
A DFT Study on FeI/FeII/FeIII Mechanism of the Cross-Coupling between Haloalkane and Aryl Grignard Reagent Catalyzed by Iron-SciOPP Complexes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
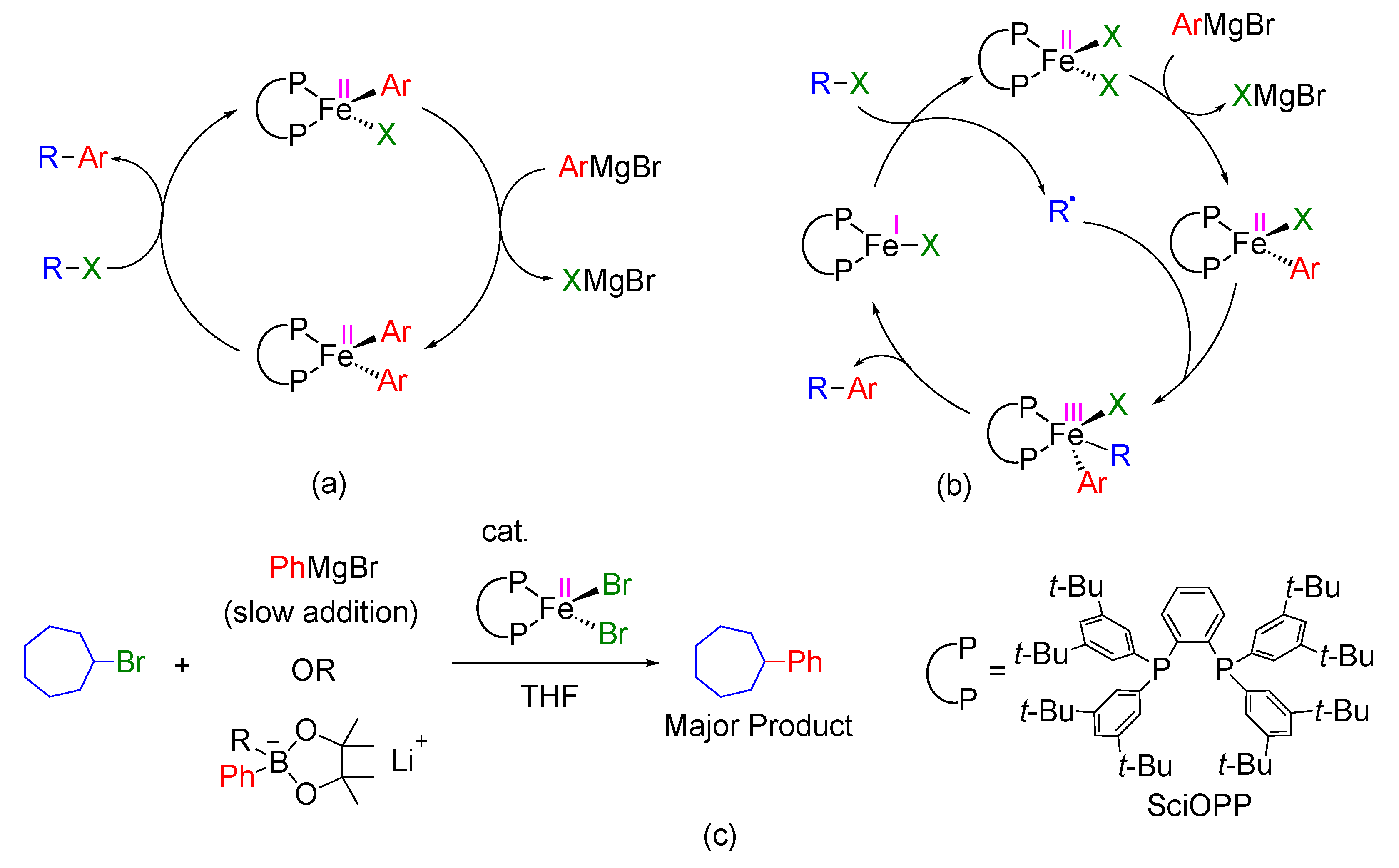
2. Results and Discussion
2.1. Reaction of FeIIBrPh(SciOPP) with Bromocycloheptane
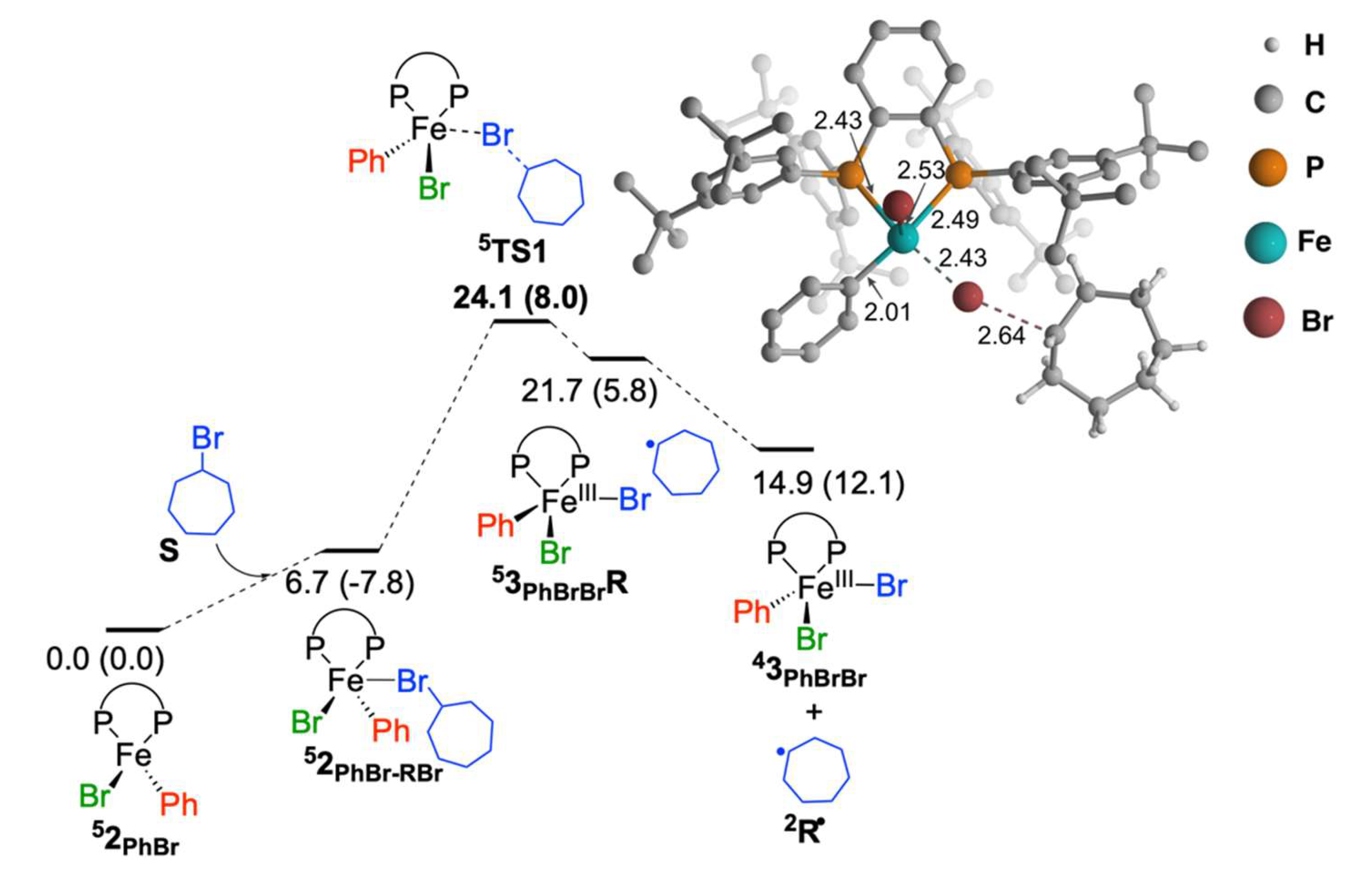
2.1.1. Cycloheptyl Radical Generation via C–Br Activation by Iron(II)-Species
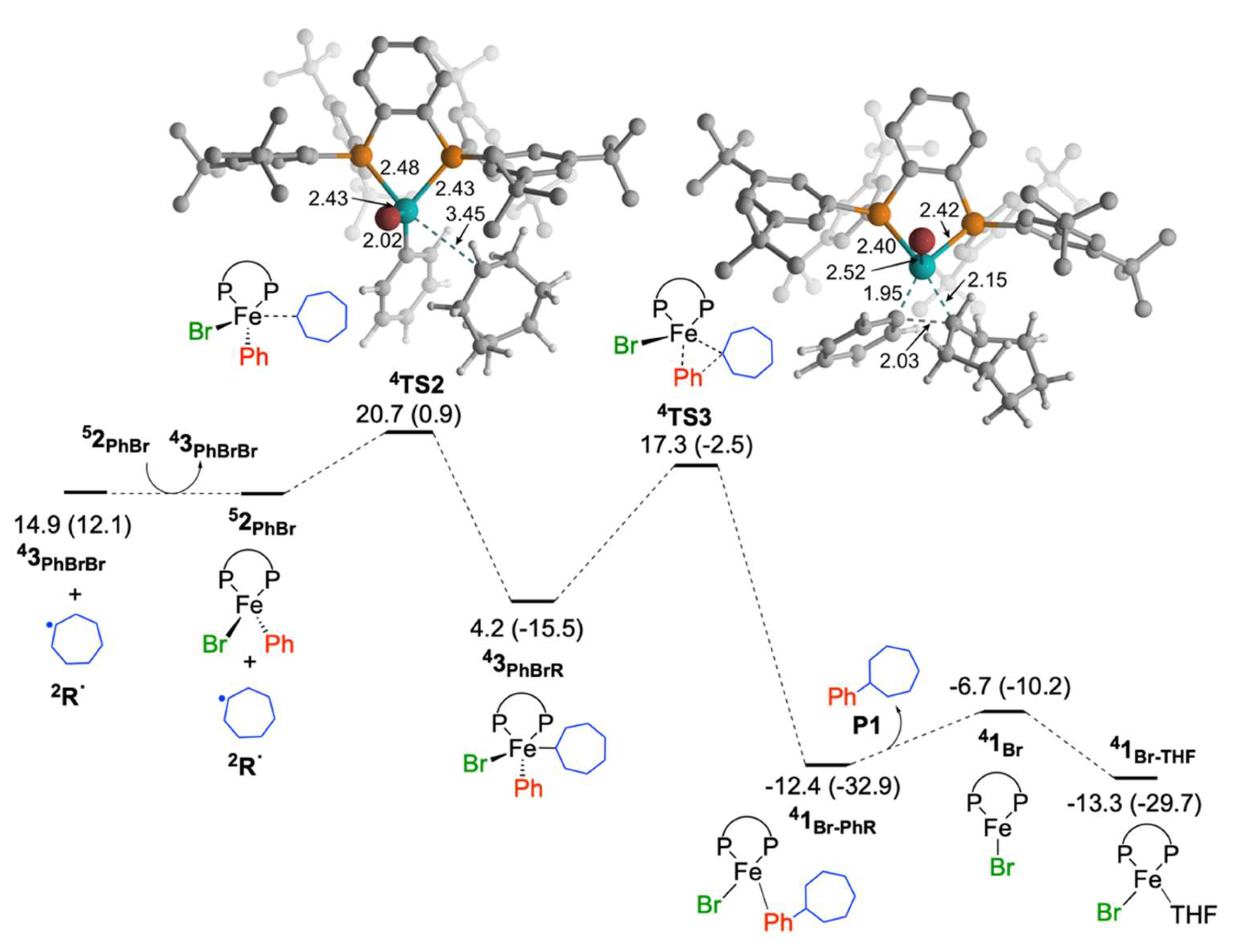
2.1.2. Reaction of Cycloheptyl Radical with Iron(II)-Species: C–C Bond Formation and Generation of Iron(I) Species
2.1.3. Reaction of Cycloheptyl Radical with FeIIIBr2Ph(SciOPP) (In-Cage Mechanism)
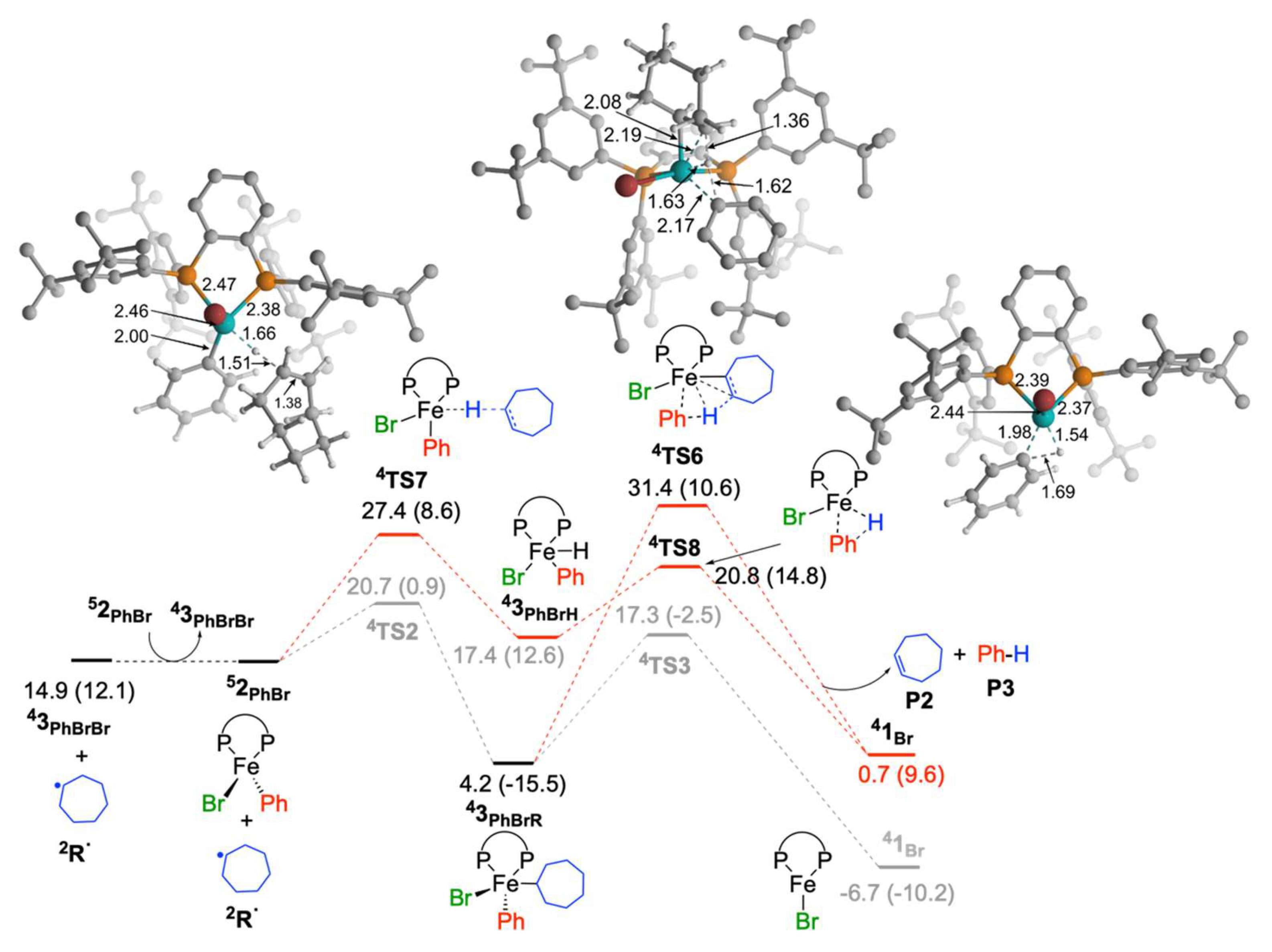
2.1.4. Reaction of Cycloheptyl Radical with Iron(II)-Species: Alkene Byproduct Formation
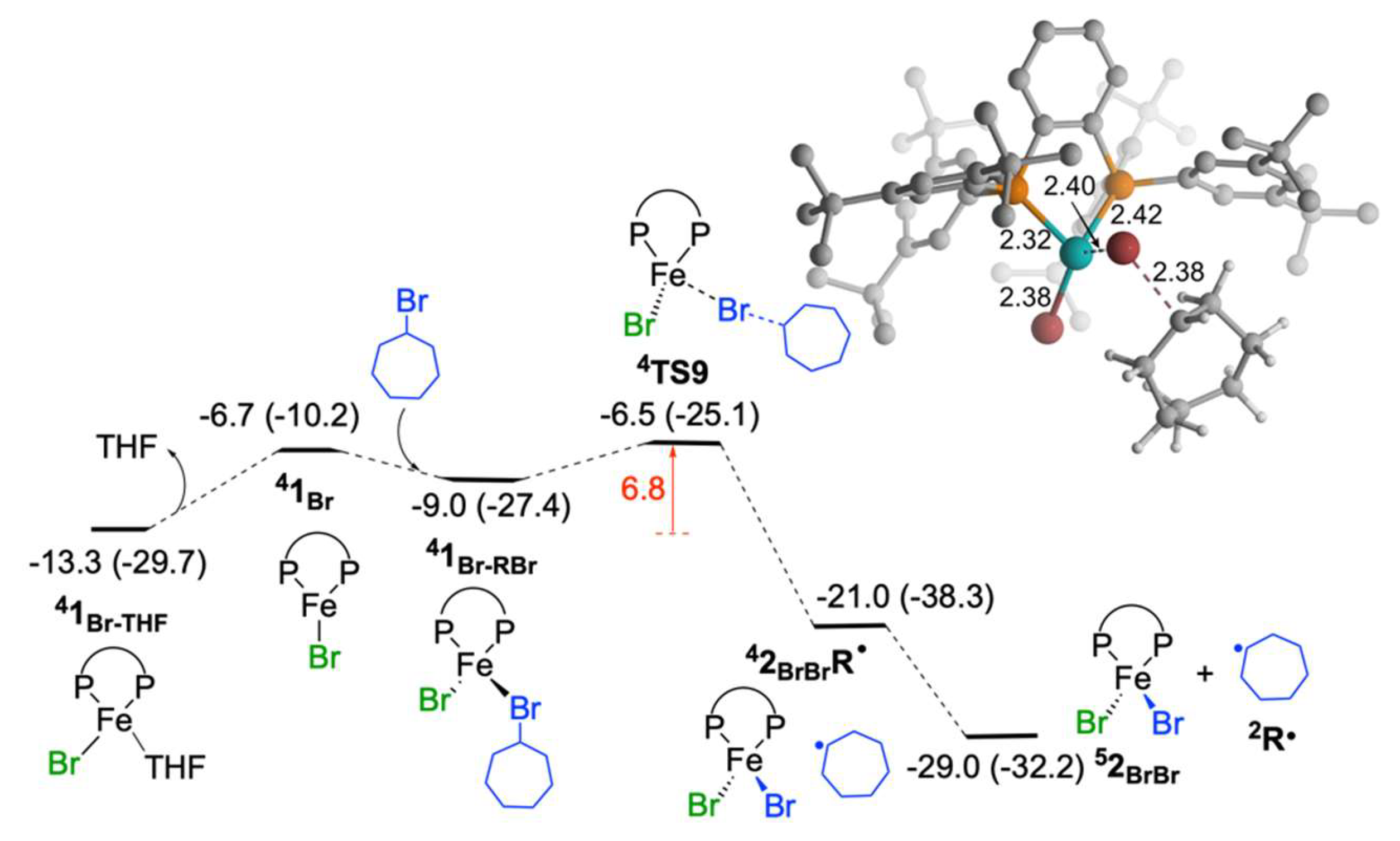
2.1.5. Reaction of Iron(I) Species with Bromocycloheptane: Regeneration of FeIIBr2(SciOPP) and Cycloheptyl Radical
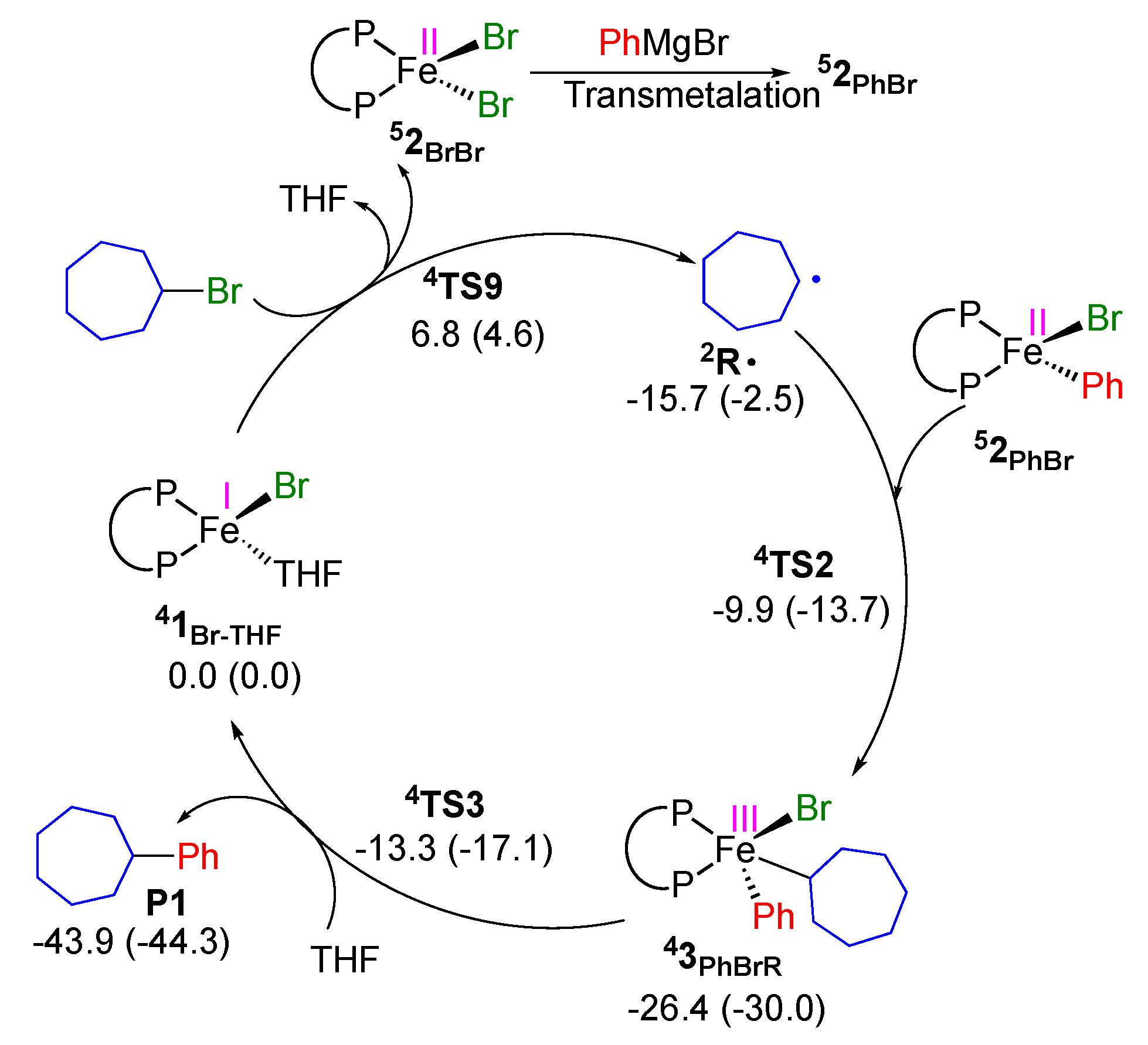
2.2. Proposed Catalytic Cycle Based on FeI/FeII/FeIII Mechanism
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Guðmundsson, A.; Bäckvall, J.-E. On the use of iron in organic chemistry. Molecules 2020, 25, 1349. [Google Scholar] [CrossRef] [PubMed]
- Fürstner, A. Iron catalysis in organic synthesis: A critical assessment of what it takes to make this base metal a multitasking champion. ACS Cent. Sci. 2016, 2, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Bauer, I.; Knölker, H.-J. Iron catalysis in organic synthesis. Chem. Rev. 2015, 115, 3170–3387. [Google Scholar] [CrossRef]
