
Piceatannol, a Natural Analog of Resveratrol, Exerts Anti-angiogenic Efficiencies by Blockage of Vascular Endothelial Growth Factor Binding to Its Receptor


Abstract

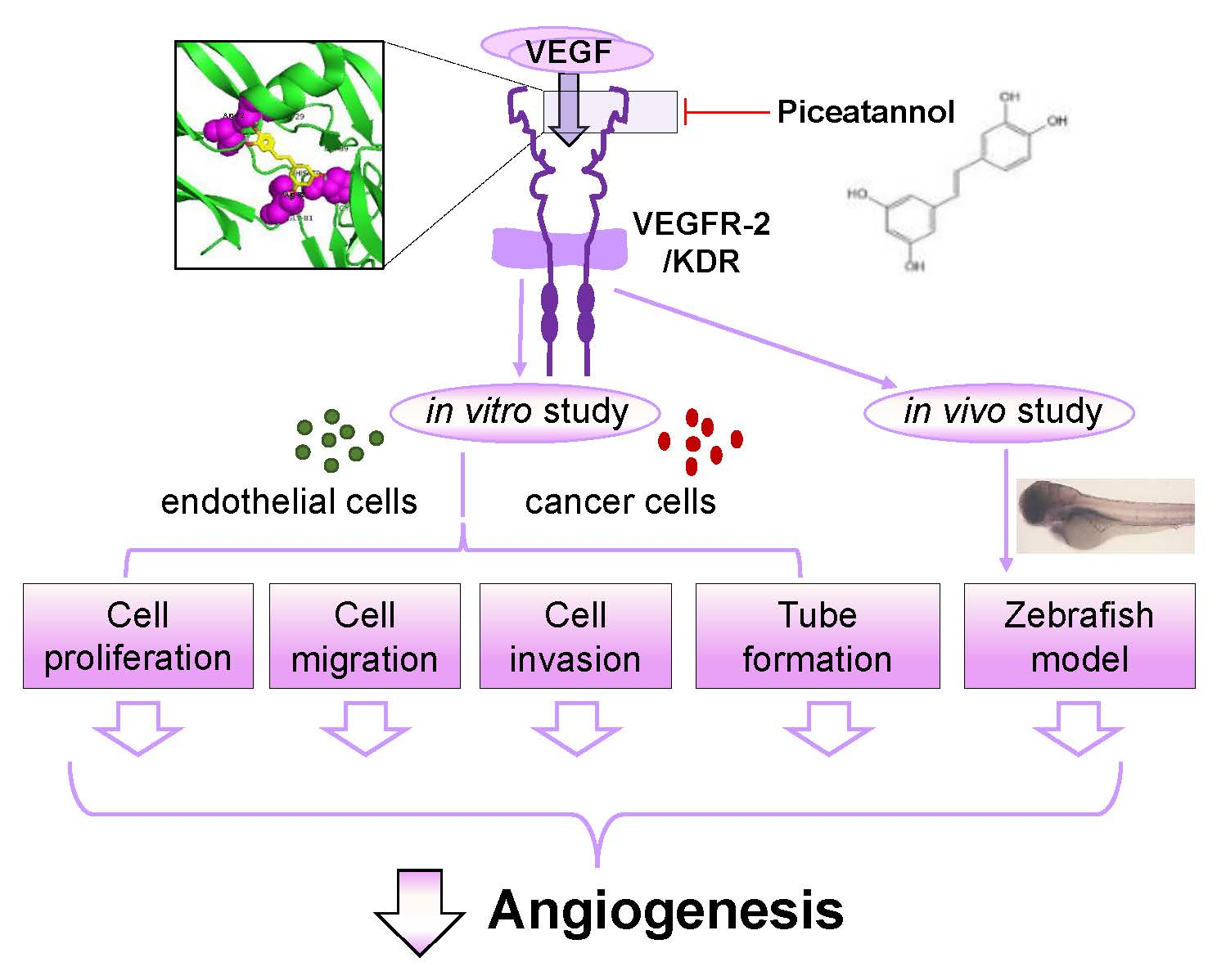{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
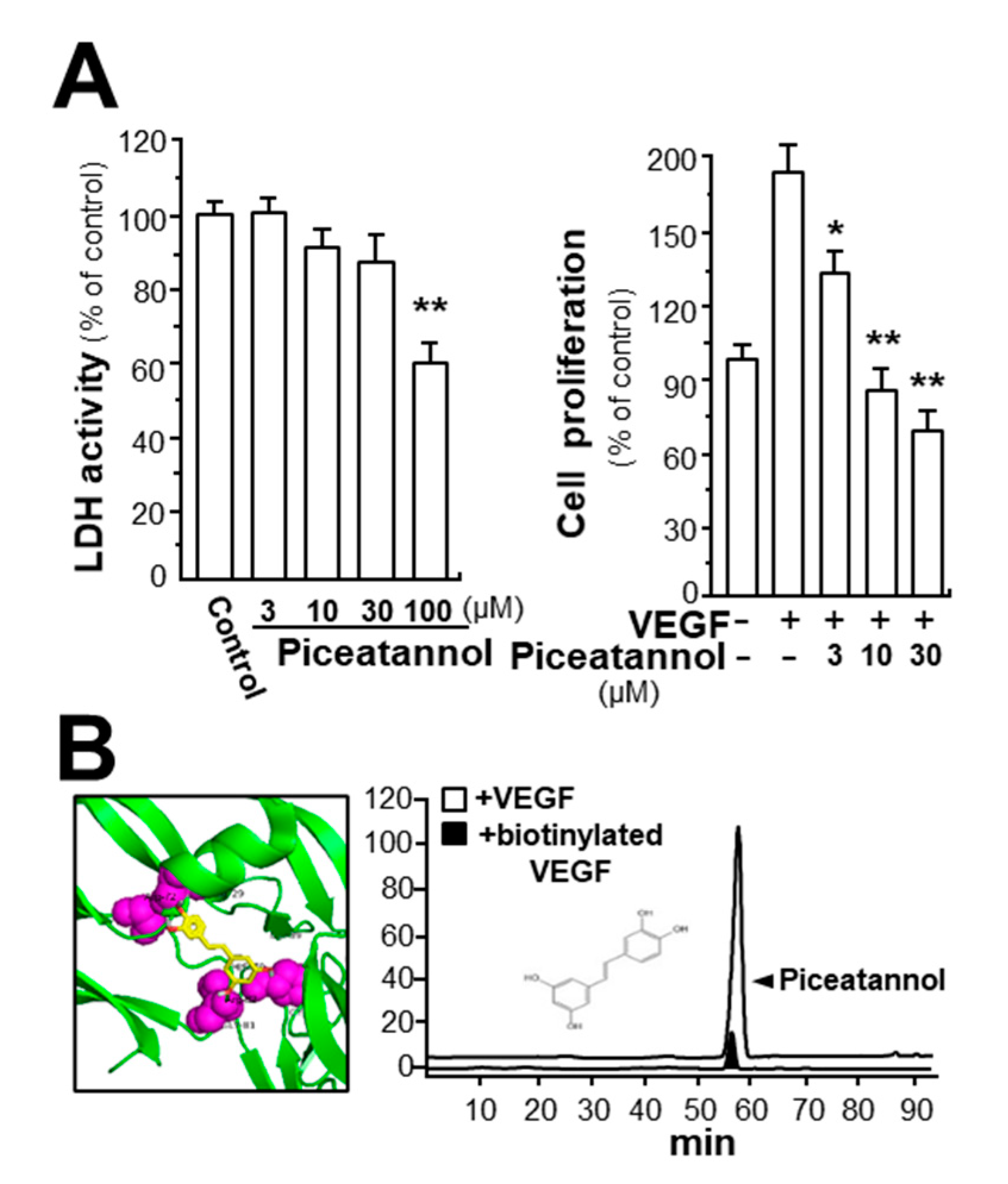
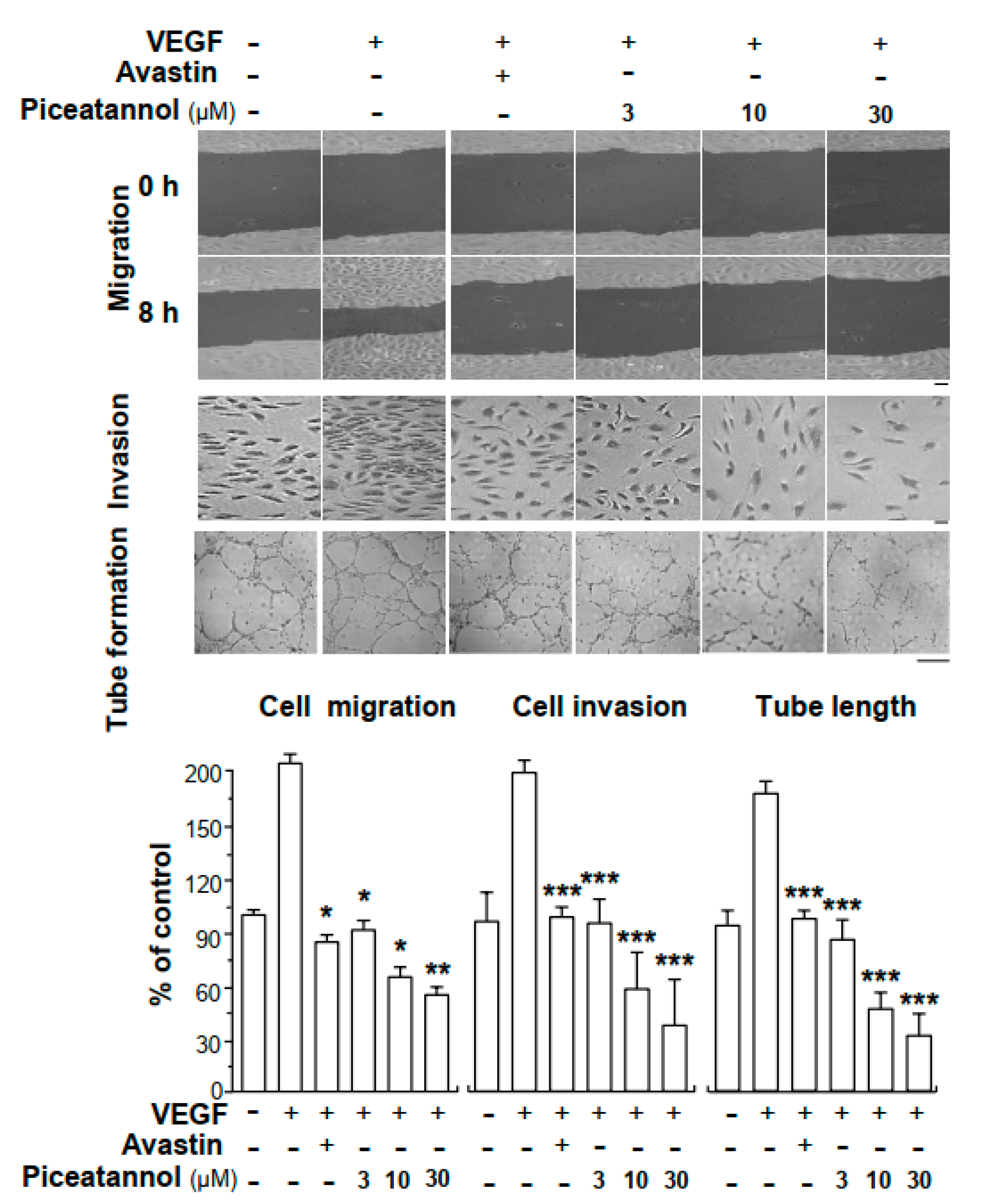
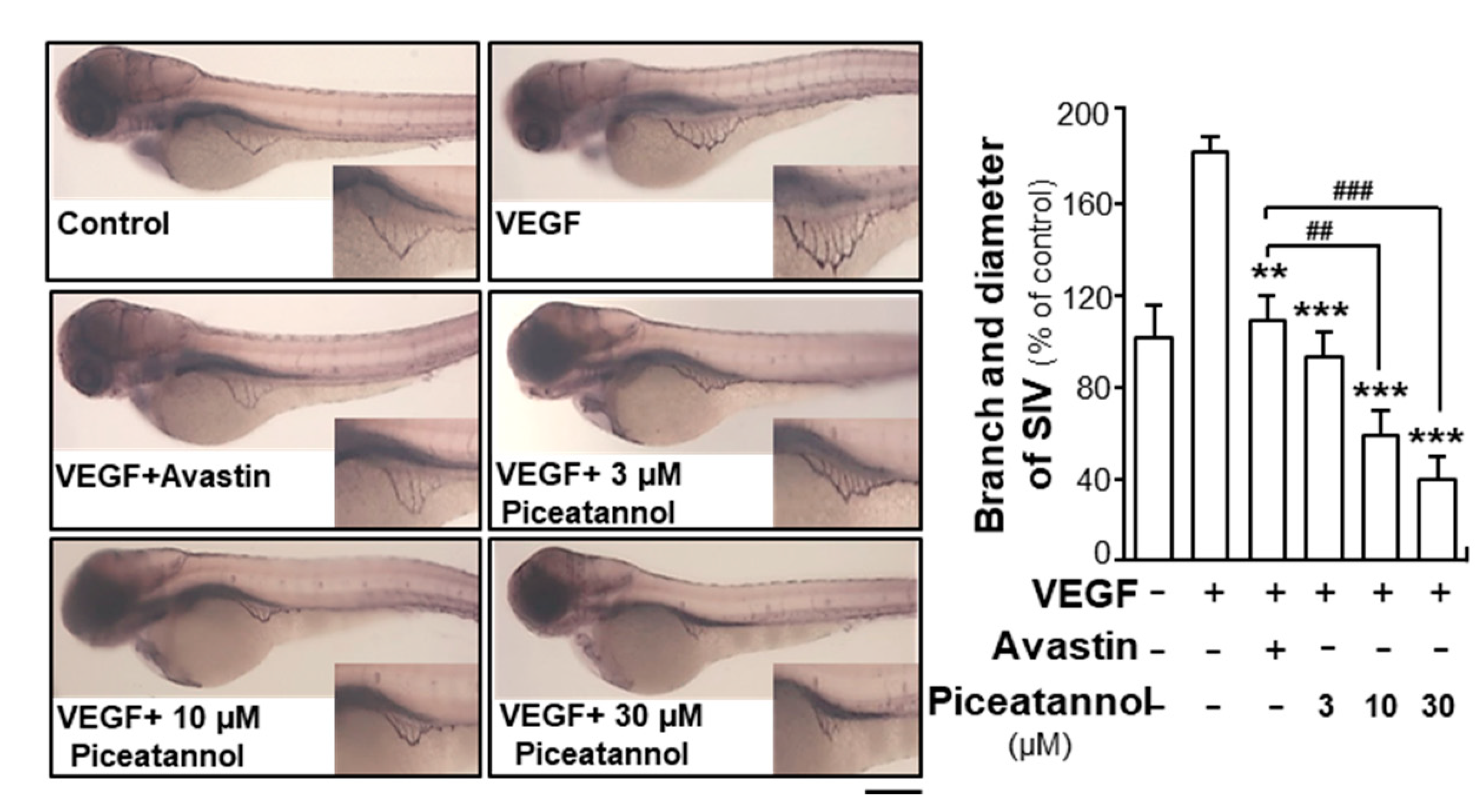
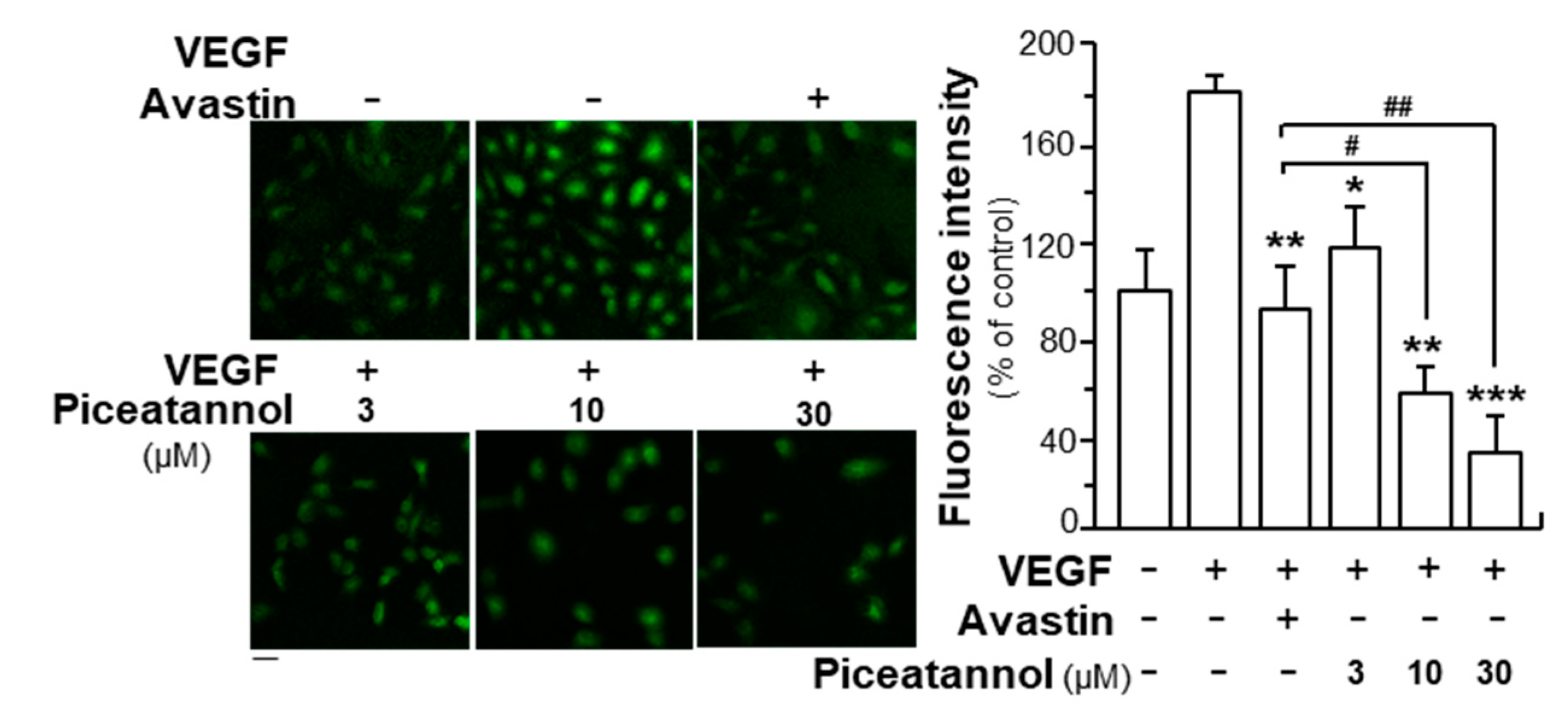
2.1. Piceatannol Suppresses VEGF-Mediated Angiogenesis
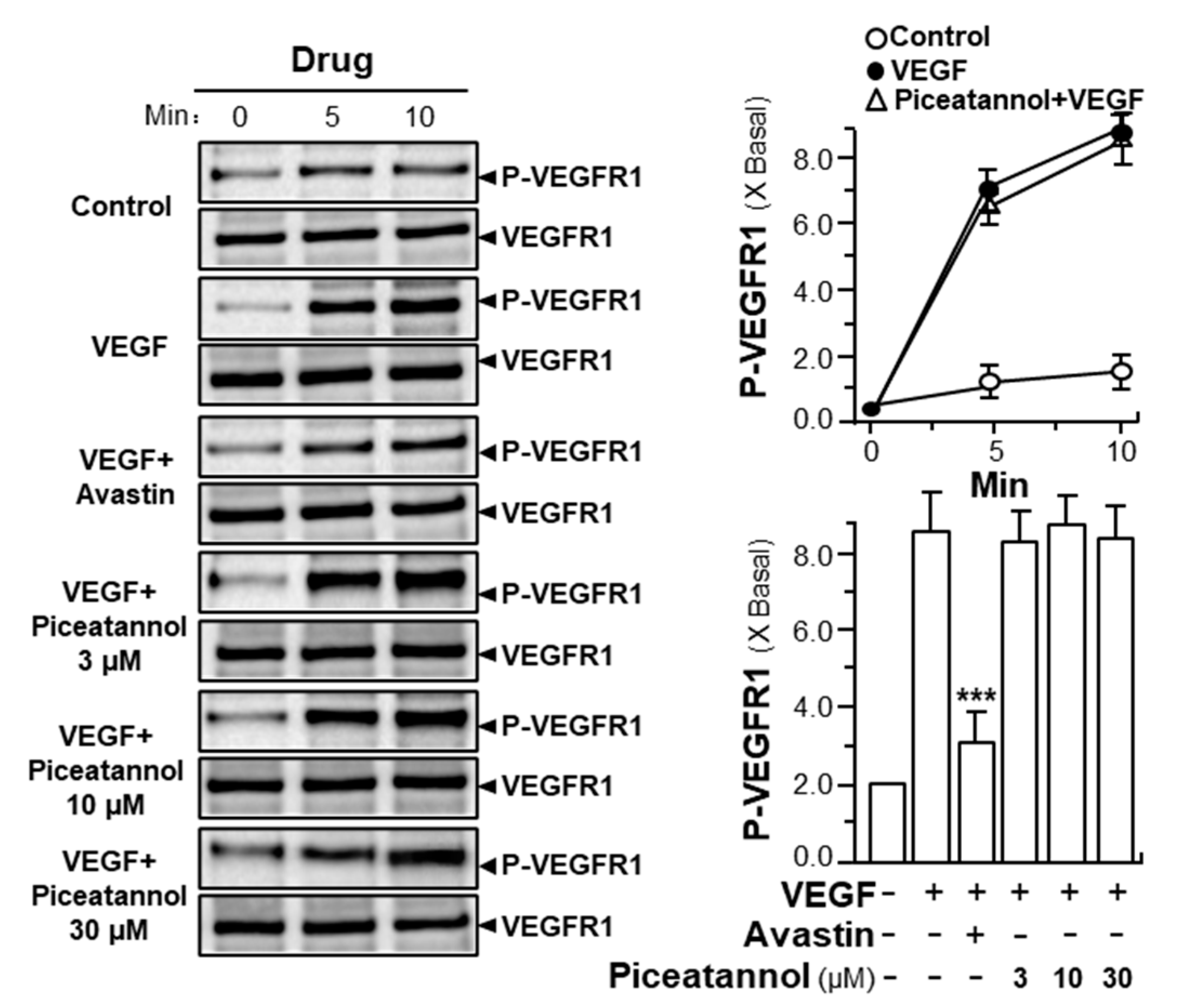
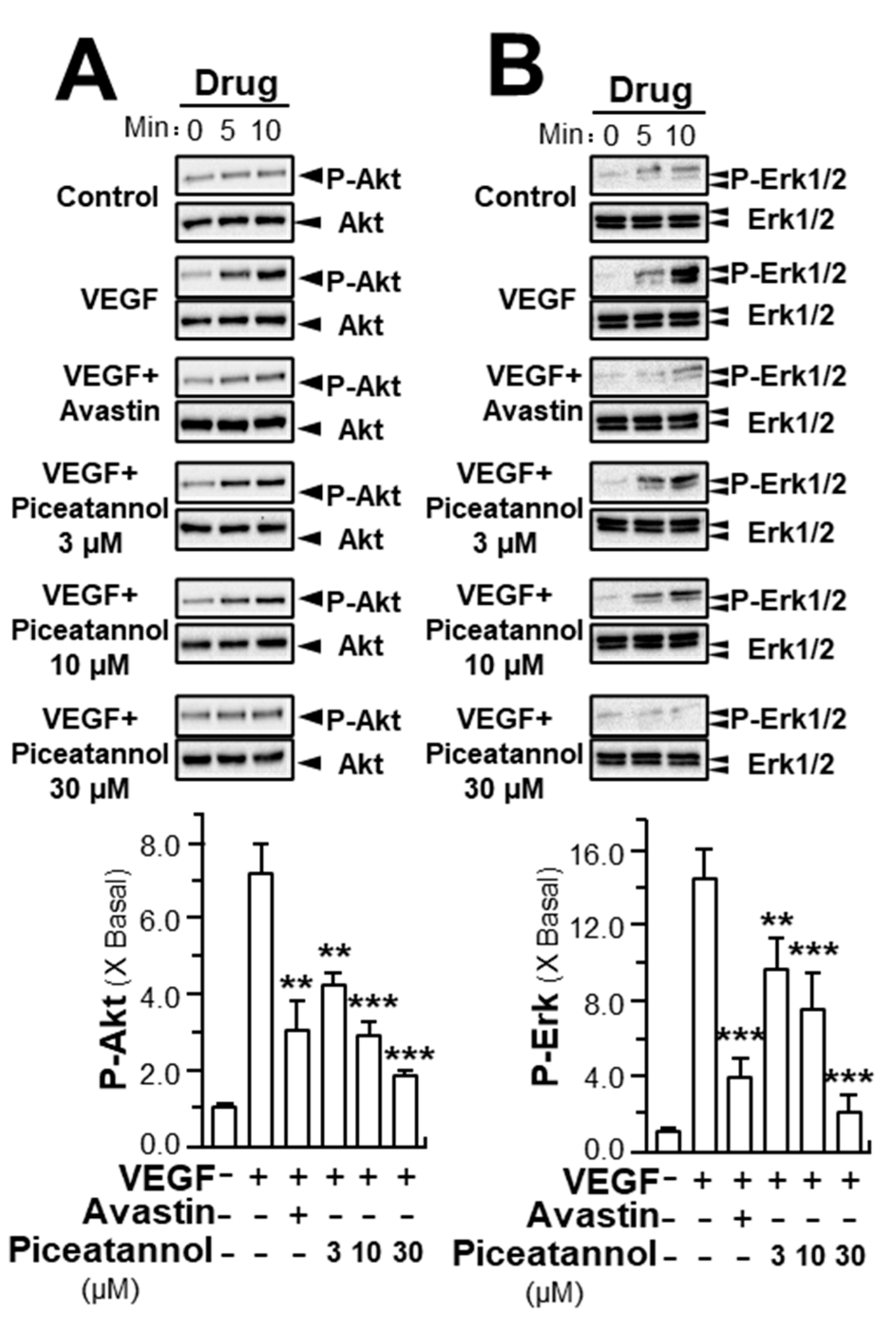
2.2. Piceatannol Inhibits VEGF-Mediated Signaling
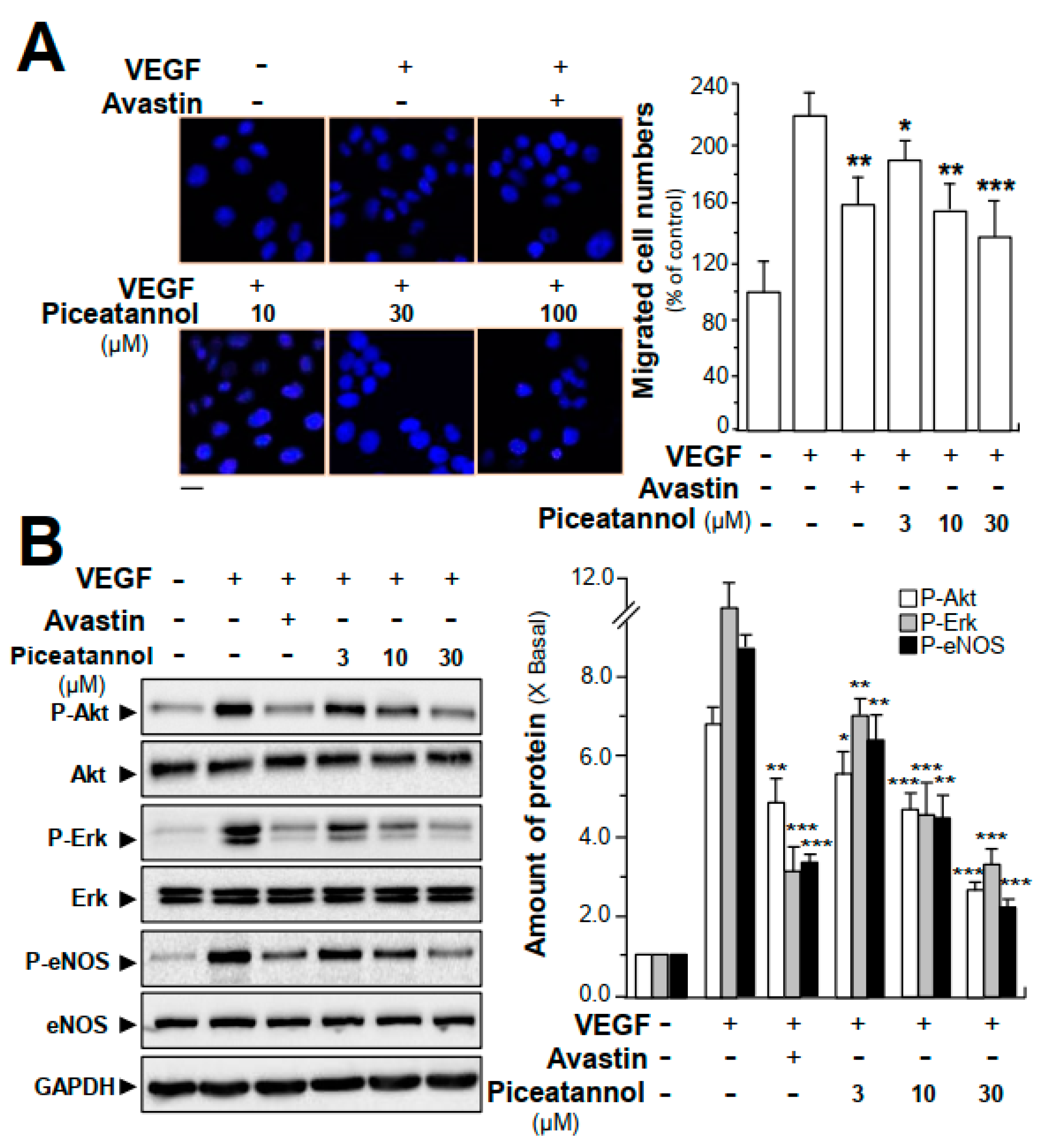
2.3. Piceatannol Suppresses VEGF-Triggered Colon Cancer Cell Proliferation and Migration
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Chemicals
4.2. Cell Viability Assay
4.3. Endothelial Cell Migration Assay
4.4. Endothelial Cell Invasion Assay
4.5. Endothelial Cell Tube Formation Assay
4.6. Zebrafish Angiogenesis Assay
4.7. Alkaline Phosphatase-based Vascular Staining
4.8. Molecular Docking
4.9. Immunoprecipitation Assay
4.10. Western Blot Analysis
4.11. Measurement of Reactive Oxygen Species
4.12. Transwell® Motility Assay
4.13. Other Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| VEGFs | vascular endothelial growth factors |
| HUVECs | human umbilical vein endothelial cells |
| VEGF | vascular endothelial growth factor |
| VEGFR1 | VEGF receptor 1 |
| VEGFR2 | VEGF receptor 2 |
| VEGFR3 | VEGF receptor 3 |
| Erk | extracellular signal-regulated kinase |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
References
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar]
- Bagherpoorfard, M.; Soheili, A.R. A moving mesh method for mathematical model of capillary formation in tumor angiogenesis. Iran. J. Sci. Technol. Trans. A. Sci. 2019, 43, 1745–1753. [Google Scholar] [CrossRef]
