. Neurosci. 2006, 23, 830–834. [Google Scholar] [CrossRef] [PubMed]
- Vanderah, T.W. Delta and Kappa Opioid Receptors as Suitable Drug Targets for Pain. Clin. J. Pain 2010, 26, S10–S15. [Google Scholar] [CrossRef] [PubMed]
- Aceto, M.D.; May, E.L.; Harris, L.S.; Bowman, E.R.; Cook, C.D. Pharmacological studies with a nonpeptidic, delta-opioid (−)-(1R,5R,9R)-5,9-dimethyl-2′-hydroxy-2-(6-hydroxyhexyl)-6,7-benzomorphan hydrochloride ((−)-NIH 11082). Eur. J. Pharmacol. 2007, 566, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Le Bourdonnec, B.; Windh, R.T.; Ajello, C.W.; Leister, L.K.; Gu, M.; Chu, G.-H.; Tuthill, P.A.; Barker, W.M.; Koblish, M.; Wiant, D.D.; et al. Potent, Orally Bioavailable Delta Opioid Receptor Agonists for the Treatment of Pain: Discovery of N,N-Diethyl-4-(5-hydroxyspiro[chromene-2,4′-piperidine]-4-yl)benzamide (ADL5859). J. Med. Chem. 2008, 51, 5893–5896. [Google Scholar] [CrossRef]
- Saitoh, A.; Sugiyama, A.; Nemoto, T.; Fujii, H.; Wada, K.; Oka, J.-I.; Nagase, H.; Yamada, M. The novel δ opioid receptor agonist KNT-127 produces antidepressant-like and antinociceptive effects in mice without producing convulsions. Behav. Brain Res. 2011, 223, 271–279. [Google Scholar] [CrossRef]
- Jones, P.; Griffin, A.M.; Gawell, L.; Lavoie, R.; Delorme, D.; Roberts, E.; Brown, W.; Walpole, C.; Xiao, W.; Boulet, J.; et al. N,N-Diethyl-4-[(3-hydroxyphenyl)(piperidin-4-yl)amino] benzamide derivatives: The development of diaryl amino piperidines as potent delta opioid receptor agonists with in vivo anti-anociceptive activity in rodent models. Bioorganic Med. Chem. Lett. 2009, 19, 5994–5998. [Google Scholar]
- Chavkin, C. The Therapeutic Potential of [kappa]-Opioids for Treatment of Pain and Addiction. Neuropsychopharmacology 2011, 36, 369–370. [Google Scholar] [CrossRef]
- Su, X.; Castle, N.A.; Antonio, B.; Roeloffs, R.; Thomas, J.B.; Krafte, D.S.; Chapman, M.L. The effect of kappa-opioid receptor agonists on tetrodotoxin-resistant sodium channels in primary sensory neurons. Anesth. Analg. 2009, 109, 632–640. [Google Scholar] [CrossRef]
- Amir, R.; Michaelis, M.; Devor, M. Membrane Potential Oscillations in Dorsal Root Ganglion Neurons: Role in Normal Electrogenesis and Neuropathic Pain. J. Neurosci. 1999, 19, 8589–8596. [Google Scholar] [CrossRef]
- Giordano, J.; Schultea, T. Serotonin 5-HT (3) receptor mediation of pain and anti-anociception: Implications for clinical therapeutics. Pain Physician 2004, 7, 141–147. [Google Scholar] [PubMed]
- Ming-Tatt, L.; Khalivulla, S.I.; Akhtar, M.N.; Lajis, N.; Perimal, E.K.; Akira, A.; Ali, D.I.; Sulaiman, M.R. Anti-hyperalgesic effect of a benzilidine-cyclohexanone analogue on a mouse model of chronic constriction injury-induced neuropathic pain: Participation of the κ-opioid receptor and KATP. Pharmacol. Biochem. Behav. 2013, 114, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, V.P.; Zambelli, V.O.; Picolo, G.; Chacur, M.; Sampaio, S.C.; Brigatte, P.; Konno, K.; Cury, Y. The peripheral L-arginine–nitric oxide–cyclic GMP pathway and ATP-sensitive K+ channels are involved in the antinociceptive effect of crotalphine on neuropathic pain in rats. Behav. Pharmacol. 2012, 23, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Chia, J.S.M.; Izham, N.A.M.; Farouk, A.A.O.; Sulaiman, M.R.; Mustafa, S.; Hutchinson, M.R.; Perimal, E.K. Zerumbone Modulates α2A-Adrenergic, TRPV1, and NMDA NR2B Receptors Plasticity in CCI-Induced Neuropathic Pain In Vivo and LPS-Induced SH-SY5Y Neuroblastoma In Vitro Models. Front. Pharmacol. 2020, 11, 92. [Google Scholar] [CrossRef] [PubMed]
- Stone, L.S.; Macmillan, L.B.; Kitto, K.F.; Limbird, L.E.; Wilcox, G.L. The α2a Adrenergic Receptor Subtype Mediates Spinal Analgesia Evoked by α2 Agonists and Is Necessary for Spinal Adrenergic–Opioid Synergy. J. Neurosci. 1997, 17, 7157–7165. [Google Scholar] [CrossRef]
- Fairbanks, C.A.; Stone, L.S.; Kitto, K.F.; Nguyen, H.O.; Posthumus, I.J.; Wilcox, G.L. α2C-Adrenergic Receptors Mediate Spinal Analgesia and Adrenergic-Opioid Synergy. J. Pharmacol. Exp. Ther. 2002, 300, 282–290. [Google Scholar] [CrossRef]
- Ossipov, M.H.; Lopez, Y.; Bian, D.; Nichols, M.L.; Porreca, F. Synergistic Antinociceptive Interactions of Morphine and Clonidine in Rats with Nerve-ligation Injury. Anesthesiology 1997, 86, 196–204. [Google Scholar] [CrossRef]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [CrossRef]