- Bolm, C.; Legros, J.; Le Paih, J.; Zani, L. Iron-catalyzed reactions in organic synthesis. Chem. Rev. 2004, 104, 6217–6254. [Google Scholar] [CrossRef] [PubMed]
- Piontek, A.; Bisz, E.; Szostak, M. Iron-catalyzed cross-couplings in the synthesis of pharmaceuticals: In pursuit of sustainability. Angew. Chem. Int. Ed. 2018, 57, 11116–11128. [Google Scholar] [CrossRef]
- Legros, J.; Figadere, B. Iron-promoted C–C bond formation in the total synthesis of natural products and drugs. Nat. Prod. Rep. 2015, 32, 1541–1555. [Google Scholar] [CrossRef]
- Cahiez, G.; Knochel, P.; Kuzmina, O.; Steib, A.; Moyeux, A. Recent advances in iron-catalyzed Csp2–Csp2 cross-couplings. Synthesis 2015, 47, 1696–1705. [Google Scholar] [CrossRef]
- Ilies, L.; Nakamura, E. Iron-catalyzed cross-coupling reactions. PATAI’S Chem. Funct. Groups 2012, 83, 1–209. [Google Scholar] [CrossRef]
- Guérinot, A.; Cossy, J. Iron-catalyzed C–C cross-couplings using organometallics. Top. Curr. Chem. 2016, 374, 49. [Google Scholar] [CrossRef]
- Tamura, M.; Kochi, J.K. Vinylation of grignard reagents. Catalysis by iron. J. Am. Chem. Soc. 1971, 93, 1487–1489. [Google Scholar] [CrossRef]
- Reddy, C.K.; Knochel, P. New cobalt and iron-catalyzed reactions of organozinc compounds. Angew. Chem. Int. Ed. 1996, 35, 1700–1701. [Google Scholar] [CrossRef]
- Cahiez, G.; Avedissian, H. Highly stereo and chemoselective iron-catalyzed alkenylation of organomagnesium compounds. Synthesis 1998, 1998, 1199–1205. [Google Scholar] [CrossRef]
