
Synthesis of Thiol Derivatives of Biological Active Compounds for Nanotechnology Application

 , ,
, , 
Abstract
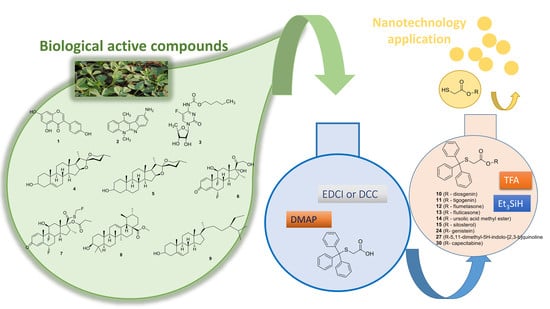
{kind=link}
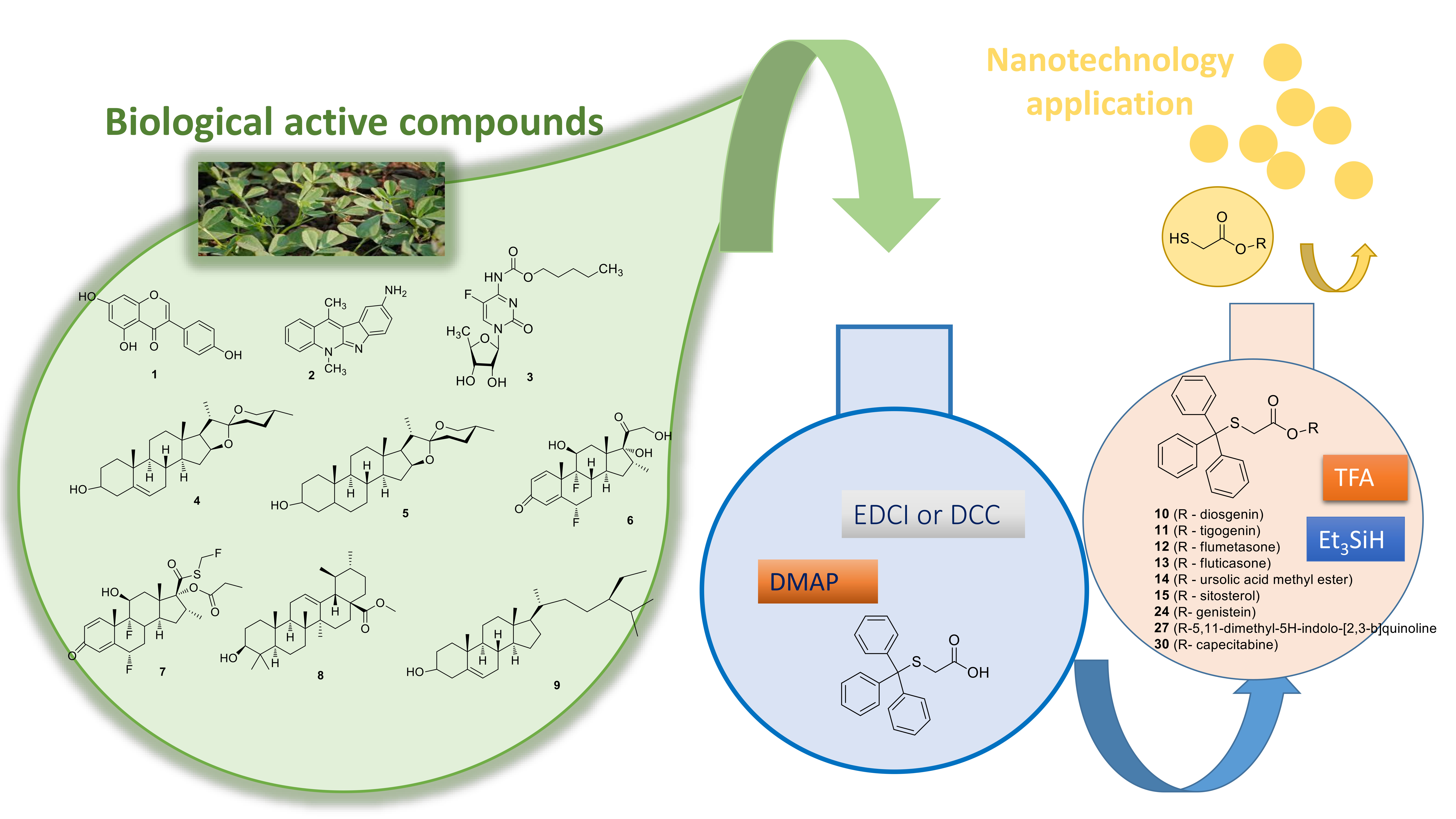
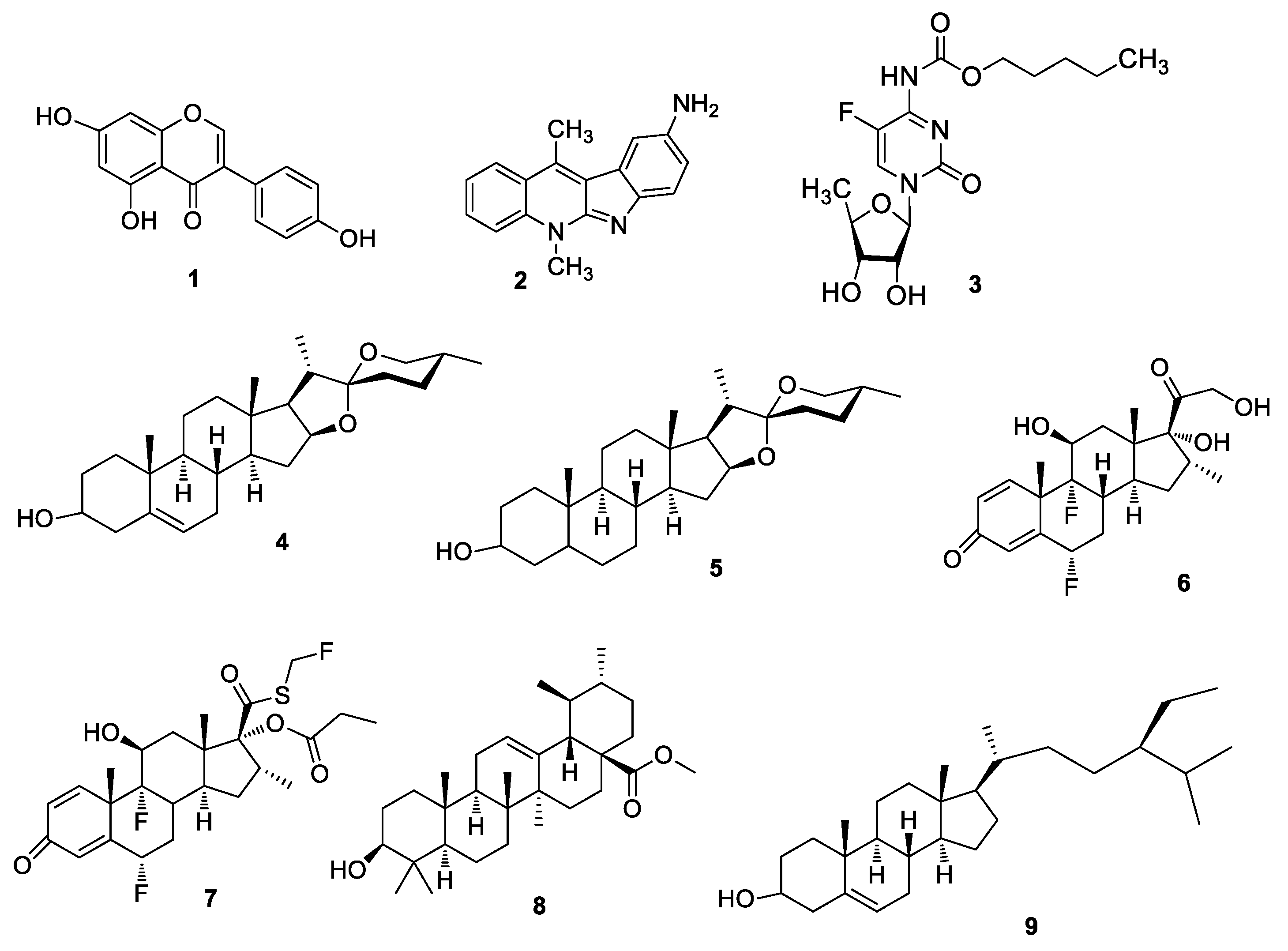{kind=link}
{kind=link}
{kind=link}
{kind=link}
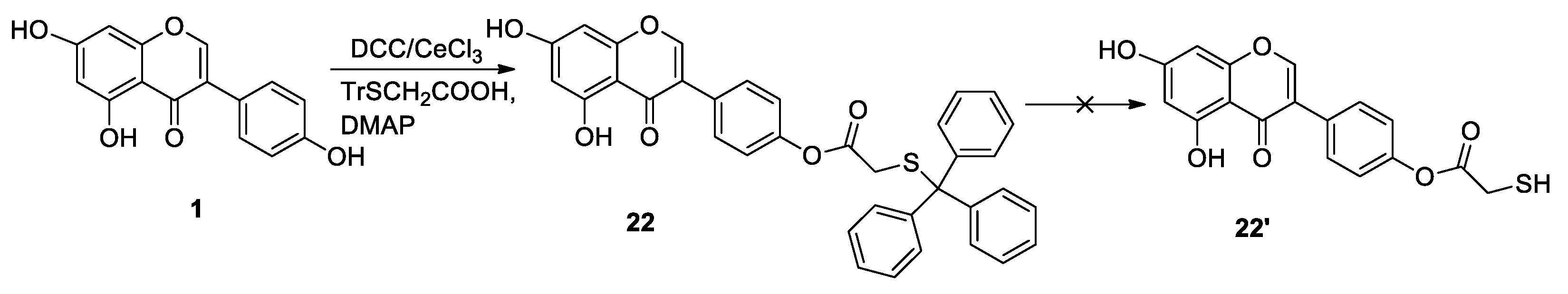{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
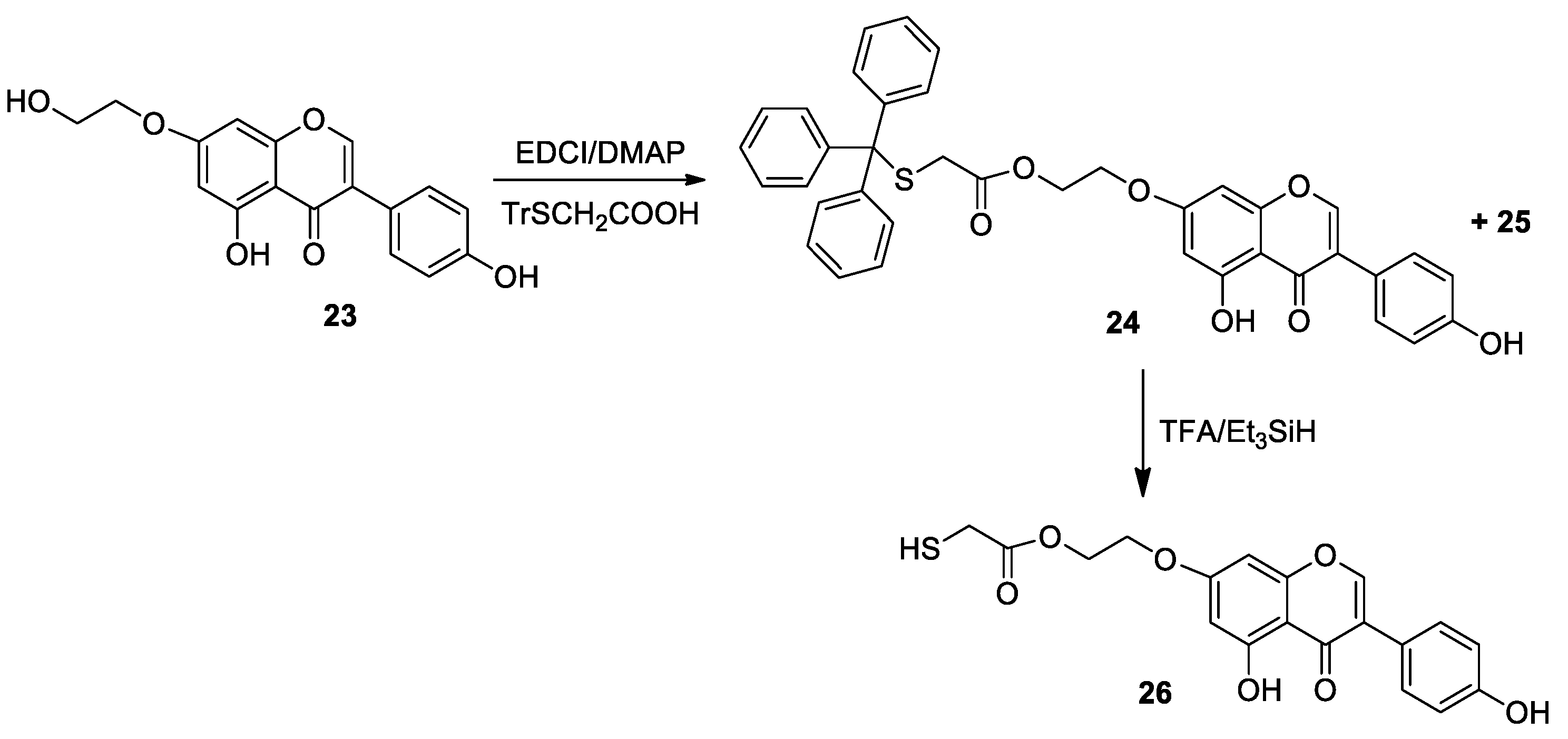
2.1. Chemistry
2.2. Theoretical Models of Synthesis of Thiol-Genistein Derivatives
3. Materials and Methods
3.1. General
3.2. General Procedure for the Synthesis of Compounds 10–15, 24, 27, 30
3.3. General Procedure for the Synthesis of Compounds 16–21, 26, 28
3.4. Theoretical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Feng, M.; Tang, B.; Liang, S.H.; Jiang, X. Sulfur containing scaffolds in drugs: Synthesis and application in medicinal chemistry. Curr. Top. Med. Chem. 2016, 16, 1200–1216. [Google Scholar] [CrossRef] [PubMed]
- Lanterna, A.E.; Coronado, E.A.; Granados, A.M. When nanoparticle size and molecular geometry matter: Analyzing the degree of surface functionalization of gold nanoparticles with sulfur heterocyclic compounds. J. Phys. Chem. C 2012, 116, 6520–6529. [Google Scholar] [CrossRef]
- Pensa, E.; Cortes, E.; Corthey, G.; Carro, P.; Vericat, C.; Fonticelli, M.H.; Benitez, G.; Rubert, A.A.; Salvarezza, R.C. The Chemistry of the sulfur gold interface: In search of a unified model. Acc. Chem. Res. 2012, 45, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Stolarczyk, E.U.; Sidoryk, K.; Cybulski, M.; Kubiszewski, M.; Stolarczyk, K. Design of therapeutic self-assembled monolayers of thiolated abiraterone. Nanomaterials 2018, 8, 1018. [Google Scholar] [CrossRef]
