Synthesis and Biological Activity of New 7-Amino-oxazolo[5,4-d]Pyrimidine Derivatives

 ,
,
Abstract
1. Introduction
2. Results and Discussion
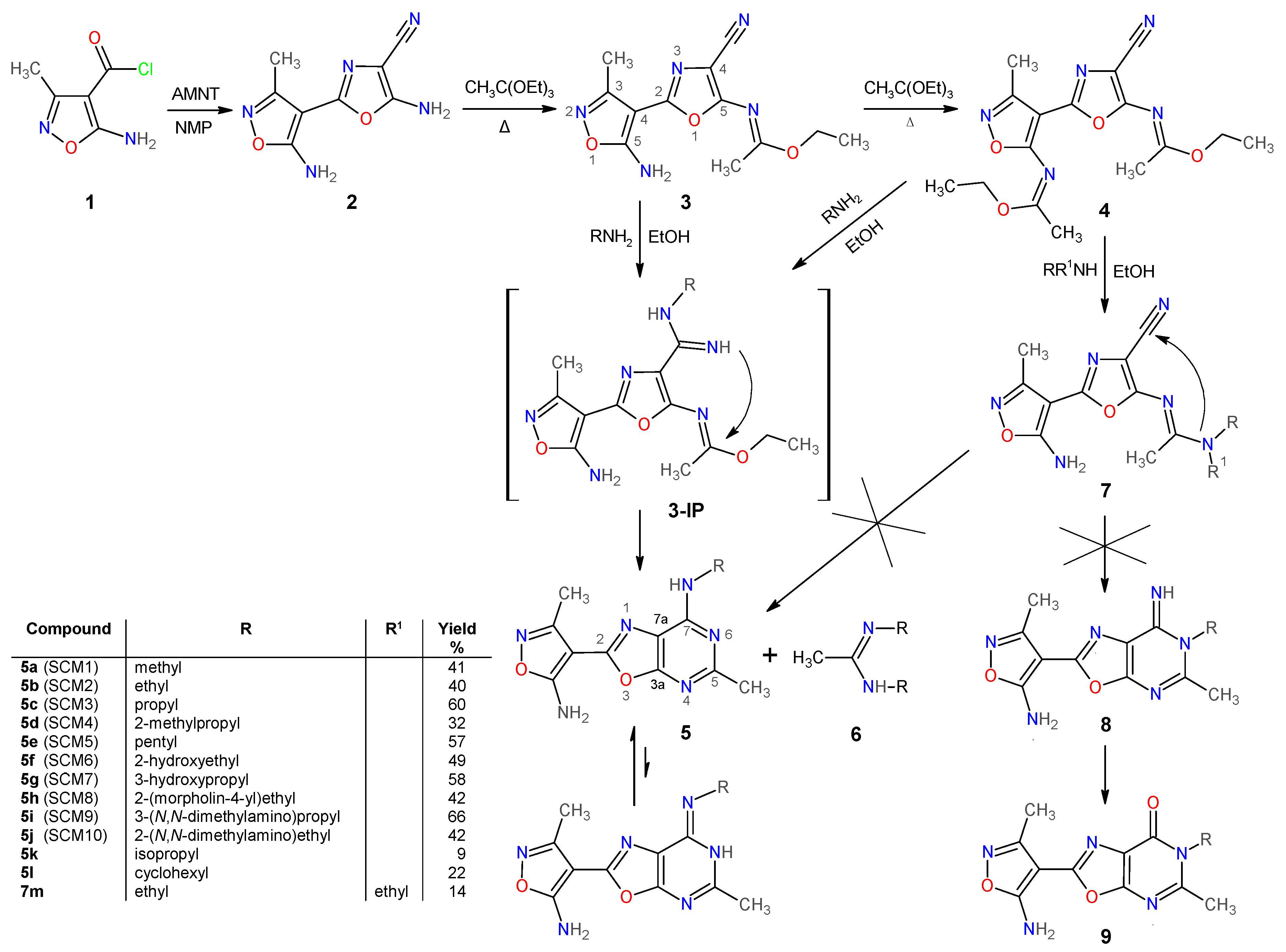
2.1. Chemistry
2.1.1. Synthesis and Some Physicochemical Properties of New 7-Aminooxazolo[5,4-d]Pyrimidines 5
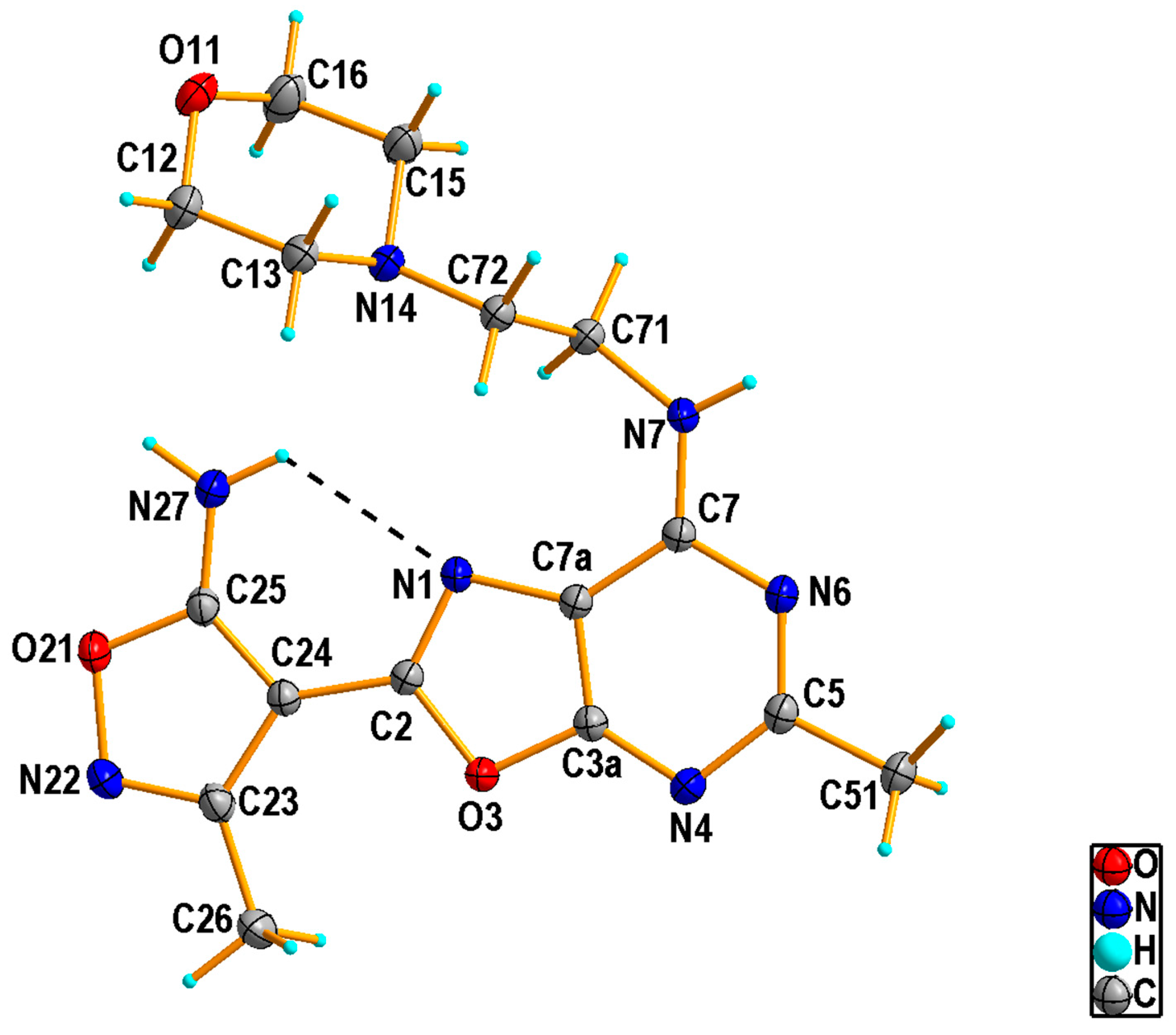
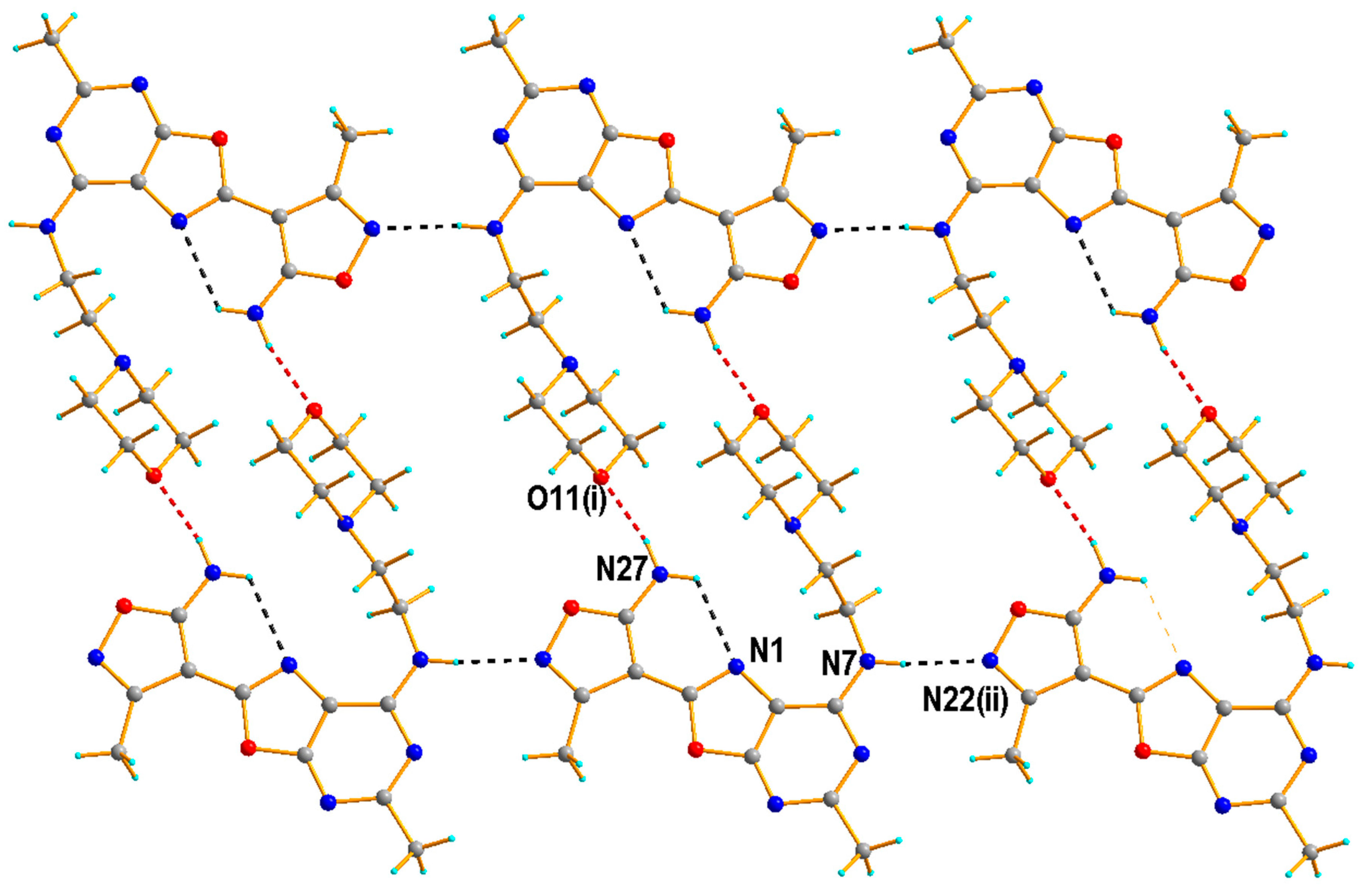
2.1.2. Crystal Structure of the Compound 5h
2.2. Biological Activity of Some New 7-Aminooxazolo[5,4-d]Pyrimidines 5a–5j (SCM1–10)
2.2.1. Effects of Tested Compounds on Viability of A-549 Reference Cell Line, Lymphocyte Proliferation and Growth of Tumor Cell Lines
Estimation of Cytotoxicity of Tested Compounds on A-549 Reference Cell Line
Influence of the Tested Compounds on Lymphocyte Proliferation
Influence of the Tested Compounds on Growth of Tumor Cell Lines
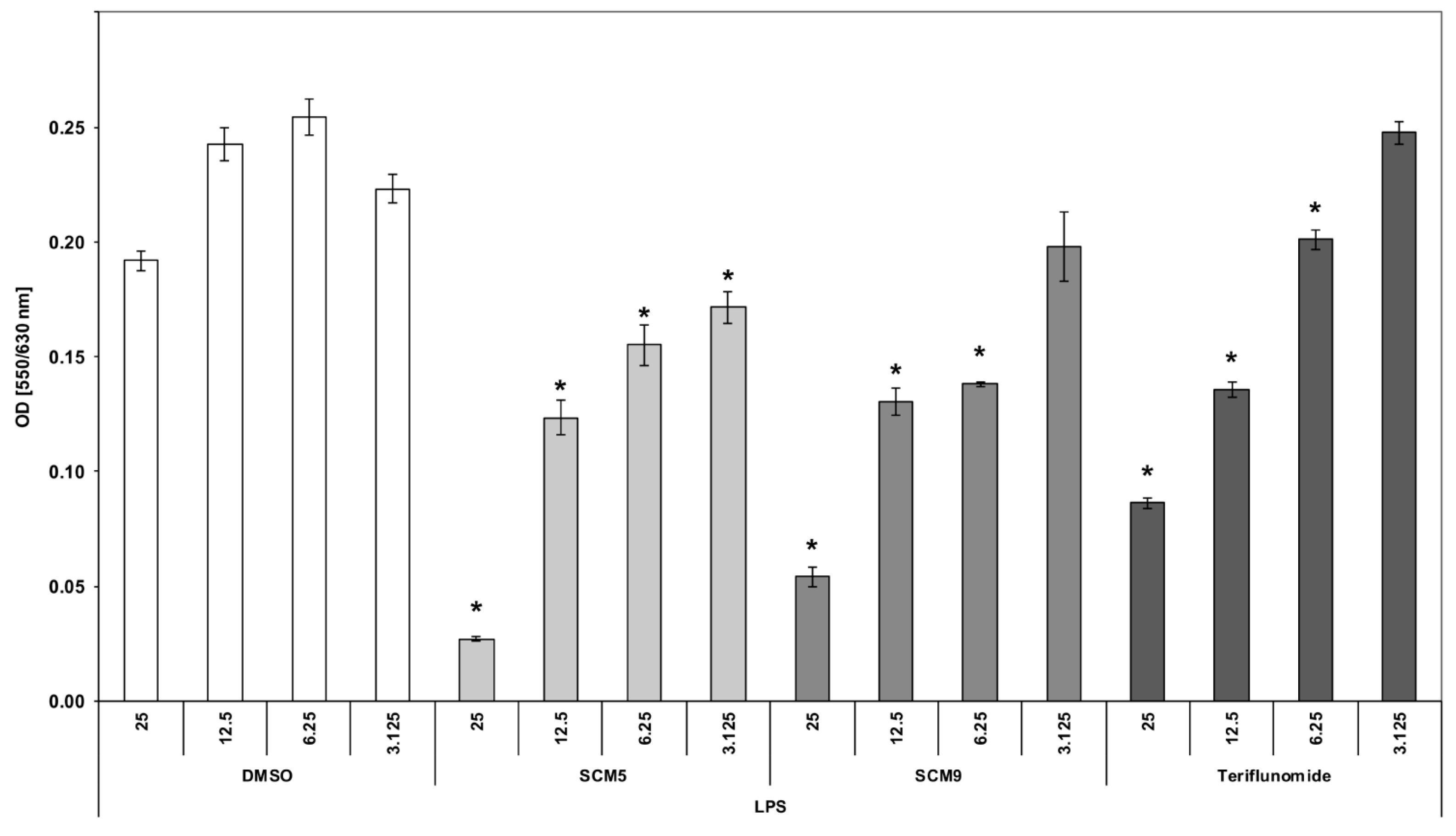
2.2.2. Effect of the Compounds on TNF α Production by LPS-Stimulated Human Whole Blood Cultures
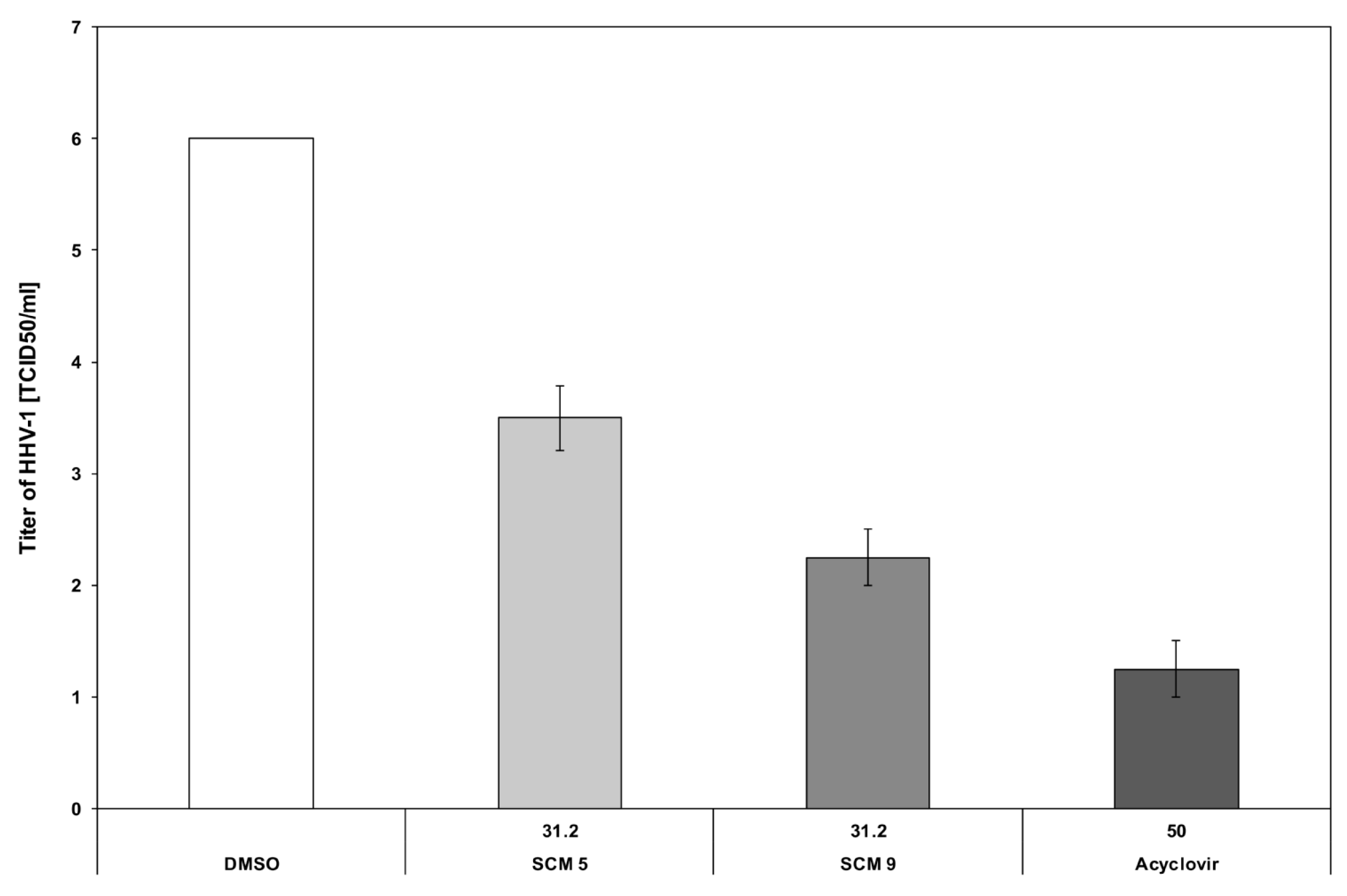
2.2.3. Antiviral Properties of the Compounds
2.2.4. Influence of the Compounds on Expression of Signaling Molecules in Cell Lines
3. Experimental
3.1. Chemistry
3.1.1. Materials and Methods
3.1.2. Preparation and Experimental Properties of Compounds 2–5 and 7
5-Amino-2-(5-Amino-3-Methyl-Isoxazol-4-yl)-Oxazole-4-Carbonitrile (2)
Ethyl N-{4-Cyano-2-[5-(1-Ethoxyethylidene)Amino-3-Methylisoxazol-4-yl]-Oxazol-5-yl}Ethanimidate (4)
Ethyl N-[2-(5-Amino-3-Methylisoxazol-4-yl)-4-Cyanooxazol-5-yl]Ethanimidate (3)
2-(5-Amino-3-Methylisoxazol-4-yl)-7-N-Methylamino-5-Methyloxazolo[5,4-d]Pyrimidine (5a, SCM1)
2-(5-Amino-3-Methylisoxazol-4-yl)-7-N-Ethylamino-5-Methyloxazolo[5,4-d]Pyrimidine (5b, SCM2)
2-(5-Amino-3-Methylisoxazol-4-yl)-7-N-Propylamino-5-Methyloxazolo[5,4-d]Pyrimidine (5c, SCM3)
2-(5-Amino-3-Methylisoxazol-4-yl)-7-N-(2-Methylpropyl)Amino-5-Methyloxazolo[5,4-d]Pyrimidine (5d, SCM4)
2-(5-Amino-3-Methylisoxazol-4-yl)-7-N-Pentylamino-5-Methyloxazolo[5,4-d]Pyrimidine (5e, SCM5)
2-(5-Amino-3-Methylisoxazol-4-yl)-7-N-(2-Hydroxyethyl)Amino-5-Methyloxazolo[5,4-d]Pyrimidine (5f, SCM6)
2-(5-Amino-3-Methylisoxazol-4-yl)-7-N-(3-Hydroxypropyl)Amino-5-Methyloxazolo[5,4-d]Pyrimidine (5g, SCM7)
2-(5-Amino-3-Methylisoxazol-4-yl)-7-N-[2-(Morpholin-4-yl)Ethyl]amino-5-Methyloxazolo[5,4-d]Pyrimidine (5h, SCM8)
2-(5-Amino-3-Methylisoxazol-4-yl)-7-N-[3-(N,N-Dimethylamino)Propyl]Amino-5-Methyloxazolo[5,4-d]Pyrimidine (5i, SCM9)
2-(5-Amino-3-Methylisoxazol-4-yl)-7-N-[2-(N,N-Dimethylamino)Ethyl]amino-5-Methyloxazolo[5,4-d]Pyrimidine (5j, SCM10)
2-(5-Amino-3-Methylisoxazol-4-yl)-7-N-Ispropylamino-5-Methyloxazolo[5,4-d]Pyrimidine (5k)