- Fürstner, A.; Leitner, A.; Méndez, M.; Krause, H. Iron-catalyzed cross-coupling reactions. J. Am. Chem. Soc. 2002, 124, 13856–13863. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, T.; Nakamura, M. Iron-catalyzed selective biaryl coupling: Remarkable suppression of homocoupling by the fluoride anion. J. Am. Chem. Soc. 2007, 129, 9844–9845. [Google Scholar] [CrossRef] [PubMed]
- Bedford, R.B. How low does iron go? Chasing the active species in Fe-catalyzed cross-coupling reactions. Acc. Chem. Res. 2015, 48, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Neidig, M.L.; Carpenter, S.H.; Curran, D.J.; DeMuth, J.C.; Fleischauer, V.E.; Iannuzzi, T.E.; Neate, P.G.N.; Sears, J.D.; Wolford, N.J. Development and evolution of mechanistic understanding in iron-catalyzed cross-coupling. Acc. Chem. Res. 2018, 52, 140–150. [Google Scholar] [CrossRef]
- Parchomyk, T.; Koszinowski, K. Iron-catalyzed cross-coupling: Mechanistic insight for rational applications in synthesis. Synthesis 2017, 49, 3269–3280. [Google Scholar] [CrossRef]
- Mako, T.L.; Byers, J.A. Recent advances in iron-catalysed cross coupling reactions and their mechanistic underpinning. Inorg. Chem. Front. 2016, 3, 766–790. [Google Scholar] [CrossRef]
- Noda, D.; Sunada, Y.; Hatakeyama, T.; Nakamura, M.; Nagashima, H. Effect of TMEDA on iron-catalyzed coupling reactions of ArMgX with Alkyl halides. J. Am. Chem. Soc. 2009, 131, 6078–6079. [Google Scholar] [CrossRef]