- Matsuoka, A.; Mizumoto, Y.; Ono, M.; Kagami, K.; Obata, T.; Terakawa, J.; Maida, Y.; Nakamura, M.; Daikoku, T.; Fujiwara, H. Novel strategy of ovarian cancer implantation: Pre-invasive growth of fibrin-anchored cells with neovascularization. Cancer Sci. 2019, 110, 2658–2666. [Google Scholar] [CrossRef]
- Folkman, J.; Shing, Y. Angiogenesis. J. Biol. Chem. 1992, 267, 10931–10934. [Google Scholar]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef]
- Chung, A.S.; Ferrara, N. Developmental and pathological angiogenesis. Annu. Rev. Cell Dev. Biol. 2011, 27, 563–584. [Google Scholar] [CrossRef]
- Ferrara, N.; Davis-Smyth, T. The biology of vascular endothelial growth factor. Endocr. Rev. 1997, 18, 4–25. [Google Scholar] [CrossRef] [PubMed]
- Nicosia, R.F. What is the role of vascular endothelial growth factor-related molecules in tumor angiogenesis? Am. J. Pathol. 1998, 153, 11–16. [Google Scholar] [CrossRef]
- Shibuya, M. Role of VEGF-Flt receptor system in normal and tumor angiogenesis. Adv. Cancer Res. 1995, 67, 281–316. [Google Scholar] [PubMed]
- Veikkola, T.; Karkkainen, M.; Claesson-Welsh, L.; Alitalo, K. Regulation of angiogenesis via vascular endothelial growth factor receptors. Cancer Res. 2000, 60, 203–212. [Google Scholar] [PubMed]
- Waltenberger, J.; Claesson-Welsh, L.; Siegbahn, A.; Shibuya, M.; Heldin, C.H. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J. Biol. Chem. 1994, 269, 26988–26995. [Google Scholar] [PubMed]
- Fong, G.H.; Rossant, J.; Gertsenstein, M.; Breitman, M.L. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995, 376, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Terman, B.I.; Dougher-Vermazen, M.; Carrion, M.E.; Dimitrov, D.; Armellino, D.C.; Gospodarowicz, D.; Böhlen, P. Identification of the KDR tyrosine kinase as a receptor for vascular endothelial cell growth factor. Biochem. Biophys. Res. Commun. 1992, 187, 1579–1586. [Google Scholar] [CrossRef]