2-(5-Amino-3-Methylisoxazol-4-yl)-7-N-Cyclohexylamino-5-Methyloxazolo[5,4-d]Pyrimidine (5l)
N’-[2-(5-Amino-3-Methylisoxazol-4-yl)-4-Cyanooxazol-5-yl]-N,N-Diethylacetamidine (7m)
3.1.3. Single Crystal X-ray Diffraction Measurement of the Compound 5h
3.1.4. Computational Details
- (a)
- 5a, 5c and their respective theoretical isoforms 8a, 8c;
- (b)
- 5h and its imine tautomer;
- (c)
- N′-Cyanooxazolylacetamidine 7m and its corresponding theoretical structurally isomeric 7-N,N-diethyloxazolo[5,4-d]pyrimidine;
- (d)
- Theoretical N′-cyanooxazolylacetamidine 7 (where R = Me, R1 = H) (see Scheme 1)
3.2. Experimental Biological Section
3.2.1. General Information Concerning Preparation for Tests and Statistics
Cell Lines and Reagents
Mice
Preparation of the Oxazolo[5,4-d]Pyrimidine SCM Derivatives for In Vitro Experiments
Statistics
3.2.2. Biological Tests
Determination of the Toxicity of the SCM Compounds against the A-549 Cell Line
Isolation of the Peripheral Blood Mononuclear Cells (PBMCs)
PHA-Induced Proliferation of Human PBMC
LPS-Induced Proliferation of Mouse Splenocytes
LPS-Induced TNF-α Production in Whole Blood Cell Cultures
Growth Inhibition of Tumor Cell Lines
Colorimetric MTT Assay for Cell Growth and Kill
Cultures of Jurkat and WEHI-231 Cells and Total RNA Isolation
Reverse Transcription
Quantification of Gene Expression by Real Time PCR
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Johnson, T.B. Researches on pyrimidines: 2-ethylmercapto-5-amino-6-oxypyrimidine. Am. Chem. J. 1905, 34, 191–204. [Google Scholar]
- Block, M.H.; Harrison, A.; Hargreaves, R.B. Preparation of Furyl-Substituted Purines, Oxazolopyrimidines and Pteridines as Adenosine Antagonist. European Patent EP544445A2, 2 June 1993. [Google Scholar]
- Martin-Kohler, A.; Widmer, J.; Bold, G.; Meyer, T.; Séquin, U.; Traxler, P. Furo [2,3-d] pyrimidines and oxazolo [5,4-d] pyrimidines as inhibitors of receptor tyrosine kinases (RTK). Helv. Chim. Acta 2004, 87, 956–975. [Google Scholar] [CrossRef]
- Bauser, M.; Delapierre, G.; Hauswald, M.; Flessner, T.; D’Urso, D.; Hermann, A.; Beyreuther, B.; De Vry, J.; Spreyer, P.; Reissmüller, E.; et al. Discovery and opitimalization of 2-aryl oxazolo-pyrimidines as adenosine kinase inhibitors using liquid phase paralel synthesis. Bioorg. Med. Chem. Lett. 2004, 14, 1997–2000. [Google Scholar] [CrossRef] [PubMed]
- Robertus, J.; Kerwin, S.M.; Yan, X. Ricin Inhibitors and Methods for Use Thereof. WO Patent WO2001073438A1, 4 October 2001. [Google Scholar]
- Miller, D.J.; Ravikumar, K.; Shen, H.; Suh, J.-K.; Kerwin, S.M.; Robertus, J.D. Structure-based design and characterization of novel platforms for ricin and Shiga toxin inhibition. J. Med. Chem. 2002, 45, 90–98. [Google Scholar] [CrossRef] [PubMed]