- Tiwari, P.M.; Vig, K.; Dennis, V.A.; Singh, S.R. Functionalized gold nanoparticles and their biomedical applications. Nanomaterials 2011, 1, 31–63. [Google Scholar] [CrossRef]
- Stolarczyk, E.U.; Leś, A.; Łaszcz, M.; Kubiszewski, M.; Strzempek, W.; Menaszek, E.; Fussaro, M.; Sidoryk, K.; Stolarczyk, K. The ligand exchange of citrates to thioabiraterone on gold nanoparticles for prostate cancer therapy. Int. J. Pharm. 2020, 583, 119319. [Google Scholar] [CrossRef]
- Petros, R.A.; DeSimone, J.M. Strategies in the design of nanoparticles for therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar] [CrossRef]
- Truong, L.; Zaikova, T.; Baldock, B.L.; Balik-Meisner, M.; Kimberly To, K.; Reif, D.M.; Kennedy, Z.C.; Hutchison, J.E.; Tangua, R.L. Systematic determination of the relationship between nanoparticle core diameter and toxicity for a series of structurally analogous gold nanoparticles in zebrafish. Nanotoxicology 2019, 13, 879–893. [Google Scholar] [CrossRef]
- Javier, D.J.; Castellanos-Gonzalez, A.; Weigum, S.E.; Clinton White, A., Jr.; Richards-Kortum, R. Oligonucleotide-gold nanoparticle networks for detection of Cryptosporidium parvum Heat Shock Protein 70 mRNA. J. Clin. Microbiol. 2009, 47, 4060–4066. [Google Scholar] [CrossRef]
- Li, X.; Li, H.; Yang, W.; Zhuang, J.; Li, H.; Wang, W. A mild and selective protecting and reversed modification of thiols. Tetrahedron Lett. 2016, 57, 2660–2663. [Google Scholar] [CrossRef]
- Sidoryk, K.; Świtalska, M.; Jaromin, A.; Cmoch, P.; Bujak, I.; Kaczmarska, M.; Wietrzyk, J.; Dominguez, E.G.; Żarnowski, R.; Andes, D.R.; et al. The synthesis of indolo[2,3-b]quinoline derivatives with a guanidine group: Highly selective cytotoxic agents. Eur. J. Med. Chem. 2015, 105, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Sidoryk, K.; Świtalska, M.; Wietrzyk, J.; Jaromin, A.; Piętka-Ottlik, M.; Cmoch, P.; Zagrodzka, J.; Szczepek, W.J.; Kaczmarek, Ł.; Peczyńska-Czoch, W. Synthesis and biological evaluation of new amino acid and dipeptide derivatives of neocryptolepine as anticancer agents. J. Med. Chem. 2012, 55, 5077–5087. [Google Scholar] [PubMed]
- Michalak, O.; Krzeczyński, P.; Cieślak, M.; Cmoch, P.; Cybulski, M.; Królewska-Golińska, K.; Kaźmierczak-Barańska, J.; Trzaskowski, B.; Ostrowska, K. Synthesis and anti–tumour, immunomodulating activity of diosgenin and tigogenin conjugates. J. Steroid Biochem. Mol. Biol. 2020, 198, 105573. [Google Scholar] [CrossRef] [PubMed]
- Sidoryk, K.; Jaromin, A.; Edward, J.A.; Świtalska, M.; Stefańska, J.; Cmoch, P.; Zagrodzka, J.; Szczepek, W.; Peczyńska-Czoch, W.; Wietrzyk, J.; et al. Searching for new derivatives of neocryptolepine: Synthesis, antiproliferative, antimicrobial and antifungal activities. Eur. J. Med. Chem. 2014, 78, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Nascimento do, P.G.G.; Lemos, T.L.G.; Bizerra, A.M.C.; Arriaga, Â.M.C.; Ferreira, D.A.; Santiago, G.M.P.; Braz-Filho, R.; Galberto, M.; Costa, J. Antibacterial and antioxidant activities of ursolic acid derivatives. Molecules 2014, 19, 1317–1327. [Google Scholar] [CrossRef]
- Saeidnia, S.; Manayi, A.; Gohari, A.R.; Abdollahi, M. The Story of Beta-sitosterol—A Review. Eur. J. Med. Plants. 2014, 4, 590–609. [Google Scholar] [CrossRef]
- Panaretto, B.A.; Wallace, A.L.C. The administration of flumethasone, by three different routes, its measurement in the plasma and some effects on wool growth in merino wethers. Aust. J. Biol. Sci. 1978, 31, 601–619. [Google Scholar] [CrossRef]