- Fundytus, M.E. Glutamate Receptors and Nociception. CNS Drugs 2001, 15, 29–58. [Google Scholar] [CrossRef]
- Trivedi, M.; Shaikh, S.; Gwinnut, C. Pharmacology of opioids. Anaesthesia 2011, 118–124. [Google Scholar]
- Zakaria, Z.; Rahim, M.H.A.; Roosli, R.A.J.; Sani, M.H.M.; Marmaya, N.H.; Omar, M.H.; Teh, L.K.; Salleh, M.Z. Antinociceptive Activity of Petroleum Ether Fraction of Clinacanthus nutans Leaves Methanolic Extract: Roles of Nonopioid Pain Modulatory Systems and Potassium Channels. BioMed. Res. Int. 2019, 2019, 6593125. [Google Scholar] [CrossRef] [PubMed]
- Klein, A.H.; Mohammad, H.K.; Ali, R.; Peper, B.; Wilson, S.P.; Raja, S.N.; Ringkamp, M.; Sweitzer, S. Overexpression of µ-Opioid Receptors in Peripheral Afferents, but Not in Combination with Enkephalin, Decreases Neuropathic Pain Behavior and Enhances Opioid Analgesia in Mouse. Anesthesiology 2018, 128, 967–983. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.J.; Pitcher, M.H.; Stone, L.S.; Tarum, F.; Niu, G.; Chen, X.; Kiesewetter, D.O.; Schweinhardt, P.; Bushnell, M.C. Chronic neuropathic pain reduces opioid receptor availability with associated anhedonia in rat. Pain 2018, 159, 1856–1866. [Google Scholar] [CrossRef] [PubMed]
- Le Guen, S.; Gestreau, C.; Besson, J.-M. Morphine withdrawal precipitated by specific mu, delta or kappa opioid receptor antagonists: A c-Fos protein study in the rat central nervous system. Eur. J. Neurosci. 2003, 17, 2425–2437. [Google Scholar] [CrossRef] [PubMed]
- Gestreau, C.; Besson, J.-M. Is there tonic activity in the endogenous opioid systems? A c-Fos study in the rat central nervous system after intravenous injection of naloxone or naloxone-methiodide. J. Comp. Neurol. 2000, 427, 285–301. [Google Scholar] [CrossRef]
- Galeotti, N.; Ghelardini, C.; Caldari, B.; Bartolini, A. Effect of potassium channel modulators in mouse forced swimming test. Br. J. Pharmacol. 1999, 126, 1653–1659. [Google Scholar] [CrossRef]
- Welch, S.P.; Dunlow, L.D. Antinociceptive activity of intrathecally administered potassium channel openers and opioid agonists: A common mechanism of action? J. Pharmacol. Exp. Ther. 1993, 267, 390–399. [Google Scholar]
- Bennett, G.J.; Xie, Y.-K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 1988, 33, 87–107. [Google Scholar] [CrossRef]
- Gopalsamy, B.; Sambasevam, Y.; Zulazmi, N.A.; Chia, J.S.M.; Farouk, A.A.O.; Sulaiman, M.R.; Mohamad, T.A.S.T.; Perimal, E.K. Experimental Characterization of the Chronic Constriction Injury-Induced Neuropathic Pain Model in Mice. Neurochem. Res. 2019, 44, 2123–2138. [Google Scholar] [CrossRef]
- Martinov, T.; Mack, M.R.; Sykes, A.; Chatterjea, D. Measuring changes in tactile sensitivity in the hind paw of mice using an electronic von Frey apparatus. J. Vis. Exp. 2013, 82, e51212. [Google Scholar] [CrossRef]
- Hargreaves, K.; Dubner, R.; Brown, F.; Flores, C.; Joris, J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 1988, 32, 77–88. [Google Scholar] [CrossRef]
Sample Availability: Samples of zerumbone are available from the authors. |









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gopalsamy, B.; Chia, J.S.M.; Farouk, A.A.O.; Sulaiman, M.R.; Perimal, E.K. Zerumbone-Induced Analgesia Modulated via Potassium Channels and Opioid Receptors in Chronic Constriction Injury-Induced Neuropathic Pain. Molecules 2020, 25, 3880. https://doi.org/10.3390/molecules25173880
Gopalsamy B, Chia JSM, Farouk AAO, Sulaiman MR, Perimal EK. Zerumbone-Induced Analgesia Modulated via Potassium Channels and Opioid Receptors in Chronic Constriction Injury-Induced Neuropathic Pain. Molecules. 2020; 25(17):3880. https://doi.org/10.3390/molecules25173880
Chicago/Turabian StyleGopalsamy, Banulata, Jasmine Siew Min Chia, Ahmad Akira Omar Farouk, Mohd Roslan Sulaiman, and Enoch Kumar Perimal. 2020. "Zerumbone-Induced Analgesia Modulated via Potassium Channels and Opioid Receptors in Chronic Constriction Injury-Induced Neuropathic Pain" Molecules 25, no. 17: 3880. https://doi.org/10.3390/molecules25173880
APA StyleGopalsamy, B., Chia, J. S. M., Farouk, A. A. O., Sulaiman, M. R., & Perimal, E. K. (2020). Zerumbone-Induced Analgesia Modulated via Potassium Channels and Opioid Receptors in Chronic Constriction Injury-Induced Neuropathic Pain. Molecules, 25(17), 3880. https://doi.org/10.3390/molecules25173880