
Asymmetric Preparation of α-Quaternary Fluorinated β-keto Esters. Review


Abstract

1. Introduction
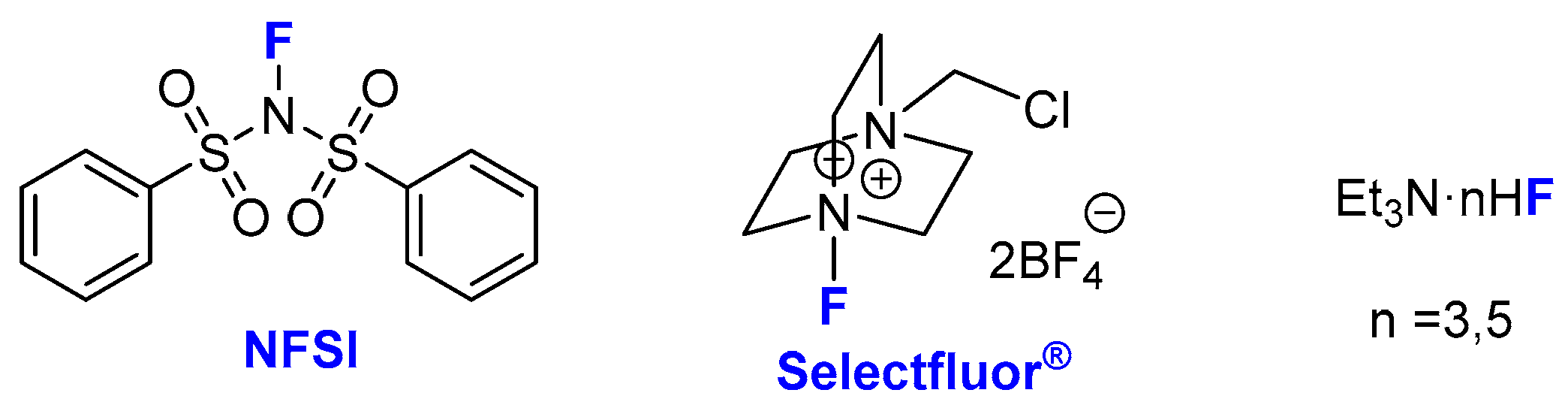
2. Asymmetric Preparation of Quaternary C-F Stereocentres on β-keto Esters
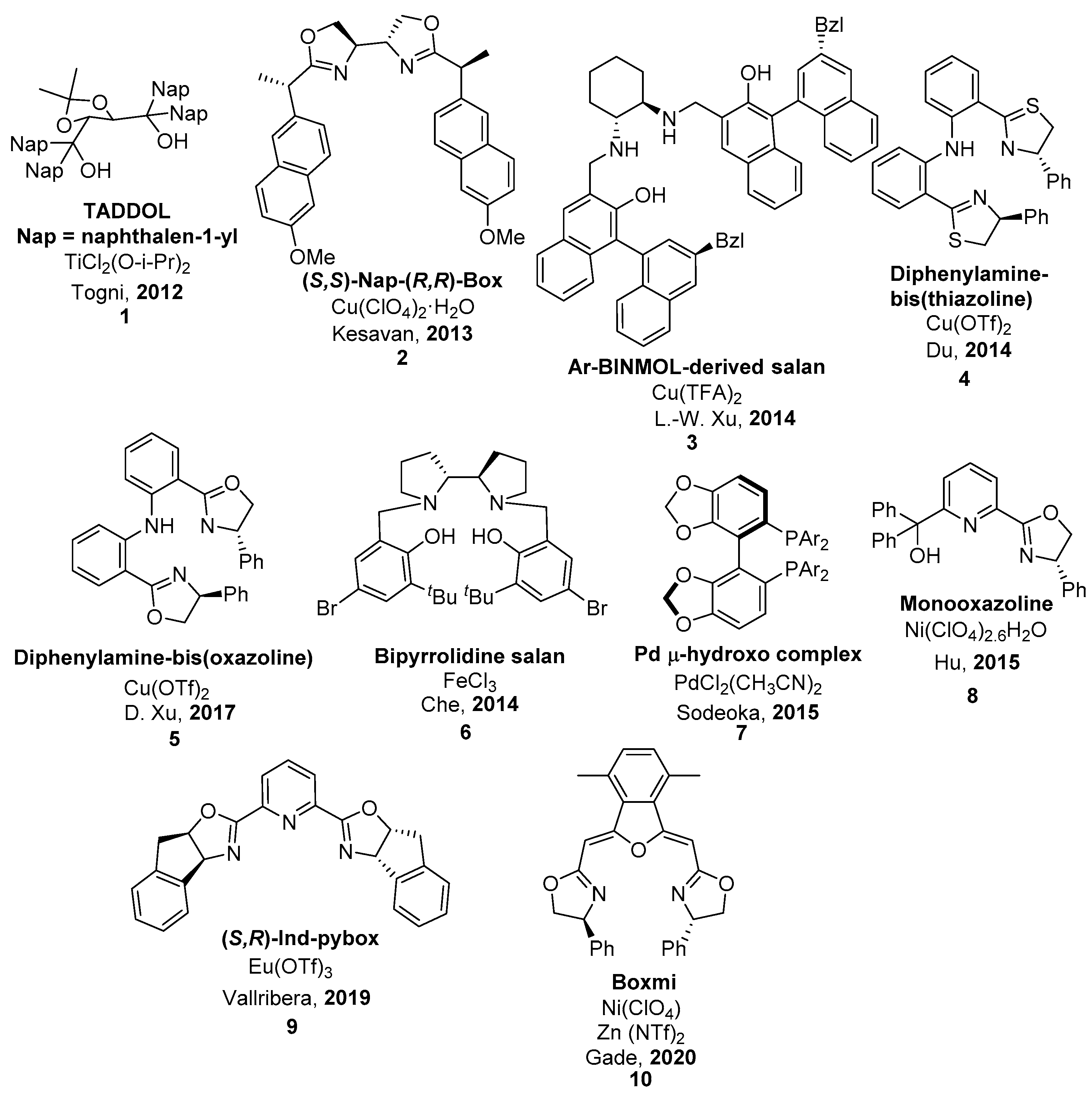
2.1. Metal-Catalyzed Methods

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
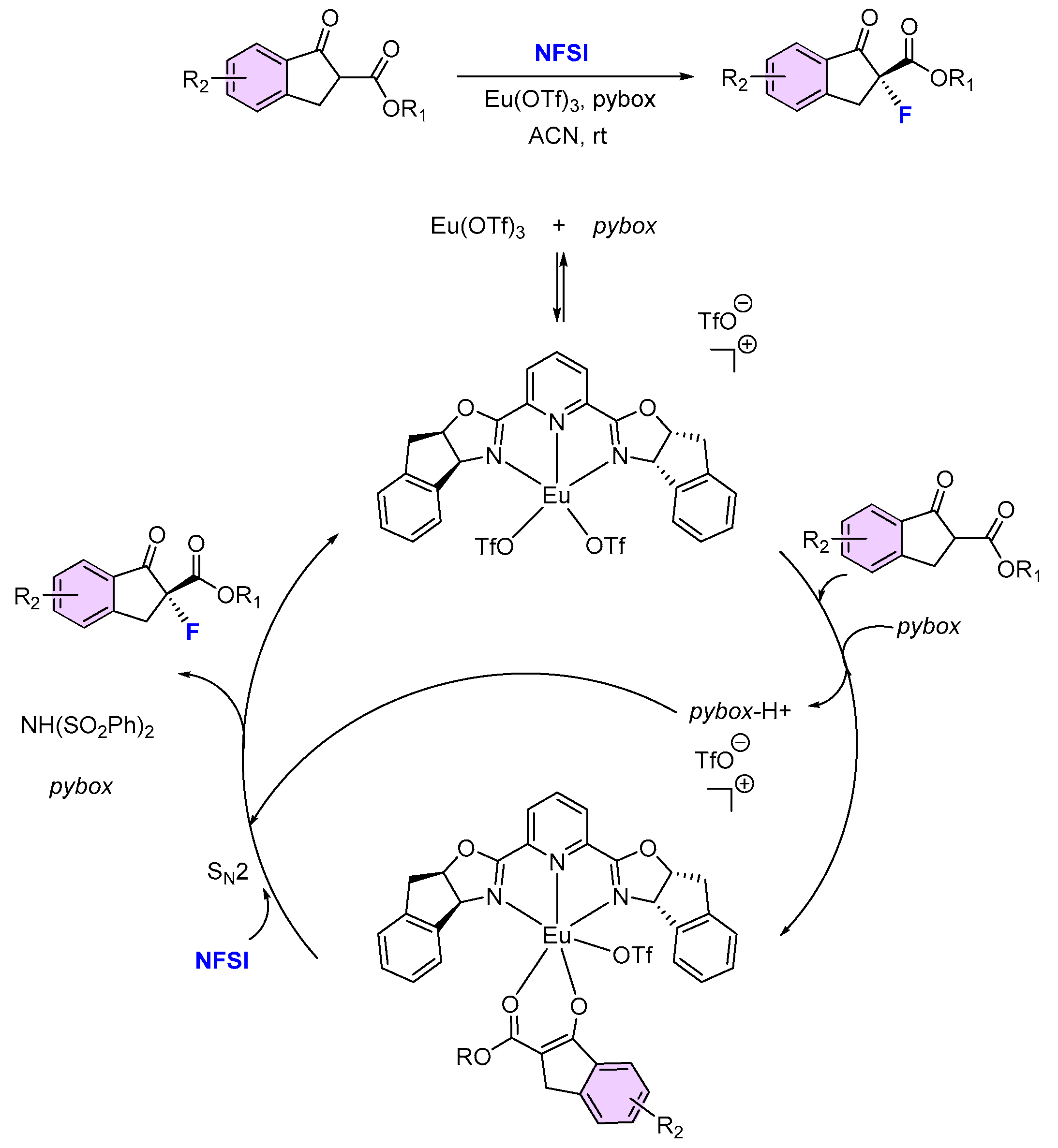{kind=link}
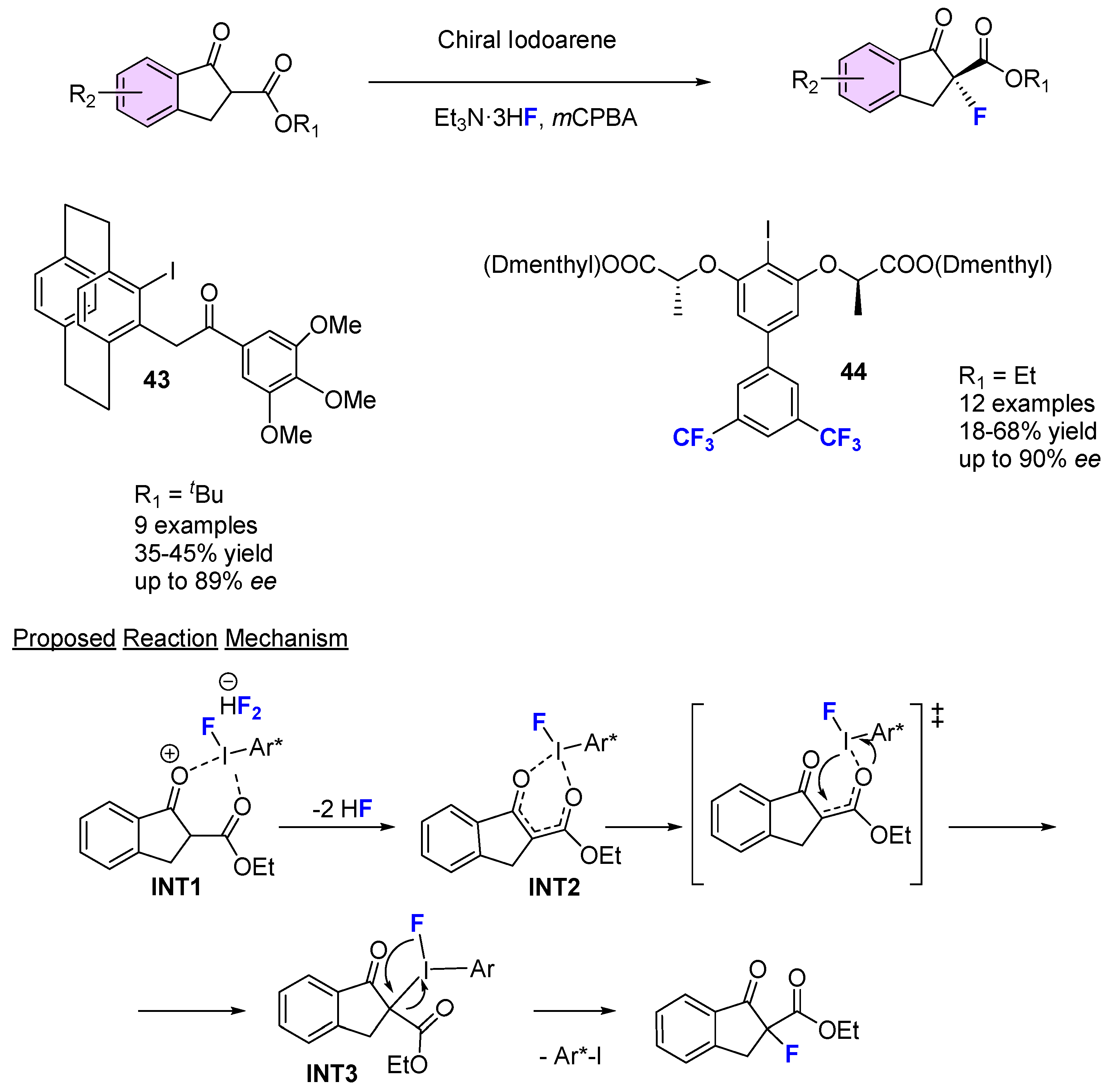{kind=link}
{kind=link}
{kind=link}
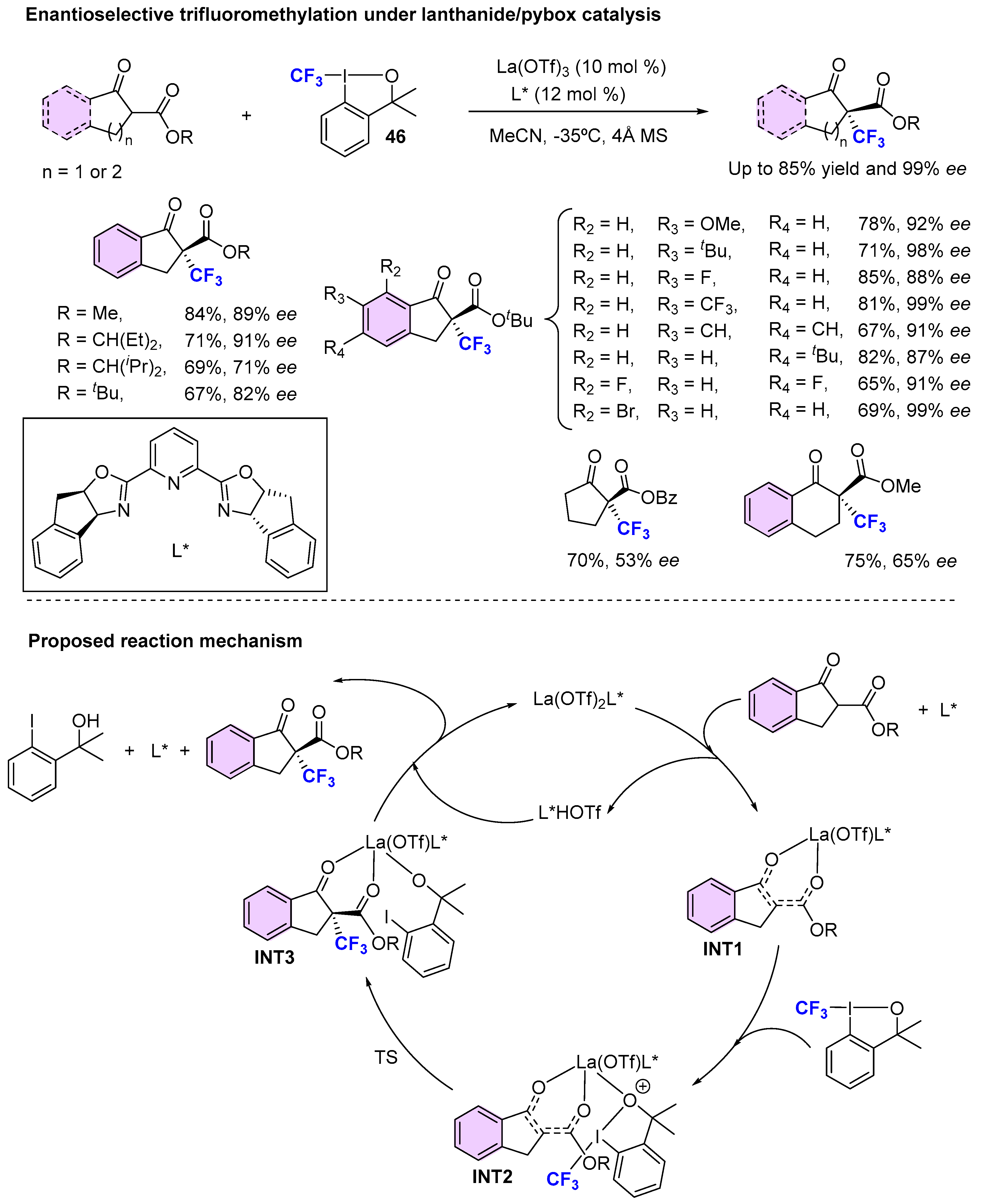{kind=link}