- De Jonghe, S.; Gao, L.-J.; Herdewijn, P.; Herman, J.; Jang, M.; Leyssen, P.; Louat, T.; Neyts, J.; Pannecouque, C.; Vanderhoydonck, B. Preparation of Bicyclic Heterocycles as Antiviral Agents. WO Patent WO2011147753A1, 1 December 2011. [Google Scholar]
- Liu, J.; Deng, Y.H.; Yang, L.; Chen, Y.; Lawali, M.; Sun, L.P.; Liu, Y. CPU-12, a novel synthesized oxazolo [5,4-d] pyrimidine derivative, showed superior anti-angiogenic activity. J. Pharmacol. Sci. 2015, 129, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.-Y.; Lin, Y.; De Jonghe, S.; Gao, L.-J.; Vanderhoydonck, B.; Froeyen, M.; Rozenski, J.; Herman, J.; Louat, T.; Van Belle, K.; et al. Discovery of 7-N-piperazinylthiazolo [5,4-d] pyrimidine analogues as a novel class of immunosuppressive agents with in vivo biological activity. J. Med. Chem. 2011, 54, 655–668. [Google Scholar] [CrossRef]
- Cai, S.X.; Kemnitzer, W.E.; Sirisoma, S.; Zhang, H.-Z. N-Aryl-Isoxazolopyrimidin-4-Amines and Related Compounds as Activators of Caspases and Inducers of Apoptosis and the Use Thereof. WO Patent WO2008057402A2, 15 May 2008. [Google Scholar]
- Claiborne, C.F.; Critchley, S.; Langston, S.P.; Olhava, E.J.; Peluso, S.; Weatherhead, G.S.; Vyskocil, S.; Visiers, I.; Mizutani, H.; Cullis, C. Heteroaryl Compounds Useful as Inhibitors of E1 Activating Enzymes. WO Patent WO2008019124A1, 14 February 2008. [Google Scholar]
- Regiec, A.; Wojciechowski, P.; Pietraszko, A.; Mączyński, M. Infrared spectra and other properties predictions of 5-amino-3-methyl-4-isoxazolecarbohydrazide with electric field simulation using CPC model. J. Mol. Struct. 2018, 1161, 320–338. [Google Scholar] [CrossRef]
- Regiec, A.; Machoń, Z.; Międzybrodzki, R.; Szymaniec, S. New isothiazole derivatives: Synthesis, reactivity, physicochemical properties and pharmacological activity. Arch. Pharm. Chem. Life Sci. 2006, 339, 401–413. [Google Scholar] [CrossRef]
- Lipnicka, U.; Regiec, A.; Sułkowski, E.; Zimecki, M. New amides of 5-acylamino-3-methyl-4-isothiazolecarboxylic acid and their immunotropic activity. Arch. Pharm. Chem. Life Sci. 2005, 338, 322–328. [Google Scholar] [CrossRef]
- Lipnicka, U.; Regiec, A.; Machoń, Z. Synthesis and cytotoxic activity of new isothiazolo [5,4-d] pyrimidine derivatives. Pharmazie 1994, 40, 642–646. [Google Scholar]
- Regiec, A.; Mastalarz, H.; Międzybrodzki, R.; Śmietańska, K.; Jasztold-Howorko, R. N-Arylamides of 4-amino-1-methyl-5-imidazolecarboxylic acid-their synthesis and biological activity. Lett. Drug Des. Discov. 2006, 3, 192–199. [Google Scholar] [CrossRef]