- Seetharam, L.; Gotoh, N.; Maru, Y.; Neufeld, G.; Yamaguchi, S.; Shibuya, M. A unique signal transduction from FLT tyrosine kinase, a receptor for vascular endothelial growth factor VEGF. Oncogene 1995, 10, 135–147. [Google Scholar]
- Boehm, T.; Folkman, J.; Browder, T.; O’Reilly, M.S. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature 1997, 390, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Assaraf, Y.G.; Brozovic, A.; Gonçalves, A.C.; Jurkovicova, D.; Line, A.; Machuqueiro, M.; Saponara, S.; Sarmento-Ribeiro, A.B.; Xavier, C.P.R.; Vasconcelos, M.H. The multi-factorial nature of clinical multidrug resistance in cancer. Drug Resist. Updat. 2019, 46, 100645. [Google Scholar] [CrossRef]
- Jayson, G.C.; Kerbel, R.; Ellis, L.M.; Harris, A.L. Antiangiogenic therapy in oncology: Current status and future directions. Lancet 2016, 388, 518. [Google Scholar] [CrossRef]
- Garcia, J.; Hurwitz, H.I.; Sandler, A.B.; Miles, D.; Coleman, R.L.; Deurloo, R.; Chinot, O.L. Bevacizumab (Avastin®) in cancer treatment: A review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 2020, 86, 102017. [Google Scholar] [CrossRef]
- Collot, T.; Fumet, J.D.; Klopfenstein, Q.; Vincent, J.; Bengrine, L.; Ghiringhelli, F. Bevacizumab-based chemotherapy for poorly-differentiated neuroendocrine tumors. Anticancer Res. 2018, 38, 5963–5968. [Google Scholar] [CrossRef]
- Bavaresco, L. Role of viticultural factors on stilbene concentrations of grapes and wine. Drugs Exp. Clin. Res. 2003, 29, 181–187. [Google Scholar]
- Rimando, A.M.; Kalt, W.; Magee, J.B.; Dewey, J.; Ballington, J.R. Resveratrol, pterostilbene, and piceatannol in vaccinium berries. J. Agric. Food Chem. 2004, 52, 4713–4719. [Google Scholar] [CrossRef] [PubMed]