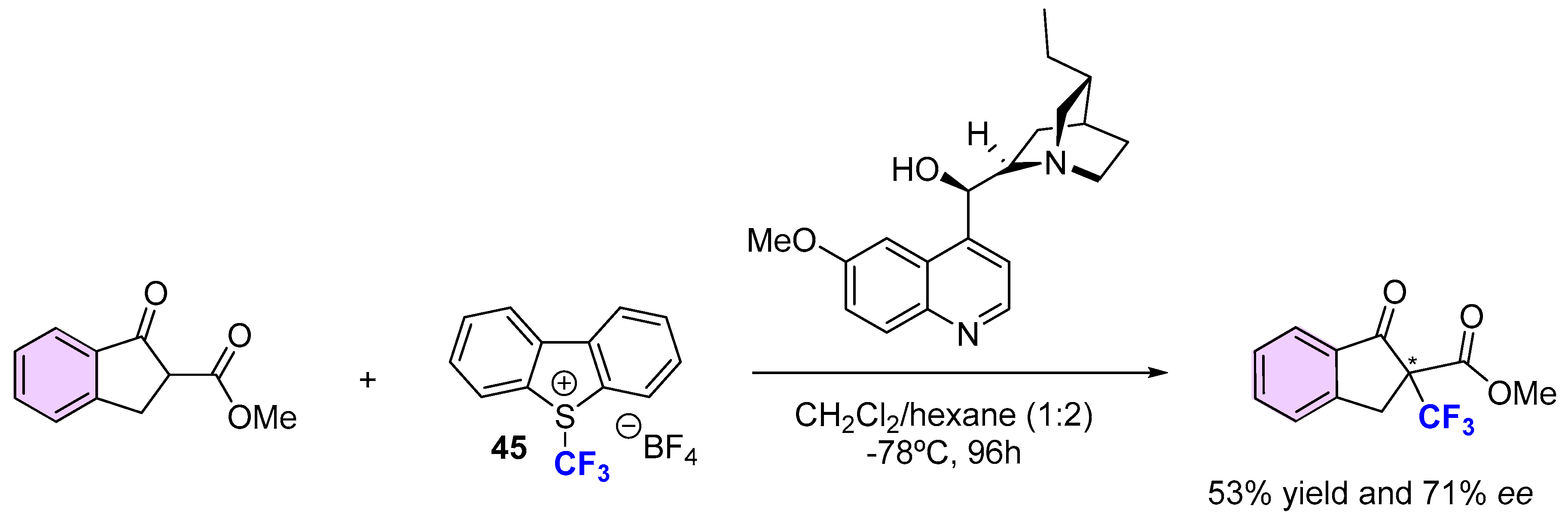{kind=link}
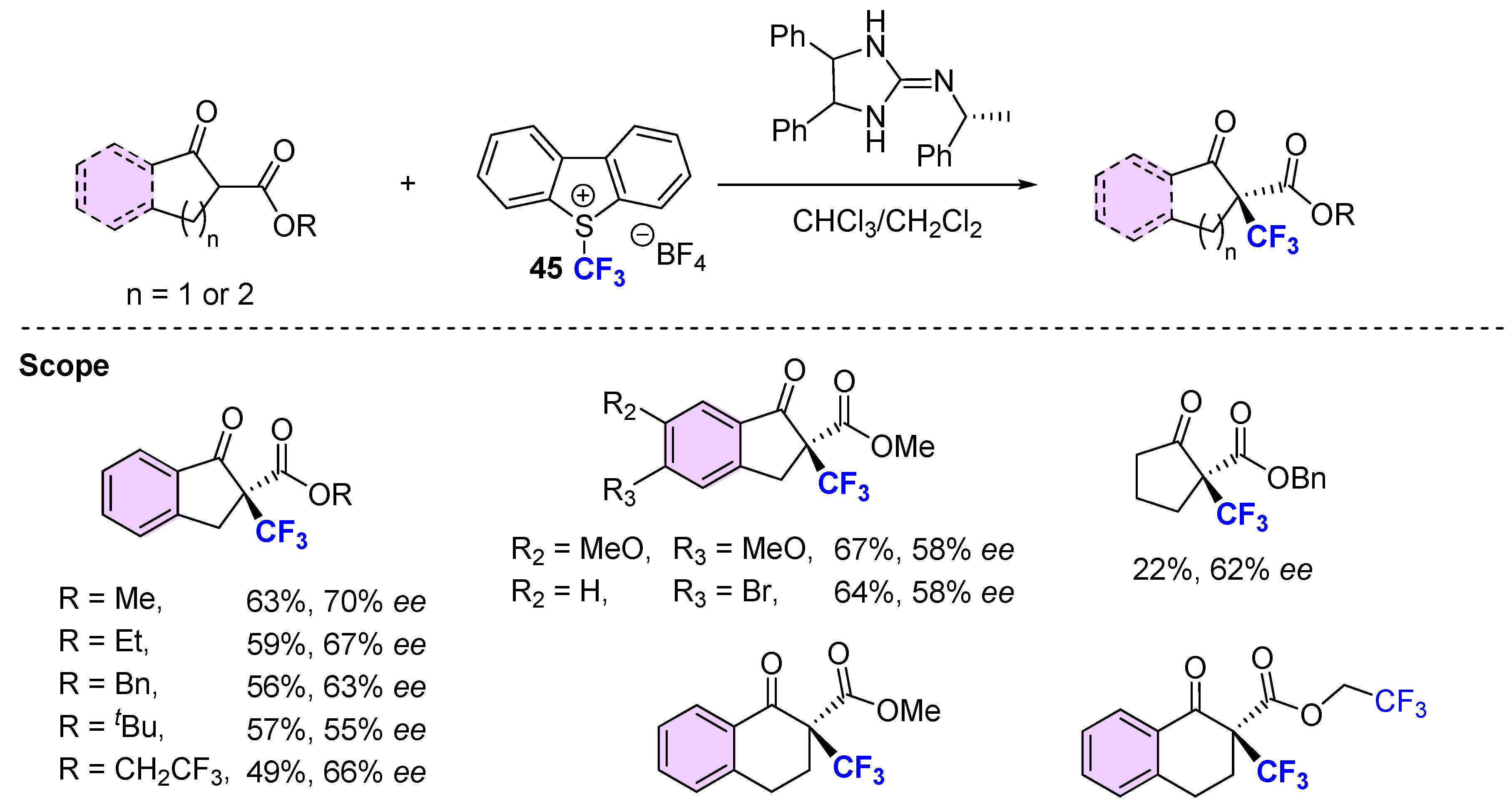{kind=link}
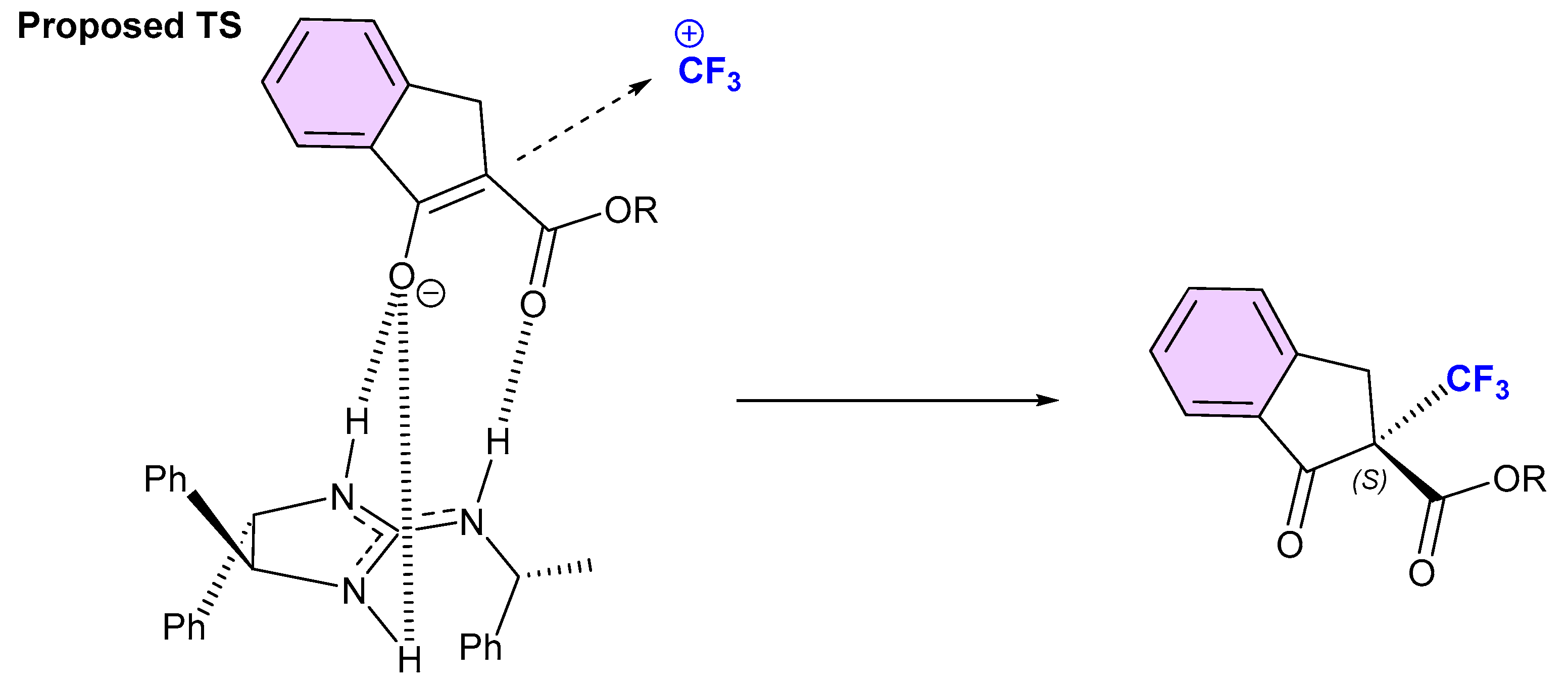{kind=link}
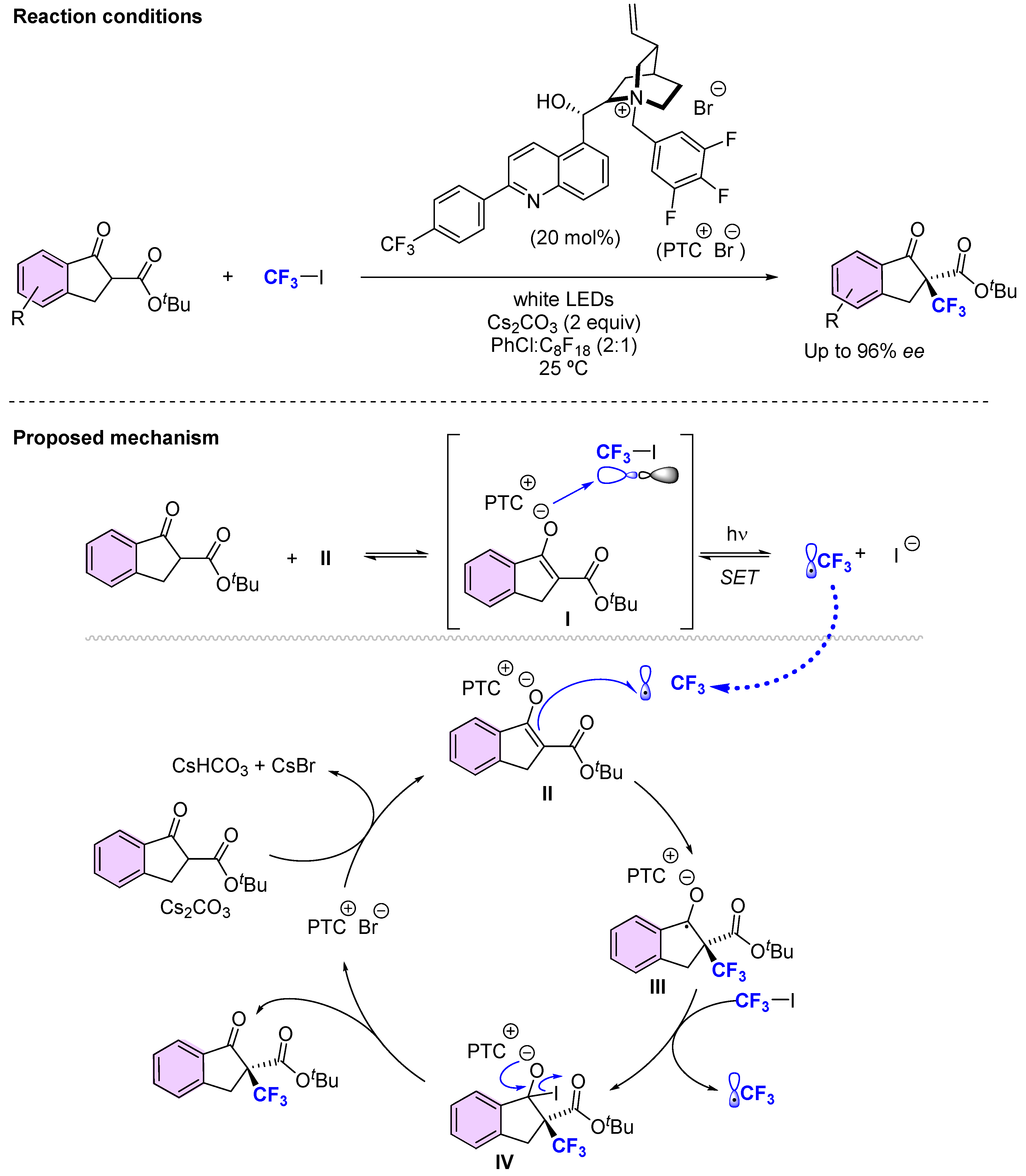{kind=link}
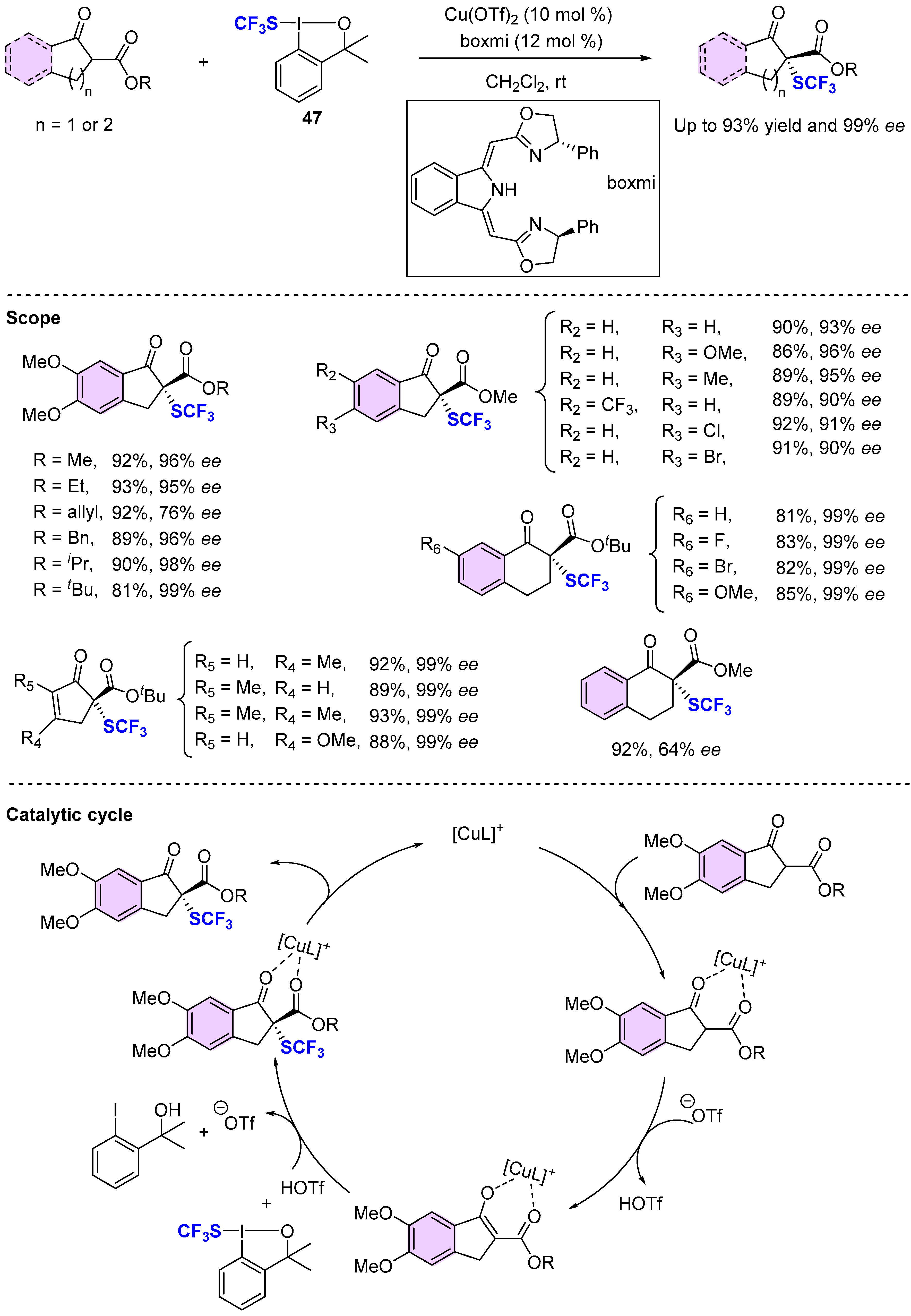{kind=link}
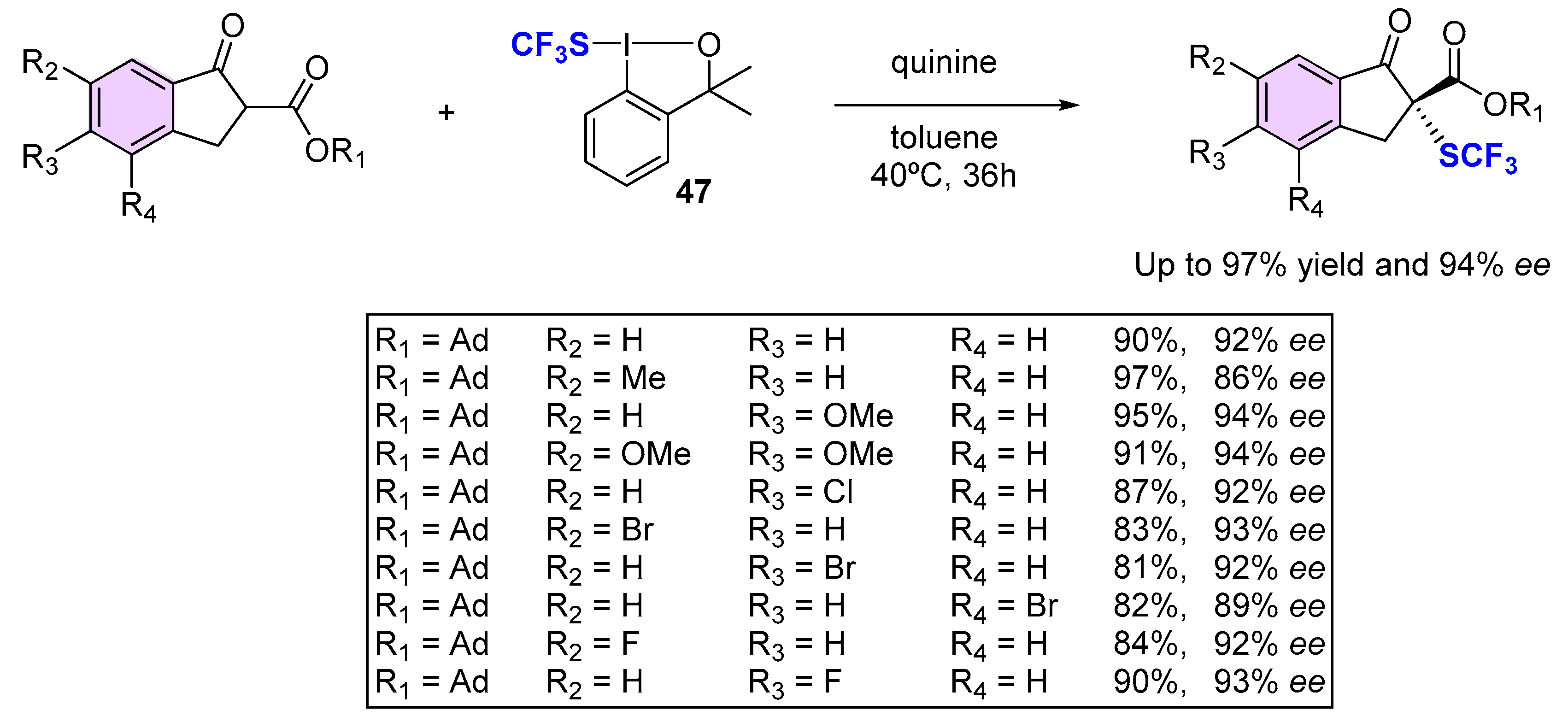{kind=link}
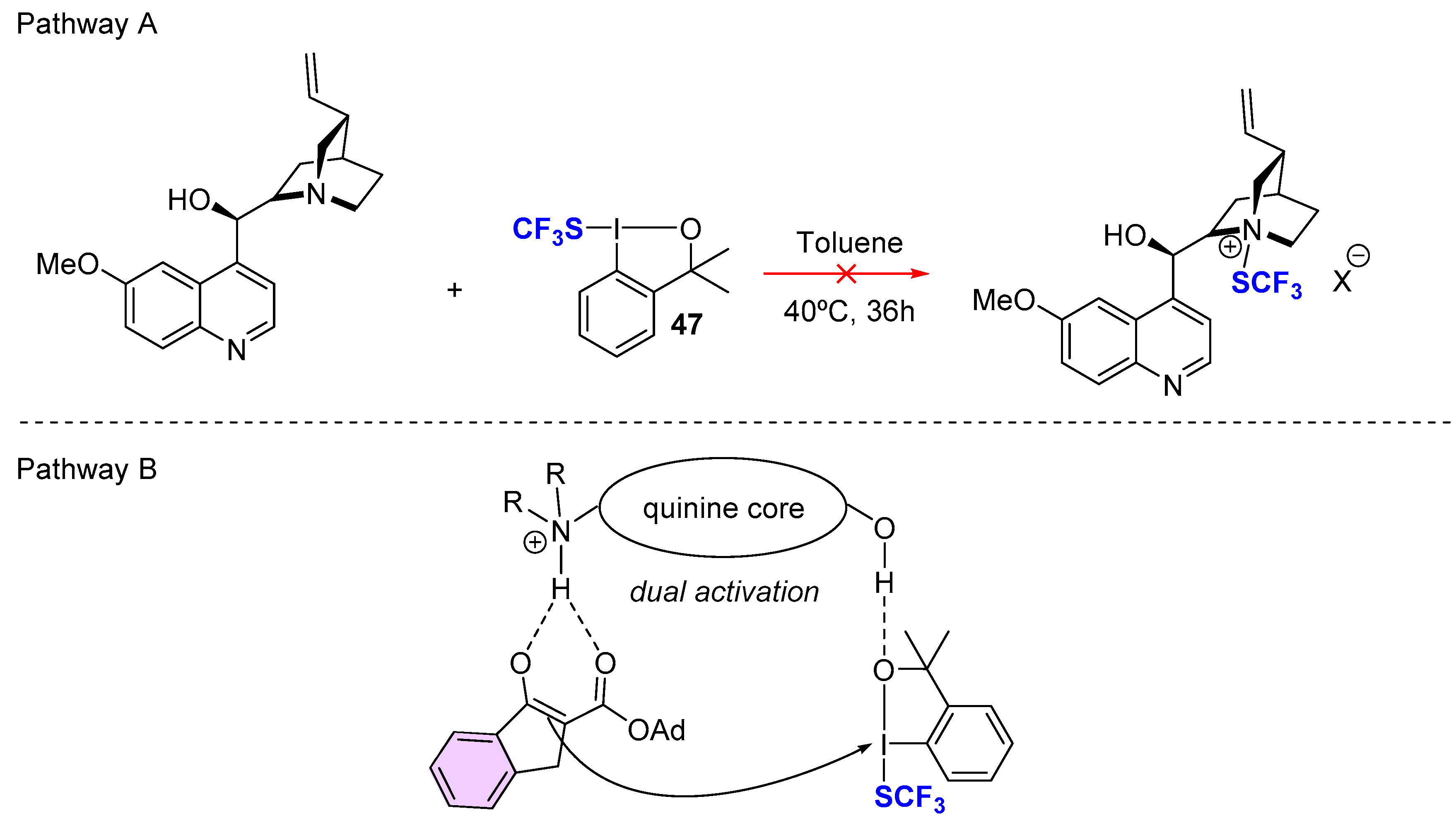{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
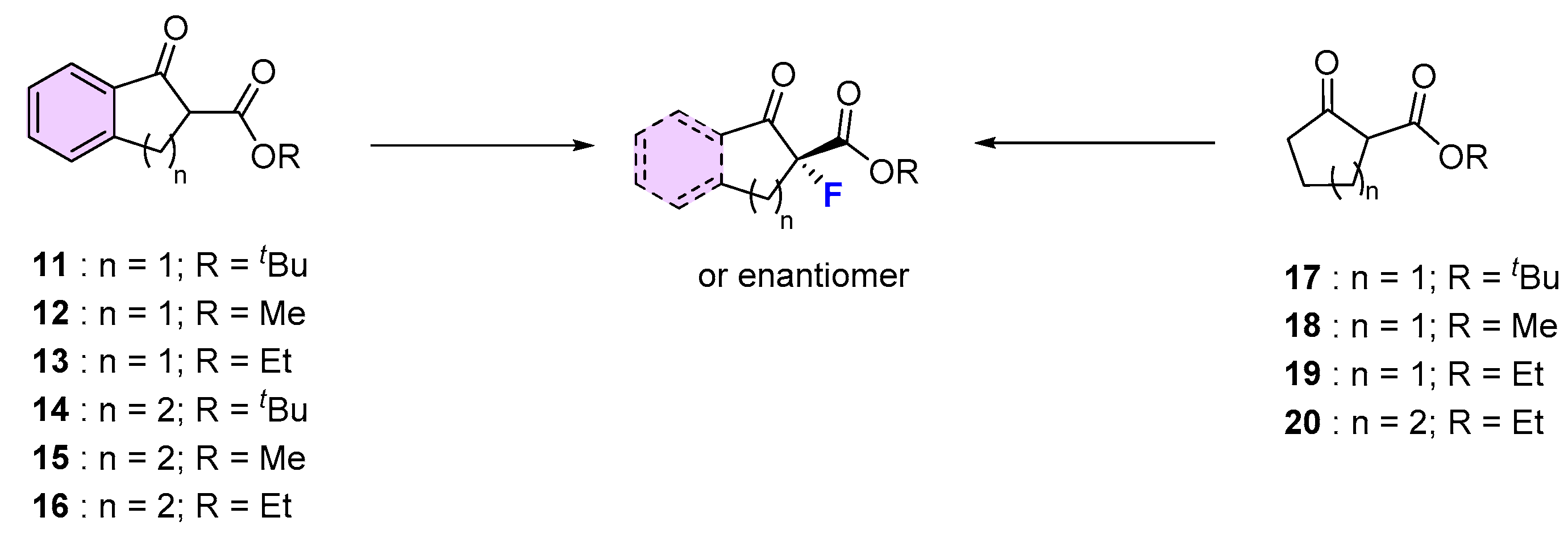
| Entry | Substrate | Fluorinating Reagent | Pre-Catalyst (see Figure 2) | Yield (%) | ee (%) | Ref |