- Mączyński, M.; Zimecki, M.; Taraszkiewicz, M.; Ryng, S. Synthesis, immunological activity and computational study of 5-amino-3-methyl-4-isoxazolecarboxylic acid semicarbazides and thiosemicarbazides. Acta Pol. Pharm. 2008, 65, 543–549. [Google Scholar] [PubMed]
- Mączyński, M.; Ryng, S.; Artym, J.; Kocięba, M.; Zimecki, M.; Brudnik, K.; Jodkowski, J.T. New lead structures in the isoxazole system: Relationship between quantum chemical parameters and immunological activity. Acta Pol. Pharm. 2014, 71, 71–83. [Google Scholar] [PubMed]
- Drynda, A.; Mączyński, M.; Ryng, S.; Obmińska-Mrukowicz, B. In vitro immunomodulatory effects of 5-amino-3-methyl-4-isoxazolecarboxylic acid hydrazide on the cellular immune response. Immunopharmacol. Immunotoxicol. 2014, 36, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Drynda, A.; Obmińska-Mrukowicz, B.; Mączyński, M.; Ryng, S. The effect of 5-amino-3-methyl-4-isoxazolecarboxylic acid hydrazide on lymphocyte subsets and humoral immune response in SRBC-immunized mice. Immunopharmacol. Immunotoxicol. 2015, 37, 148–157. [Google Scholar] [CrossRef]
- Mączyński, M.; Artym, J.; Kocięba, M.; Sochacka-Ćwikła, A.; Drozd-Szczygieł, E.; Ryng, S.; Zimecki, M. Synthesis and immunoregulatory properties of selected 5-amino-3-methyl-4 isoxazolecarboxylic acid benzylamides. Acta Pol. Pharm. 2016, 73, 1201–1211. [Google Scholar]
- Zimecki, M.; Artym, J.; Kocięba, M.; Obmińska-Mrukowicz, B.; Mączyński, M.; Ryng, S. Restoration of immune system function is accelerated in immunocompromised mice by the B-cell-tropic isoxazole R-11. Pharmacol. Rep. 2012, 64, 403–411. [Google Scholar] [CrossRef]
- Zimecki, M.; Mączyński, M.; Artym, J.; Ryng, S. Closely related isoxazoles may exhibit opposite immunological activities. Acta Pol. Pharm. 2008, 65, 793–794. [Google Scholar]
- Mączyński, M.; Artym, J.; Kocięba, M.; Kochanowska, I.; Ryng, S.; Zimecki, M. Anti-inflammatory properties of an isoxazole derivative—MZO-2. Pharmacol. Rep. 2016, 68, 894–902. [Google Scholar] [CrossRef]
- Zimecki, M.; Ryng, S.; Mączyński, M.; Chodaczek, G.; Kocięba, M.; Kuryszko, J.; Kaleta, K. Immunosuppressory activity of an isoxazolo [5,4-e] triazepine-compound RM-33 II. Effects on the carrageenan-induced inflammation. Pharmacol. Rep. 2006, 58, 236–241. [Google Scholar]
- De Coen, L.M.; Roman, B.I.; Movsisyan, M.; Heugebaert, T.S.A.; Stevens, C.V. Synthesis and biological activity of oxazolopyrimidines. Eur. J. Org. Chem. 2018, 19, 2148–2166. [Google Scholar] [CrossRef]
- Shaw, G.; Sugowdz, G. The hydrogenation of 5-aminoizooxazoles. A new synthesis of pyrimidines. J. Chem. Soc. 1954, 665–668. [Google Scholar] [CrossRef]