- Edin, H.M.; Andersen, L.B.; Schoaf, L.; Scott-Wilson, C.A.; Ho, S.Y.; Ortega, H.G. Effects of fluticasone propionate and salmeterol hydrofluoroalkane inhalation aerosol on asthma-related quality of life. Ann. Alergy Asthma Immunol. 2009, 102, 323–327. [Google Scholar] [CrossRef]
- Cmoch, P.; Krzeczyński, P.; Leś, A. Multinuclear NMR measurements and DFT calculations for Capecitabine tautomeric form assignment in a solution. Molecules 2018, 23, 161. [Google Scholar] [CrossRef]
- Walko, C.M.; Lindley, C. Capecitabine: A review. Clin. Ther. 2005, 27, 23–44. [Google Scholar] [CrossRef]
- Grynkiewicz, G.; Achmatowicz, O.; Pucko, W. Bioactive isoflavone–genistein; synthesis and prospective applications. Herbal Pol. 2000, 46, 151–160. [Google Scholar]
- Tuli, H.S.; Tuorkey, M.J.; Thakral, F.; Sak, K.; Kumar, M.; Kumar Sharma, A.; Sharma, U.; Jain, A.; Aggarwal, V.; Bishayee, A. Molecular Mechanisms of Action of Genistein in Cancer: Recent Advances. Front. Pharmacol. 2019, 10, 1336. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Wang, S.; Li, X.; Zou, T.; Huang, X.; Zhang, W.; Chen, Y.; Yang, C.; Pan, Q.; Liu, H.-F. Prospects of and limitations to the clinical applications of genistein. Discov. Med. 2019, 27, 177–188. [Google Scholar] [PubMed]
- Stolarczyk, E.U.; Stolarczyk, K.; Łaszcz, M.; Kubiszewski, M.; Maruszak, W.; Olejarz, W.; Bryk, D. Synthesis and characterization of genistein conjugated with gold nanoparticles and the study of their cytotoxic properties. Eur. J. Pharm. Sci. 2017, 96, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Stolarczyk, E.U.; Łaszcz, M.; Leś, A.; Kubiszewski, M.; Kuziak, K.; Sidoryk, K.; Stolarczyk, K. Design and Molecular Modeling of Abiraterone-Functionalized Gold Nanoparticles. Nanomaterials 2018, 8, 641. [Google Scholar] [CrossRef]