|---|---|---|---|---|---|---|
| 1 | 11 | NFSI | Fe(III)/salan (6) 0 °C, MeCN, AgClO4 (2 mol.%) | 96 | 94 | [50] |
| 2 | 11 | NFSI | Ni(II)-monooxazoline (8) | 89 | 0 | [52] |
| 3 | 11 | NFSI | Cu(II)/diphenylamine-bis(oxazoline) (5), Ball mill | 99 | 95 | [49] |
| 4 | 11 | NFSI | Eu(III)/(R,S)-ind-pybox (9) | 78 | 96 (S) | [53] |
| 5 | 11 | NFSI | Cu(II)/diphenylamine-linked bis(thiazoline) (4), CHCl3, rt | 93 | 99 (S) | [48] |
| 6 | 12 | NFSI | Fe(III)/salan (6)0 °C, MeCN, AgClO4 (2 mol.%) | 99 | 46 | [50] |
| 7 | 12 | NFSI | Ni(II)-monooxazoline (8) | 90 | 75 | [52] |
| 8 | 12 | NFSI | Cu(II)/diphenylamine-bis(oxazoline), (5), Ball mill | 97 | 92 | [49] |
| 9 | 12 | NFSI | La(III)/(R,S)-ind-pybox (9) | 80 | 62 (S) | [53] |
| 10 | 12 | NFSI | Cu/Ar-BINMOL-derived salan (3) Xylene, 0 °C | 99 | 82 (S) | [47] |
| 11 | 13 | NFSI | Cu(II)/(S,S)-Nap-(R,R)-Box (2) 0 °C, toluene, HFIP | 98 | 34 | [45] |
| 12 | 13 | NFSI | Fe(III)/salan (6) 0 °C, MeCN, AgClO4 (2 mol.%) | 99 | 59 | [50] |