- Cantos, E.; Espin, J.C.; Fernandez, M.J.; Oliva, J.; Tomas-Barberan, F.A. Postharvest UV-C-irradiated grapes as a potential source for producing stilbene-enriched red wines. J. Agric. Food Chem. 2003, 51, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Kita, Y.; Miura, Y.; Yagasaki, K. Antiproliferative and antiinvasive effect of piceatannol, a polyphenol present in grapes and wine, against hepatoma AH109A cells. J. Biomed. Biotechnol. 2012, 672416. [Google Scholar]
- Potter, G.A.; Patterson, L.H.; Wanogho, E.; Perry, P.J.; Butler, P.C.; Ijaz, T.; Ruparelia, K.C.; Lamb, J.H.; Farmer, P.B.; Stanley, L.A.; et al. The cancer preventative agent resveratrol is converted to the anticancer agent piceatannol by the cytochrome P450 enzyme CYP1B1. Br. J. Cancer 2002, 86, 774–778. [Google Scholar] [CrossRef] [PubMed]
- Setoguchi, Y.; Oritani, Y.; Ito, R.; Inagaki, H.; Maruki-Uchida, H.; Ichiyanagi, T.; Ito, T. Absorption and metabolism of piceatannol in rats. J. Agric. Food Chem. 2014, 62, 2541–2548. [Google Scholar] [CrossRef]
- Piotrowska, H.; Kucinska, M.; Murias, M. Biological activity of piceatannol: Leaving the shadow of resveratrol. Mutat. Res. 2012, 750, 60–82. [Google Scholar] [CrossRef]
- Tang, Y.L.; Chan, S.W. A review of the pharmacological effects of piceatannol on cardiovascular diseases. Phytother. Res. 2014, 28, 1581–1588. [Google Scholar] [CrossRef]
- Seyed, M.A.; Jantan, I.; Bukhari, S.N.; Vijayaraghavan, K. A comprehensive review on the chemotherapeutic potential of piceatannol for cancer treatment, with mechanistic insights. J. Agric. Food Chem. 2016, 64, 725–737. [Google Scholar] [CrossRef]