- Stolarczyk, E.U.; Stolarczyk, K.; Łaszcz, M.; Kubiszewski, M.; Leś, A.; Michalak, O. Pemetrexed conjugated with gold nanoparticles—Synthesis, characterization and a study of noncovalent interactions. Eur. J. Pharm. Sci. 2017, 109, 13–20. [Google Scholar] [CrossRef]
- Okawara, M.; Hashimoto, F.; Todo, H.; Sugibayashi, K.; Tokudome, Y. Effect of liquid crystals with cyclodextrin on the bioavailability of a poorly water-soluble compound, diosgenin, after its oral administration to rats. Int. J. Pharm. 2014, 472, 257–261. [Google Scholar] [CrossRef]
- Baan, J.; MEM Bos, M.; U Gonesh-Kisoensingh, S.; Meynaar, I.A.; Alsma, J.; Meijer, E.; GVulto, A. Capecitabine-induced Toxicity: An Outcome Study into Drug Safety. J. Integr. Oncol. 2014, 3. [Google Scholar] [CrossRef]
- Du, Y.; Xia, L.; Jo, A.; Davis, R.M.; Bissel, P.; Ehrich, M.; Kingston, D.G.I. Synthesis and Evaluation of Doxorubicin-Loaded Gold Nanoparticles for Tumor-Targeted Drug Delivery. Bioconjugate Chem. 2018, 29, 420–430. [Google Scholar] [CrossRef]
- Anwar, A.; Siddiqui, R.; Raza Shah, M.; Khan, N.A. Gold Nanoparticles Conjugation Enhances Antiacanthamoebic Properties of Nystatin, Fluconazole and Amphotericin B. J. Microbiol. Biotechnol. 2019, 29, 171–177. [Google Scholar] [CrossRef]
- Singh, M.; Hamid, A.A.; Maurya, A.K.; Prakash, O.; Khan, F.; Kumar, A.; Aiyelaagbe, O.O.; Negi, A.S.; Bawankule, D.U. Synthesis of diosgenin analogues as potential anti-inflammatory agents. J. Steroid Biochem. Mol. Biol. 2014, 143, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.Z.; Xin, G.; Ma, L.M.; Wei, Z.L.; Shen, Y.; Zhang, R.; Zheng, H.J.; Zhang, X.H.; Niu, H.; Huang, W. Synthesis, characterization, and biological studies of diosgenyl analogs. J. Asian Nat. Prod. Res. 2017, 19, 272–298. [Google Scholar] [CrossRef] [PubMed]