- Regiec, A.; Gadziński, P. New Methods for Preparing of Esters of 2-Cyano-3-Alkoxy-2-Butenoate Acids. Polish Patent PL216770, 30 May 2014. (Chemical Abstracts CAN 165:274203). [Google Scholar]
- Freeman, F.; Kim, D.S.H.L. Preparation of 2-alkyl- and 2-aryl-5-amino-4-cyano-1,3-oxazoles. Tetrahedron Lett. 1989, 30, 2631–2632. [Google Scholar] [CrossRef]
- Günther, H. NMR Spectroscopy. An Introduction; John Wiley and Sons: Chichester, UK, 1980; pp. 18 and 266. [Google Scholar]
- Ohtsuka, Y. Study on oxazolopyrimidines. VI. Formation of 3-substitutes xanthines via 7(6H)-iminooxazolopirymidines. Bull. Chem. Soc. Jpn. 1973, 46, 506–509. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comp. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.I.; Wannemuehler, M.J.; Jirillo, E.; Pritchard, D.G.; Michalek, S.M.; McGhee, J.R. LPS regulation of the immune response: Separate mechanisms for murine B cell activation by lipid A (direct) and polysaccharide (macrophage-dependent) derived from Bacteroides LPS. J. Immunol. 1984, 133, 2294–2300. [Google Scholar] [PubMed]
- Harrington, E.O.; Smeglin, A.; Parks, N.; Newton, J.; Rounds, S. Adenosine induces endothelial apoptosis by activating protein tyrosine phosphatase: A possible role of p38alpha. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L733–L742. [Google Scholar] [CrossRef]
- Wu, Y.J.; Su, T.R.; Dai, G.F.; Su, J.H.; Liu, C.I. Flaccidoxide-13-acetate-induced apoptosis in human bladder cancer cells is through activation of p38/JNK, mitochondrial dysfunction, and endoplasmic reticulum stress regulated pathway. Mar. Drugs 2019, 17, 287. [Google Scholar] [CrossRef]
- Sutherland, C.L.; Heath, A.W.; Pelech, S.L.; Young, P.R.; Gold, M.R. Differential activation of the ERK, JNK, and p38 mitogen-activated protein kinases by CD40 and the B cell antigen receptor. J. Immunol. 1996, 157, 3381–3390. [Google Scholar]
- Chaudhary, P.M.; Eby, M.T.; Jasmin, A.; Kumar, A.; Liu, L.; Hood, L. Activation of the NF-kappaB pathway by caspase 8 and its homologs. Oncogene 2000, 19, 4451–4460. [Google Scholar] [CrossRef]
- Zhao, Y.; Lei, M.; Wang, Z.; Qiao, G.; Yang, T.; Zhang, J. TCR-induced, PKC-θ-mediated NF-κB activation is regulated by a caspase-8-caspase-9-caspase-3 cascade. Biochem. Biophys. Res. Commun. 2014, 450, 526–531. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Goodwin, M.; Vu, T.; Brantley-Finley, C.; Gaarde, W.A.; Chambers, T.C. Vinblastine-induced phosphorylation of Bcl-2 and Bcl-XL is mediated by JNK and occurs in parallel with inactivation of the Raf-1/MEK/ERK cascade. J. Biol. Chem. 2000, 275, 29980–29985. [Google Scholar] [CrossRef] [PubMed]