| 13 | 13 | NFSI | Ni(II)-monooxazoline (8) | 85 | 77 | [52] |
| 14 | 13 | NFSI | Cu(II)/diphenylamine-bis(oxazoline) (5), Ball mill | 99 | 91 | [49] |
| 15 | 13 | NFSI | Cu(II)/diphenylamine-linked bis(thiazoline) (4), CHCl3, rt | 100 | 99 | [48] |
| 16 | 14 | NFSI | Fe(III)/salan (6) 0 °C, MeCN, AgClO4 (2 mol.%) | 96 | 69 | [50] |
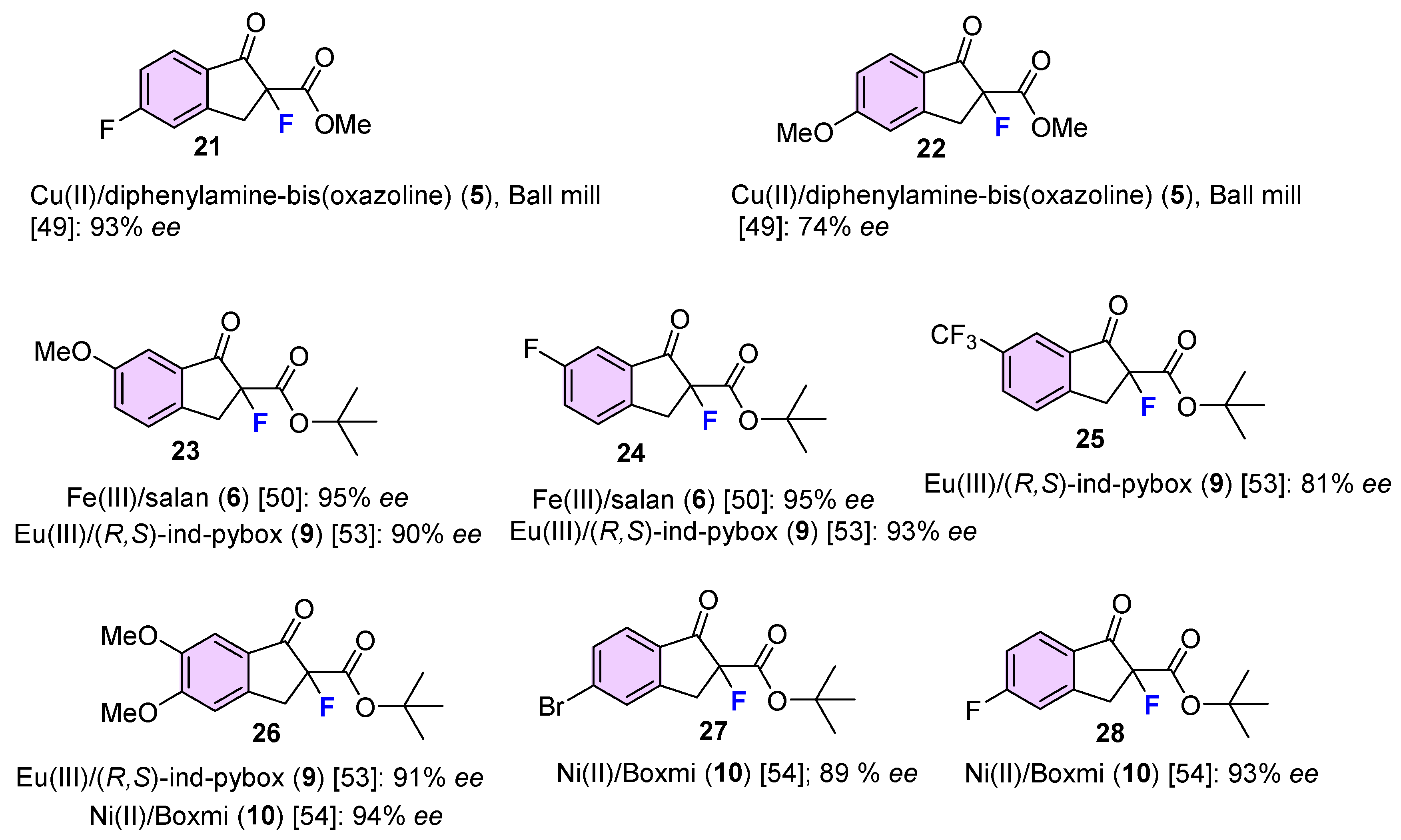
| 17 | 15 | NFSI | Cu(II)/diphenylamine-bis(oxazoline) (5), Ball mill | 93 | 56 | [49] |
| 18 | 15 | NFSI | Cu(II)/diphenylamine-linked bis(thiazoline) (4), CHCl3, rt | 100 | 93 | [48] |