- Nakamura, M.; Matsuo, K.; Ito, S.; Nakamura, E. Iron-catalyzed cross-coupling of primary and secondary Alkyl halides with Aryl grignard reagents. J. Am. Chem. Soc. 2004, 126, 3686–3687. [Google Scholar] [CrossRef]
- Nagano, T.; Hayashi, T. Iron-catalyzed grignard cross-coupling with Alkyl halides possessing β-hydrogens. Org. Lett. 2004, 6, 1297–1299. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.; Fürstner, A. Cross-coupling of Alkyl halides with Aryl grignard reagents catalyzed by a low-valent iron complex. Angew. Chem. Int. Ed. 2004, 43, 3955–3957. [Google Scholar] [CrossRef] [PubMed]
- Bedford, R.B.; Bruce, D.W.; Frost, R.M.; Goodby, J.W.; Hird, M. Iron(III) salen-type catalysts for the cross-coupling of aryl grignards with alkyl halides bearing β-hydrogens. Chem. Commun. 2004, 2822. [Google Scholar] [CrossRef] [PubMed]
- Bedford, R.B.; Huwe, M.; Wilkinson, M.C. Iron-catalysed Negishi coupling of benzylhalides and phosphates. Chem. Commun. 2009, 600–602. [Google Scholar] [CrossRef]
- Hatakeyama, T.; Kondo, Y.; Fujiwara, Y.-I.; Takaya, H.; Ito, S.; Nakamura, E.; Nakamura, M. Iron-catalysed fluoroaromatic coupling reactions under catalytic modulation with 1,2-bis (diphenylphosphino) benzene. Chem. Commun. 2009, 1216–1218. [Google Scholar] [CrossRef]
- Hatakeyama, T.; Hashimoto, T.; Kondo, Y.; Fujiwara, Y.; Seike, H.; Takaya, H.; Tamada, Y.; Ono, T.; Nakamura, M. Iron-catalyzed Suzuki−Miyaura coupling of Alkyl halides. J. Am. Chem. Soc. 2010, 132, 10674–10676. [Google Scholar] [CrossRef]