- Juo, P.; Kuo, C.J.; Yuan, J.; Blenis, J. Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade. Curr. Biol. 1998, 8, 1001–1008. [Google Scholar] [CrossRef]
- Gutierrez-Sanmartin, D.; Varela-Ledo, E.; Aguilera, A.; Romero-Yuste, S.; Romero-Jung, P.; Gomez-Tato, A.; Regueiro, B.J. Implication of p38 mitogen-activated protein kinase isoforms (alpha, beta, gamma and delta) in CD4+ T-cell infection with human immunodeficiency virus type I. J. Gen. Virol. 2008, 89, 1661–1671. [Google Scholar] [CrossRef]
- Nunn, S.; Nishikida, K. Advanced ATR Correction Algorithm; Thermo Fisher Scientific Inc.: Madison, WI, USA, 2008; pp. 1–4. [Google Scholar]
- CrysAlis, Pro; Rigaku Oxford Diffraction: Yarnton, UK, 2018.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Brandenburg, K. Diamond. Version 3.2i; Crystal Impact GbR: Bonn, Germany, 2012. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. IV. A new dynamical correlation functional and implications for exact-exchange mixing. J. Chem. Phys. 1996, 104, 1040–1046. [Google Scholar] [CrossRef]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 (Revision, A.03); Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Ochterski, J.W. Thermochemistry in Gaussian; Gaussian Inc.: Pittsburgh, PA, USA, 2000; Volume 264, pp. 1–19. [Google Scholar]
- Hansen, M.B.; Nielsen, S.E.; Berg, K. Reexamination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J. Immunol. Methods 1989, 119, 203–210. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration [µM] | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 62.5 | 31.2 | 15.6 | |||||||
| Compound | Mean | ±SE | Grade Toxicity | Mean | ±SE | Grade Toxicity | Mean | ±SE | Grade Toxicity |
| SCM1 | 6.1 | 0.8 | 0 lack | 3.0 | 1.5 | 0 lack | 2.5 | 1.4 | 0 lack |
| SCM2 | 0.5 | 0.5 | 0 lack | 0.0 | 0.0 | 0 lack | 0.0 | 0.0 | 0 lack |
| SCM3 | 15.6 | 1.9 | 1 weak | 14.3 | 1.4 | 1 weak | 6.2 | 0.3 | 0 lack |
| SCM4 | 18.0 | 1.8 | 1 weak | 1.2 | 0.8 | 0 lack | 0.5 | 0.5 | 0 lack |
| SCM5 | 31.8 | 0.5 | 2 moderate | 8.3 | 3.0 | 0 lack | 2.0 | 1.2 | 0 lack |
| SCM6 | 15.2 | 0.2 | 1 weak | 4.9 | 0.4 | 0 lack | 4.3 | 2.2 | 0 lack |
| SCM7 | 14.3 | 1.2 | 1 weak | 8.2 | 0.6 | 0 lack | 2.6 | 2.6 | 0 lack |
| SCM8 | 16.5 | 2.7 | 1 weak | 15.3 | 0.8 | 1 weak | 12.7 | 1.5 | 1 weak |
| SCM9 | 15.7 | 0.7 | 1 weak | 7.5 | 1.1 | 0 lack | 0.1 | 0.1 | 0 lack |
| SCM 10 | 1.9 | 1.0 | 0 lack | 1.2 | 0.5 | 0 lack | 0.2 | 0.2 | 0 lack |
| Compound | Concentration [µM] | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 50 | 25 | 12.5 | ||||||||||
| Optical Density | Inhibition | Optical Density | Inhibition | Optical Density | Inhibition | |||||||