| 19 | 15 | NFSI | Ni(II)-monooxazoline (8) | 86 | 13 | [52] |
| 20 | 16 | Selectfluor® | Ti/TADDOL (1), rt, MeCN | 93 | 20 | [44] |
| 21 | 16 | NFSI | Cu(II)/(S,S)-Nap-(R,R)-Box (2) 0 °C, toluene, HFIP | 90 | 16 | [45] |
| 22 | 16 | NFSI | Cu(II)/diphenylamine-linked bis(thiazoline) (4), CHCl3, rt | 99 | 93 | [48] |
| 23 | 17 | NFSI | Cu(II)/(S,S)-Nap-(R,R)-Box (2) 0 °C, toluene, HFIP | 97 | 83 | [45] |
| 24 | 17 | NFSI | Fe(III)/salan (6) O °C, MeCN, AgClO4 (2 mol.%) | 88 | 95 | [50] |
| 25 | 17 | NFSI | Pd(II)/(R)-DTBM-SEGPHOS (7) iPrOH, rt | 93 | 90 | [51] |
| 26 | 18 | NFSI | Ni(II)-monooxazoline (8) | 30 | 0 | [52] |
| 27 | 18 | NFSI | Cu(II)/(S,S)-Nap-(R,R)-Box (2) 0 °C, toluene, HFIP | 96 | 72 | [45] |
| 28 | 19 | NFSI | Cu(II)/(S,S)-Nap-(R,R)-Box (2) 0 °C, toluene, HFIP | 93 | 86 | [45] |
| 29 | 19 | NFSI | Cu(II)/diphenylamine-linked bis(thiazoline) (4), CHCl3, rt | 46 | 0 | [48] |
| 30 | 19 | NFSI | Cu(II)/diphenylamine-bis(oxazoline) (8), Ball mill | 96 | 92 | [49] |