- Hatakeyama, T.; Fujiwara, Y.-I.; Okada, Y.; Itoh, T.; Hashimoto, T.; Kawamura, S.; Ogata, K.; Takaya, H.; Nakamura, M. Kumada–Tamao–Corriu coupling of Alkyl halides catalyzed by an iron–bisphosphine complex. Chem. Lett. 2011, 40, 1030–1032. [Google Scholar] [CrossRef]
- Bauer, G.; Wodrich, M.D.; Scopelliti, R.; Hu, X. Iron pincer complexes as catalysts and intermediates in Alkyl–Aryl Kumada coupling reactions. Organometallics 2014, 34, 289–298. [Google Scholar] [CrossRef]
- Hedström, A.; Izakian, Z.; Vreto, I.; Wallentin, C.-J.; Norrby, P.-O. On the radical nature of iron-catalyzed cross-coupling reactions. Chem. A Eur. J. 2015, 21, 5946–5953. [Google Scholar] [CrossRef]
- Bedford, R.B.; Brenner, P.B.; Richards, E.; Carvell, T.W.; Cogswell, P.M.; Gallagher, T.; Harvey, J.; Murphy, D.; Neeve, E.C.; Nunn, J.; et al. Expedient iron-catalyzed coupling of Alkyl, Benzyl and Allyl halides with Arylboronic esters. Chem. A Eur. J. 2014, 20, 7935–7938. [Google Scholar] [CrossRef]
- Przyojski, J.A.; Veggeberg, K.P.; Arman, H.D.; Tonzetich, Z.J. Mechanistic studies of catalytic carbon–carbon cross-coupling by well-defined iron NHC complexes. ACS Catal. 2015, 5, 5938–5946. [Google Scholar] [CrossRef]
- Daifuku, S.L.; Al-Afyouni, M.H.; Snyder, B.E.R.; Kneebone, J.L.; Neidig, M.L. A combined Moössbauer, magnetic circular dichroism, and density functional theory approach for iron cross-coupling catalysis: Electronic structure, In Situ Formation, and reactivity of iron-Mesityl-Bisphosphines. J. Am. Chem. Soc. 2014, 136, 9132–9143. [Google Scholar] [CrossRef] [PubMed]