| Mean | ±SE | [%] | Mean | ±SE | [%] | Mean | ±SE | [%] | ||||
| DMSO | 0.364 | 0.01 | 0 | 0.388 | 0.00 | 0 | 0.385 | 0.01 | 0 | |||
| SCM1 | 0.312 | * | 0.01 | 14.3 | 0.353 | 0.01 | 9.0 | 0.401 | 0.01 | −4.2 | ||
| SCM2 | 0.259 | * | 0.01 | 28.8 | 0.370 | 0.01 | 4.6 | 0.380 | 0.01 | 1.3 | ||
| SCM3 | 0.238 | * | 0.01 | 34.6 | 0.365 | 0.01 | 5.9 | 0.382 | 0.00 | 0.8 | ||
| SCM4 | 0.193 | * | 0.00 | 47.0 | 0.341 | * | 0.01 | 12.1 | 0.383 | 0.01 | 0.5 | |
| SCM5 | 0.110 | * | 0.00 | 69.8 | 0.279 | * | 0.00 | 28.1 | 0.377 | 0.01 | 2.1 | |
| SCM6 | 0.351 | 0.00 | 3.6 | 0.383 | 0.01 | 1.3 | 0.371 | 0.01 | 3.6 | |||
| SCM7 | 0.336 | 0.01 | 7.7 | 0.365 | 0.01 | 5.9 | 0.352 | 0.01 | 8.6 | |||
| SCM8 | 0.297 | * | 0.01 | 18.4 | 0.350 | 0.00 | 9.8 | 0.361 | 0.01 | 6.2 | ||
| SCM9 | 0.143 | * | 0.00 | 60.7 | 0.278 | * | 0.00 | 28.4 | 0.322 | * | 0.01 | 16.4 |
| SCM10 | 0.228 | * | 0.01 | 37.4 | 0.360 | 0.01 | 7.2 | 0.371 | 0.01 | 3.6 | ||
| Teriflunomide | 0.226 | * | 0.01 | 37.9 | 0.228 | * | 0.00 | 41.2 | 0.291 | * | 0.00 | 24.4 |
| Concentration [μM] | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cell line | Compound | 50 | 25 | 12.5 | |||||||||
| Optical Density | Inhibition | Optical Density | Inhibition | Optical Density | Inhibition | ||||||||
| Mean | ±SE | [%] | Mean | ±SE | [%] | Mean | ±SE | [%] | |||||
| A-431 | Control | 1.020 | 0.020 | ||||||||||
| DMSO control | 0.459 | 0.016 | 0 | 0.809 | 0.024 | 0 | 0.875 | 0.020 | 0 | ||||
| SCM5 | 0.396 | 0.013 | 13.7 | 0.557 | * | 0.015 | 31.1 | 0.688 | * | 0.009 | 21.4 | ||
| SCM9 | 0.478 | 0.022 | −4.1 | 0.641 | * | 0.027 | 20.8 | 0.726 | * | 0.006 | 17.0 | ||
| Cisplatin | 0.022 | * | 0.000 | 97.8 | 0.025 | * | 0.001 | 97.6 | 0.066 | * | 0.002 | 93.5 | |
| HT-29 | Control | 1.147 | 0.031 | ||||||||||
| DMSO control | 0.685 | 0.017 | 0 | 0.871 | 0.028 | 0 | 1.131 | 0.039 | 0 | ||||
| SCM5 | 0.483 | * | 0.013 | 29.5 | 0.630 | * | 0.039 | 27.7 | 0.853 | * | 0.034 | 24.6 | |
| SCM9 | 0.301 | * | 0.010 | 56.1 | 0.533 | * | 0.021 | 38.8 | 0.789 | * | 0.004 | 30.2 | |
| Cisplatin | 0.197 | * | 0.009 | 82.8 | 0.451 | * | 0.012 | 60.7 | 0.675 | * | 0.026 | 41.2 | |
| L-1210 | Control | 0.769 | 0.008 | ||||||||||
| DMSO control | 1.172 | 0.029 | 0 | 0.910 | 0.036 | 0 | 1.160 | 0.051 | 0 | ||||
| SCM5 | 0.308 | * | 0.007 | 73.7 | 0.834 | 0.050 | 8.4 | 1.008 | * | 0.010 | 13.1 | ||
| SCM9 | 0.411 | * | 0.015 | 64.9 | 0.894 | 0.018 | 1.8 | 1.043 | 0.028 | 10.1 | |||
| Cisplatin | 0.018 | * | 0.000 | 97.7 | 0.020 | * | 0.000 | 97.4 | 0.030 | * | 0.001 | 96.1 | |
| Compound | Concentration [µM] | % Inhibition | ||
|---|---|---|---|---|