- Wakasugi, K.; Nakamura, A.; Tanabe, Y. Me2NSO2Cl and N,N-dimethylamines; a novel and efficient agent for esterification, amidation between carboxylic acids, and equimolar amounts of alcohols and amines. Tetrahedron Lett. 2001, 42, 7427–7430. [Google Scholar] [CrossRef]
- Kawabata, T.; Mizugaki, T.; Ebitani, K.; Kaneda, K. Highly efficient esterification of carboxylic acids with alcohols by montmorillonite-enwrapped titanium as a heterogeneous acid catalyst. Tetrahedron Lett. 2003, 44, 9205–9208. [Google Scholar] [CrossRef]
- Lee, S.G.; Park, J.H. Metallic Lewis acids-catalyzed acetylation of alcohols with acetic anhydride and acetic acid in ionic liquids: Study on reactivity and reusability of the catalysts. J. Mol. Cat. A Chem. 2003, 194, 49–52. [Google Scholar] [CrossRef]
- Chen, C.T.; Munot, Y.S. Direct Atom-Efficient Esterification between Carboxylic Acids and Alcohols Catalyzed by Amphoteric, Water-Tolerant TiO(acac)2. J. Org. Chem. 2005, 70, 8625–8627. [Google Scholar] [CrossRef]
- Ishihara, K.; Nakagawa, S.; Sakakura, A. Bulky Diarylammonium Arenesulfonates as Selective Esterification Catalysts. J. Am. Chem. Soc. 2005, 127, 4168–4169. [Google Scholar] [CrossRef]
- Khalafi-Nezhad, A.; Parhami, A.; Zare, A.; Moosavi Zare, A.R. Efficient method for the direct preparation of aryl carboxylates from carboxylic acids using tosyl chloride under solvent-free conditions. J. Iran. Chem. Soc. 2008, 5, 413–419. [Google Scholar] [CrossRef]
- Gilles, V.; Vieira, M.A.; Lacerda, V., Jr.; Castro, E.V.R.; Santos, R.B.; Orestes, E.; Carneiro, J.W.M.; Greco, S.J. A new, simple and efficient method of steglich esterification of Juglone with long-fatty acids: Synthesis of a new class of non-polymeric wax deposition inhibitors for crude oil. J. Braz. Chem. Soc. 2015, 26, 74–83. [Google Scholar] [CrossRef]