- Daifuku, S.L.; Kneebone, J.L.; Snyder, B.E.R.; Neidig, M.L. Iron (II) active species in iron–bisphosphine catalyzed Kumada and Suzuki–Miyaura cross-couplings of Phenyl Nucleophiles and secondary Alkyl halides. J. Am. Chem. Soc. 2015, 137, 11432–11444. [Google Scholar] [CrossRef] [PubMed]
- Takaya, H.; Nakajima, S.; Nakagawa, N.; Isozaki, K.; Iwamoto, T.; Imayoshi, R.; Gower, N.J.; Adak, L.; Hatakeyama, T.; Honma, T.; et al. Investigation of organoiron catalysis in Kumada–Tamao–Corriu-type cross-coupling reaction assisted by solution-phase X-ray absorption spectroscopy. Bull. Chem. Soc. Jpn. 2015, 88, 410–418. [Google Scholar] [CrossRef]
- Adams, C.J.; Bedford, R.B.; Carter, E.; Gower, N.J.; Haddow, M.; Harvey, J.; Huwe, M.; Cartes, M.Á.; Mansell, S.M.; Mendoza, C.; et al. Iron(I) in Negishi cross-coupling reactions. J. Am. Chem. Soc. 2012, 134, 10333–10336. [Google Scholar] [CrossRef]
- Jin, M.; Adak, L.; Nakamura, M. Iron-catalyzed enantioselective cross-coupling reactions of α-chloroesters with Aryl grignard reagents. J. Am. Chem. Soc. 2015, 137, 7128–7134. [Google Scholar] [CrossRef]
- Iwamoto, T.; Okuzono, C.; Adak, L.; Jin, M.; Nakamura, M. Iron-catalysed enantioselective Suzuki–Miyaura coupling of racemic alkyl bromides. Chem. Commun. 2019, 55, 1128–1131. [Google Scholar] [CrossRef]
- Sharma, A.K.; Sameera, W.M.C.; Jin, M.; Adak, L.; Okuzono, C.; Iwamoto, T.; Kato, M.; Nakamura, M.; Morokuma, K. DFT and AFIR study on the mechanism and the origin of enantioselectivity in iron-catalyzed cross-coupling reactions. J. Am. Chem. Soc. 2017, 139, 16117–16125. [Google Scholar] [CrossRef]