- Hu, W.H.; Duan, R.; Xia, Y.T.; Xiong, Q.P.; Wang, H.Y.; Chan, G.L.; Liu, S.Y.; Dong, T.X.; Qin, Q.W.; Tsim, W.K. The binding of resveratrol to vascular endothelial growth factor (VEGF) suppresses angiogenesis by inhibiting the receptor signalling. J. Agric. Food Chem. 2019, 67, 1127–1137. [Google Scholar] [CrossRef]
- Michael, W.S. Pharmacokinetics, pharmacodynamics and pre-clinical characteristics of ophthalmic drugs that bind VEGF. Expert Rev. Clin. Pharm. 2014, 7, 167–180. [Google Scholar]
- Cai, W.; Chen, K.; Mohamedali, K.A.; Cao, Q.; Gambhir, S.S.; Rosenblum, M.G.; Chen, X. PET of vascular endothelial growth factor receptor expression. J. Nucl. Med. 2006, 47, 2048–2056. [Google Scholar] [PubMed]
- Hempel, C.; Hoyer, N.; Staalso, T.; Kurtzhals, J.A. Effects of the vascular endothelial growth factor receptor-2 (VEGFR-2) inhibitor SU5416 on in vitro cultures of Plasmodium falciparum. Malar. J. 2014, 1, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Ushio, F.M. VEGF signaling through NADPH oxidase derived ROS. Antioxid. Redox Signal. 2007, 9, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Kershaw, J.; Kim, K.H. The therapeutic potential of piceatannol, a natural stilbene, in metabolic diseases: A review. J. Med. Food 2017, 20, 427–438. [Google Scholar] [CrossRef]
- Walle, T. Bioavailability of resveratrol. Ann. N. Y. Acad. Sci. 2011, 1215, 9–15. [Google Scholar] [CrossRef]
- Kwon, G.T.; Jung, J.I.; Song, H.R.; Woo, E.Y.; Jun, J.G.; Kim, J.K.; Her, S.; Park, J.H. Piceatannol inhibits migration and invasion of prostate cancer cells: Possible mediation by decreased interleukin-6 signaling. J. Nutr. Biochem. 2012, 23, 228–238. [Google Scholar] [CrossRef]
- Kim, Y.H.; Park, C.; Lee, J.O.; Kim, G.Y.; Lee, W.H.; Choi, Y.H.; Ryu, C.H. Induction of apoptosis by piceatannol in human leukemic U937 cells through down-regulation of Bcl-2 and activation of caspases. Oncol. Rep. 2008, 19, 961–967. [Google Scholar] [CrossRef]