- Drouet, S.; Doussot, J.; Garros, L.; Mathiron, D.; Bassard, S.; Favre-Reguillon, A.; Molinie, R.; Laine, E.; Hano, C. Selective synthesis of 3-O-palmitoyl-sylibyn, a new-to-nature flavonolignan with increased protective action against oxidative damages in lipophilic media. Molecules 2018, 23, 2594. [Google Scholar] [CrossRef]
- Kicsak, M.; Bege, M.; Bereczki, I.; Csavas, M.; Herczeg, M.; Kupihar, Z.; Kovacs, L.; Borbas, A.; Herczegh, P. A tree-component reagent system for rapid and mild removal of O-, N-, S-trityl protecting groups. Org. Biomol. Chem. 2016, 14, 3190–3192. [Google Scholar] [CrossRef] [PubMed]
- Grynkiewicz, G.; Zegrocka-Stendel, O.; Pucko, W.; Ramza, J.; Kościelecka, A.; Kołodziejski, W.; Woźniak, K. X-ray and 13C CP MAS investigation of structure of two genistein derivatives. J. Mol. Struct. 2004, 694, 121–129. [Google Scholar] [CrossRef]
- Szeja, W.; Puchałka, J.; Świerk, P.; Hendrich, A.B.; Grynkiewicz, G. Selective alkylation of genistein and daidzein. Chem. Biol. Interface 2013, 3, 95–106. [Google Scholar]
- Ingle, G.K.; Mormino, M.G.; Wojtas, L.; Antilla, J.C. Chiral phosphoric acid-catalyzed addition of thiol to N-acyl imines: Access to chiral N,S-acetales. Org. Lett. 2011, 13, 4822–4825. [Google Scholar] [CrossRef]
- Nakamura, S.; Takahashi, S.; Nakane, D.; Masuda, H. Organocatalytic enantioselective addition of thiols to ketimines derived from isatins. Org. Lett. 2015, 2, 106–109. [Google Scholar] [CrossRef]
- Wang, H.Y.; Zhang, J.X.; Cao, D.D.; Zhao, G. Enantioselective addition of thiols to imines catalysed by thiourea-quaternary ammonium salts. ACS Catal. 2013, 3, 2218–2221. [Google Scholar] [CrossRef]