- Lee, W.; Zhou, J.; Gutierrez, O. Mechanism of Nakamura’s Bisphosphine-iron-catalyzed asymmetric C(sp2)–C(sp3) cross-coupling reaction: The role of spin in controlling Arylation pathways. J. Am. Chem. Soc. 2017, 139, 16126–16133. [Google Scholar] [CrossRef]
- Schley, N.D.; Fu, G.C. Nickel-catalyzed Negishi Arylations of Propargylic bromides: A mechanistic investigation. J. Am. Chem. Soc. 2014, 136, 16588–16593. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01/E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Ditchfield, R. Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724. [Google Scholar] [CrossRef]
- Dolg, M.; Wedig, U.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the first row transition elements. J. Chem. Phys. 1987, 86, 866–872. [Google Scholar] [CrossRef]
- Fukui, K. The path of chemical reactions—The IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Radoń, M. Benchmarking quantum chemistry methods for spin-state energetics of iron complexes against quantitative experimental data. Phys. Chem. Chem. Phys. 2019, 21, 4854–4870. [Google Scholar] [CrossRef]
- Siig, O.S.; Kepp, K.P. Iron(II) and Iron(III) spin crossover: Toward an optimal density functional. J. Phys. Chem. A 2018, 122, 4208–4217. [Google Scholar] [CrossRef]