- Lee, Y.M.; Lim, D.Y.; Cho, H.J.; Seon, M.R.; Kim, J.K.; Lee, B.Y.; Park, J.H. Piceatannol, a natural stilbene from grapes, induces G1 cell cycle arrest in androgen-insensitive DU145 human prostate cancer cells via the inhibition of CDK activity. Cancer Lett. 2009, 285, 166–173. [Google Scholar] [CrossRef]
- Maggiolini, M.; Recchia, A.G.; Bonofiglio, D.; Catalano, S.; Vivacqua, A.; Carpino, A.; Rago, V.; Rossi, R.; Ando, S. The red wine phenolics piceatannol and myricetin act as agonists for estrogen receptor α in human breast cancer cells. J. Mol. Endocrinol. 2005, 35, 269–281. [Google Scholar] [CrossRef]
- Song, H.; Jung, J.I.; Cho, H.J.; Her, H.; Kwon, S.H.; Yu, R.; Kang, Y.H.; Lee, K.W.; Yoon, J.H.P. Inhibition of tumor progression by oral piceatannol in mouse 4T1 mammary cancer is associated with decreased angiogenesis and macrophage infiltration. J. Nutr. Biochem. 2015, 26, 1368–1378. [Google Scholar] [CrossRef]
- Mathi, P.; Musunuru, N.; Adurthi, U.; Botlagunta, M. Comparative in vitro and in silico characterization of anticancer compounds piceatannol, biochanin-A and resveratrol on breast cancer cells. Pharmacogn. Mag. 2019, 15, 410–418. [Google Scholar]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Wood, A.C.; Elvin, P.; Hickman, J.A. Induction of apoptosis by anti-cancer drugs with disparate modes of action: Kinetics of cell death and changes in c-myc expression. Br. J. Cancer 1995, 71, 937–941. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ouyang, L.; Shi, Z.; Zhao, S.; Wang, F.T.; Zhou, T.T.; Liu, B.; Bao, J.K. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012, 45, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Florea, A.M.; Busselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef]
- Joyce, J.A. Therapeutic targeting of the tumor microenvironment. Cancer Cell 2005, 7, 513–520. [Google Scholar] [CrossRef]