| Mean | p | ±SE | ||
| SCM5 | 25 | - | - | |
| 10 | - | - | ||
| 1 | - | - | ||
| SCM9 | 25 | 21.3 | * | 2.7 |
| 10 | 6.9 | 3.1 | ||
| 1 | - | - | ||
| Compound | ERK-1 | ERK-2 | p38α | p38β | p38γ | p38δ | JNK |
|---|---|---|---|---|---|---|---|
| SCM5 | 0.09 | 0.13 | 0.84 | 0.48 | 0.33 | 0.54 | 0.35 |
| SCM9 | 0.80 | 0.68 | 1.45 | 1.40 | 0.93 | 0.67 | 2.04 |
| Compound | ERK-1 | ERK-2 | p38α | p38β | p38γ | p38δ | JNK |
|---|---|---|---|---|---|---|---|
| SCM 5 | 1.04 | 0.85 | 1.38 | 1.20 | 2.68 | 3.74 | 1.20 |
| SCM 9 | 1.01 | 0.52 | 0.65 | 1.01 | 1.36 | 1.98 | 0.65 |
| Compound | Casp-3 | Casp-8 | Casp-9 | NFκB | Bcl-2 | Fas |
|---|---|---|---|---|---|---|
| SCM 5 | 0.97 | 8.20 | 23.13 | 17.91 | 10.32 | 8.82 |
| SCM 9 | 1.16 | 0.80 | 0.86 | 0.62 | 0.74 | 0.61 |
| Grade | Toxicity | Cell Morphology |
|---|---|---|
| 0 | lack | Discrete intracytoplasmic granules, no evidence cell lysis, lack of inhibition of cell growth |
| 1 | weak | Not more than 20% of rounded, shrunk cells, separating from the substrate without densities of cytoplasm, individual cells disrupted |
| 2 | moderate | Not more than 50% of rounded cells, no evidence of granules, vast cell lysis and empty spaces between cells |
| 3 | average | Not more than 70% of rounded cells, cells underwent lysis |
| 4 | strong | Almost completely or completely damage cell culture |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sochacka-Ćwikła, A.; Regiec, A.; Zimecki, M.; Artym, J.; Zaczyńska, E.; Kocięba, M.; Kochanowska, I.; Bryndal, I.; Pyra, A.; Mączyński, M. Synthesis and Biological Activity of New 7-Amino-oxazolo[5,4-d]Pyrimidine Derivatives. Molecules 2020, 25, 3558. https://doi.org/10.3390/molecules25153558
Sochacka-Ćwikła A, Regiec A, Zimecki M, Artym J, Zaczyńska E, Kocięba M, Kochanowska I, Bryndal I, Pyra A, Mączyński M. Synthesis and Biological Activity of New 7-Amino-oxazolo[5,4-d]Pyrimidine Derivatives. Molecules. 2020; 25(15):3558. https://doi.org/10.3390/molecules25153558
Chicago/Turabian StyleSochacka-Ćwikła, Aleksandra, Andrzej Regiec, Michał Zimecki, Jolanta Artym, Ewa Zaczyńska, Maja Kocięba, Iwona Kochanowska, Iwona Bryndal, Anna Pyra, and Marcin Mączyński. 2020. "Synthesis and Biological Activity of New 7-Amino-oxazolo[5,4-d]Pyrimidine Derivatives" Molecules 25, no. 15: 3558. https://doi.org/10.3390/molecules25153558
APA StyleSochacka-Ćwikła, A., Regiec, A., Zimecki, M., Artym, J., Zaczyńska, E., Kocięba, M., Kochanowska, I., Bryndal, I., Pyra, A., & Mączyński, M. (2020). Synthesis and Biological Activity of New 7-Amino-oxazolo[5,4-d]Pyrimidine Derivatives. Molecules, 25(15), 3558. https://doi.org/10.3390/molecules25153558