- Kneebone, J.L.; Brennessel, W.W.; Neidig, M.L. Intermediates and reactivity in iron-catalyzed cross-couplings of Alkynyl grignards with Alkyl Halides. J. Am. Chem. Soc. 2017, 139, 6988–7003. [Google Scholar] [CrossRef] [PubMed]
- Swart, M. Accurate spin-state energies for iron complexes. J. Chem. Theory Comput. 2008, 4, 2057–2066. [Google Scholar] [CrossRef] [PubMed]
- Song, J.-W.; Hirosawa, T.; Tsuneda, T.; Hirao, K. Long-range corrected density functional calculations of chemical reactions: Redetermination of parameter. J. Chem. Phys. 2007, 126, 154105. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, A.K.; Nakamura, M. A DFT Study on FeI/FeII/FeIII Mechanism of the Cross-Coupling between Haloalkane and Aryl Grignard Reagent Catalyzed by Iron-SciOPP Complexes. Molecules 2020, 25, 3612. https://doi.org/10.3390/molecules25163612
Sharma AK, Nakamura M. A DFT Study on FeI/FeII/FeIII Mechanism of the Cross-Coupling between Haloalkane and Aryl Grignard Reagent Catalyzed by Iron-SciOPP Complexes. Molecules. 2020; 25(16):3612. https://doi.org/10.3390/molecules25163612
Chicago/Turabian StyleSharma, Akhilesh K., and Masaharu Nakamura. 2020. "A DFT Study on FeI/FeII/FeIII Mechanism of the Cross-Coupling between Haloalkane and Aryl Grignard Reagent Catalyzed by Iron-SciOPP Complexes" Molecules 25, no. 16: 3612. https://doi.org/10.3390/molecules25163612
APA StyleSharma, A. K., & Nakamura, M. (2020). A DFT Study on FeI/FeII/FeIII Mechanism of the Cross-Coupling between Haloalkane and Aryl Grignard Reagent Catalyzed by Iron-SciOPP Complexes. Molecules, 25(16), 3612. https://doi.org/10.3390/molecules25163612