- Imagawa, H.; Tsuchihashi, T.; Singh, R.K.; Yamamoto, H.; Sugihara, T.; Nishizawa, M. Triethyl-(or trimethyl-)silyl triflate-catalyzed reductive cleavage of triphenylmethyl (trityl) ethers with triethylsilane. Org. Lett. 2003, 5, 153–155. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford CT, UK, 2016. [Google Scholar]
- MOPAC2016, J.J.P.; Stewart, Stewart Computational Chemistry: Colorado Springs, CO, USA, 2016.
Sample Availability: Samples of the compounds are not available from the authors. |










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sidoryk, K.; Michalak, O.; Kubiszewski, M.; Leś, A.; Cybulski, M.; Stolarczyk, E.U.; Doubsky, J. Synthesis of Thiol Derivatives of Biological Active Compounds for Nanotechnology Application. Molecules 2020, 25, 3470. https://doi.org/10.3390/molecules25153470
Sidoryk K, Michalak O, Kubiszewski M, Leś A, Cybulski M, Stolarczyk EU, Doubsky J. Synthesis of Thiol Derivatives of Biological Active Compounds for Nanotechnology Application. Molecules. 2020; 25(15):3470. https://doi.org/10.3390/molecules25153470
Chicago/Turabian StyleSidoryk, Katarzyna, Olga Michalak, Marek Kubiszewski, Andrzej Leś, Marcin Cybulski, Elżbieta U. Stolarczyk, and Jan Doubsky. 2020. "Synthesis of Thiol Derivatives of Biological Active Compounds for Nanotechnology Application" Molecules 25, no. 15: 3470. https://doi.org/10.3390/molecules25153470
APA StyleSidoryk, K., Michalak, O., Kubiszewski, M., Leś, A., Cybulski, M., Stolarczyk, E. U., & Doubsky, J. (2020). Synthesis of Thiol Derivatives of Biological Active Compounds for Nanotechnology Application. Molecules, 25(15), 3470. https://doi.org/10.3390/molecules25153470