- Ellis, L.M.; Hicklin, D.J. VEGF-targeted therapy: Mechanisms of anti-tumour activity. Nat. Rev. Cancer 2008, 8, 579–591. [Google Scholar] [CrossRef]
- Fang, H.; Declerck, Y.A. Targeting the tumor microenvironment: From understanding pathways to effective clinical trials. Cancer Res. 2013, 73, 4965–4977. [Google Scholar] [CrossRef]
- Cho, H.; Shah, C.P.; Weber, M. Aflibercept for exudative AMD with persistent fluid on ranibizumab and/or bevacizumab. Br. J. Ophthalmol 2013, 97, 1032–1035. [Google Scholar] [CrossRef]
- Millauer, B.; Wizigmann-Voos, S.; Schnurch, H.; Martinez, R.; Moller, N.P.; Risau, W.; Ullrich, A. High affinity VEGF binding and developmental expression suggest Flk-1 as a major regulator of vasculogenesis and angiogenesis. Cell 1993, 72, 835–846. [Google Scholar] [CrossRef]
- Shibuya, M.; Yamaguchi, S.; Yamane, A.; Ikeda, T.; Tojo, A.; Matsushime, H.; Sato, M. Nucleotide sequence and expression of a novel human receptor-type tyrosine kinase gene (Flt) closely related to the Fms family. Oncogene 1990, 5, 519–524. [Google Scholar] [PubMed]
- Matthews, W.; Jordan, C.T.; Gavin, M.; Jenkins, N.A.; Copeland, N.G.; Lemischka, I.R. A receptor tyrosine kinase cDNA isolated from a population of enriched primitive hematopoietic cells and exhibiting close genetic linkage to c-kit. Proc. Natl. Acad. Sci. USA 1991, 88, 9026–9030. [Google Scholar] [CrossRef] [PubMed]
- Keyt, B.A.; Nguyen, H.V.; Berleau, L.T.; Duarte, C.M.; Park, J.; Chen, H.; Ferrara, N. Identification of vascular endothelial growth factor determinants for binding KDR and FLT-1 receptors: Generation of receptor-selective VEGF variants by site-directed mutagenesis. J. Biol. Chem. 1996, 271, 5638–5646. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.H.; Wang, H.Y.; Kong, X.P.; Xiong, Q.P.; Poon, K.M.; Xu, L.; Duan, R.; Chan, K.L.; Dong, T.X.; Tsim, W.K. Polydatin suppresses VEGF-induced angiogenesis through binding with VEGF and inhibiting its receptor signalling. FASEB J. 2019, 33, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. Fast docking using the CHARMM force field with EADock DSS. J. Comput. Chem. 2011, 32, 2149–2159. [Google Scholar] [CrossRef]
Sample Availability: Samples are not available from the authors. |








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, W.-H.; Dai, D.K.; Zheng, B.Z.-Y.; Duan, R.; Dong, T.T.-X.; Qin, Q.-W.; Tsim, K.W.-K. Piceatannol, a Natural Analog of Resveratrol, Exerts Anti-angiogenic Efficiencies by Blockage of Vascular Endothelial Growth Factor Binding to Its Receptor. Molecules 2020, 25, 3769. https://doi.org/10.3390/molecules25173769
Hu W-H, Dai DK, Zheng BZ-Y, Duan R, Dong TT-X, Qin Q-W, Tsim KW-K. Piceatannol, a Natural Analog of Resveratrol, Exerts Anti-angiogenic Efficiencies by Blockage of Vascular Endothelial Growth Factor Binding to Its Receptor. Molecules. 2020; 25(17):3769. https://doi.org/10.3390/molecules25173769
Chicago/Turabian StyleHu, Wei-Hui, Diana Kun Dai, Brody Zhong-Yu Zheng, Ran Duan, Tina Ting-Xia Dong, Qi-Wei Qin, and Karl Wah-Keung Tsim. 2020. "Piceatannol, a Natural Analog of Resveratrol, Exerts Anti-angiogenic Efficiencies by Blockage of Vascular Endothelial Growth Factor Binding to Its Receptor" Molecules 25, no. 17: 3769. https://doi.org/10.3390/molecules25173769
APA StyleHu, W.-H., Dai, D. K., Zheng, B. Z.-Y., Duan, R., Dong, T. T.-X., Qin, Q.-W., & Tsim, K. W.-K. (2020). Piceatannol, a Natural Analog of Resveratrol, Exerts Anti-angiogenic Efficiencies by Blockage of Vascular Endothelial Growth Factor Binding to Its Receptor. Molecules, 25(17), 3769. https://doi.org/10.3390/molecules25173769