| 31 | 20 | NFSI | Cu(II)/(S,S)-Nap-(R,R)-Box (2) 0 °C, toluene, HFIP | 90 | 52 | [45] |
| 32 | 20 | NFSI | Cu(II)/diphenylamine-linked bis(thiazoline) (4), CHCl3, rt | n. r. | - | [48] |
| 33 | 20 | NFSI | Cu(II)/diphenylamine-bis(oxazoline) (5), Ball mill | 95 | 99 | [49] |
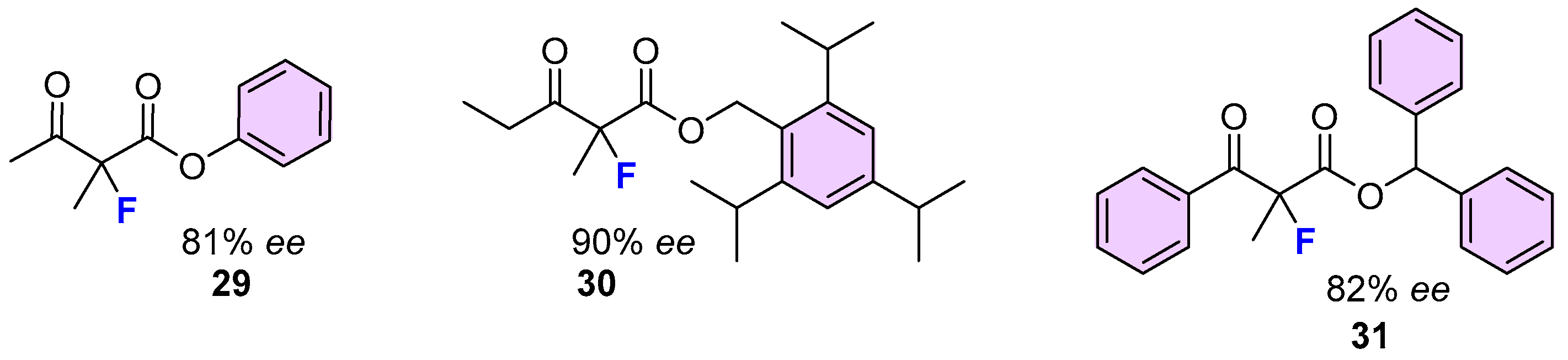
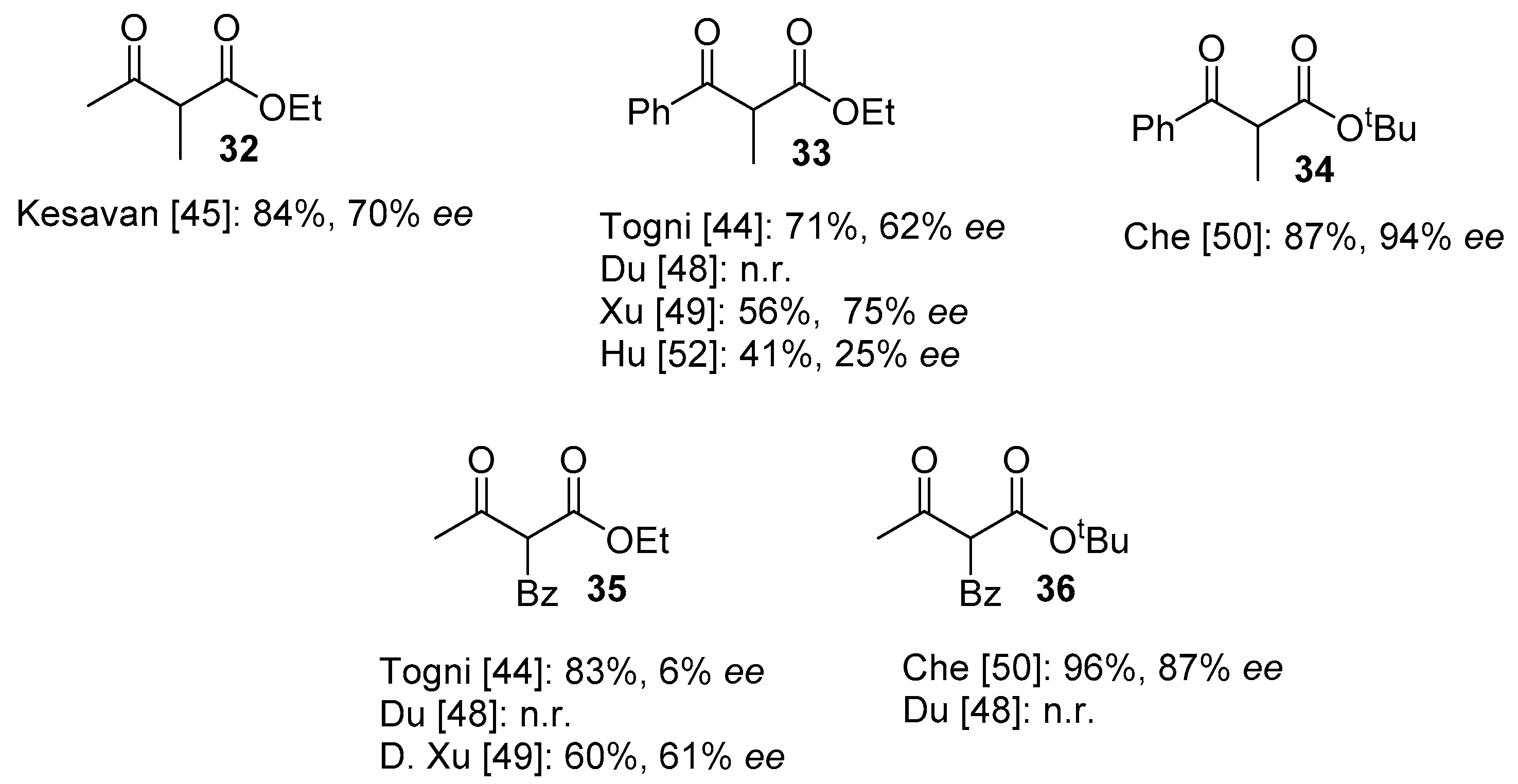
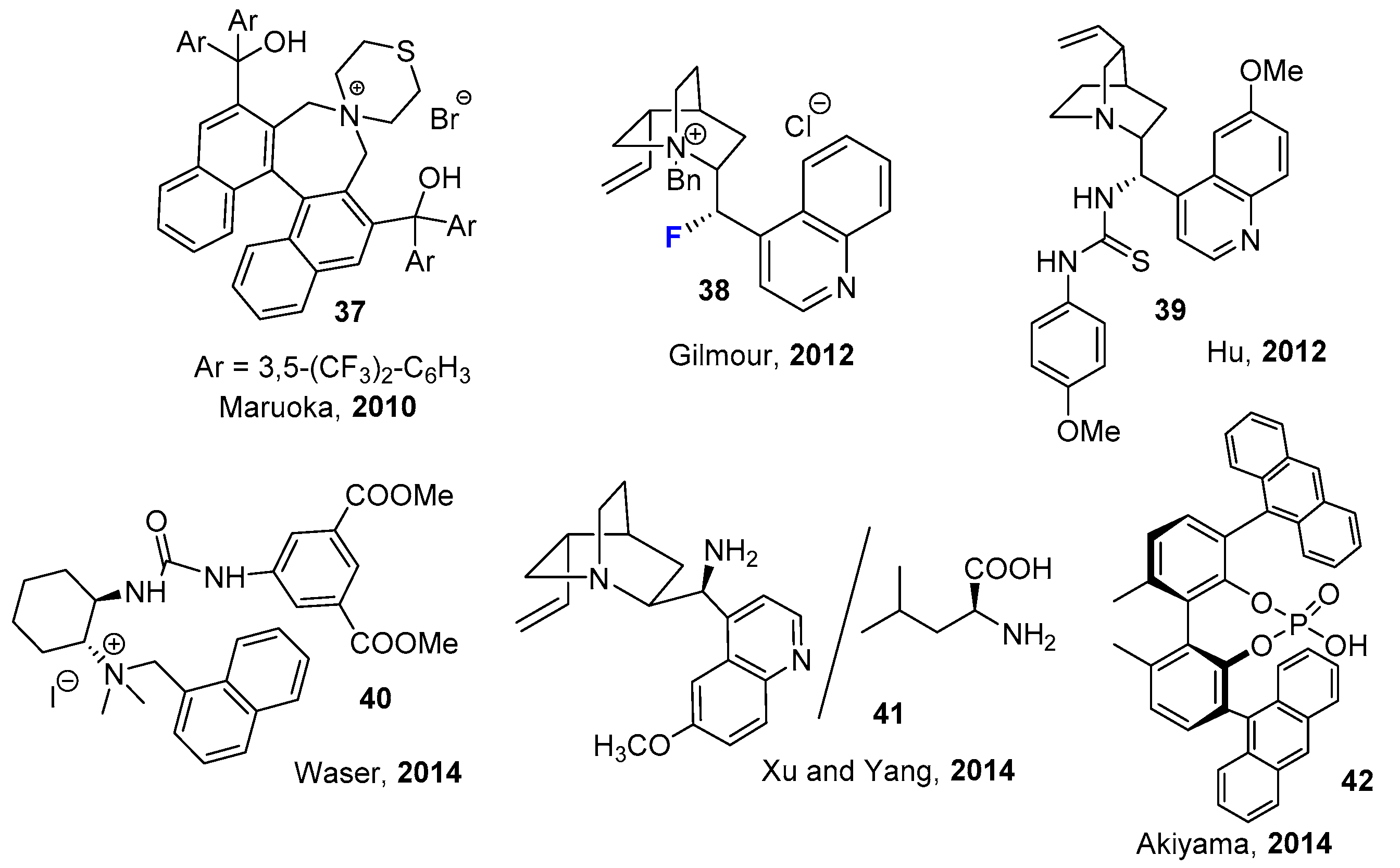
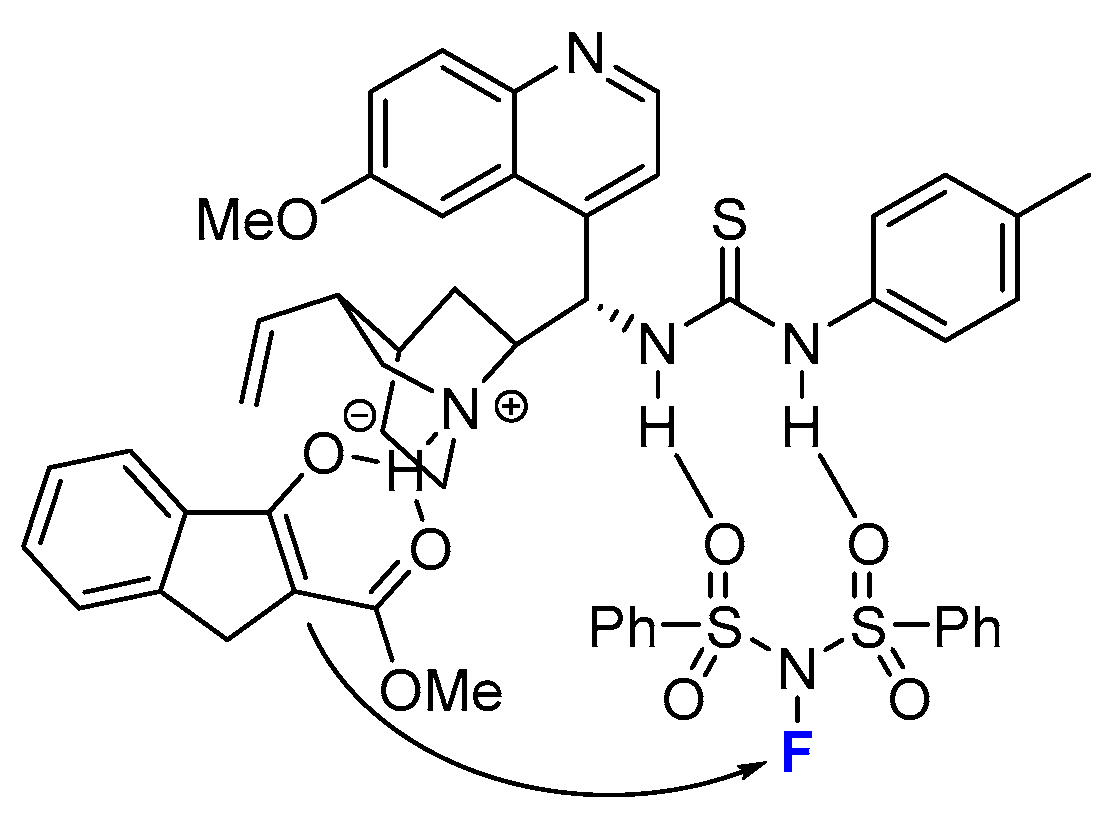
2.2. Organocatalytic Methods
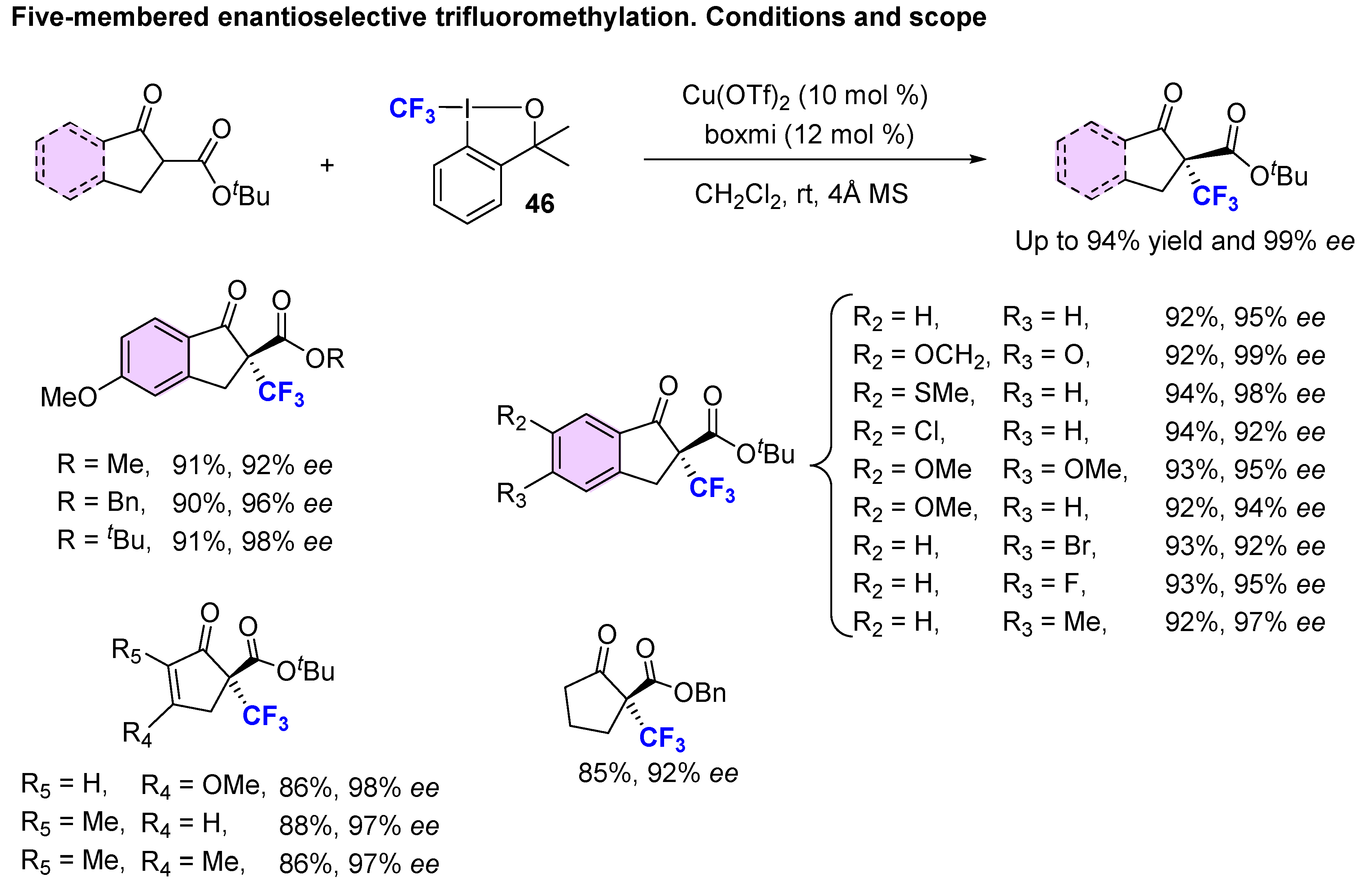
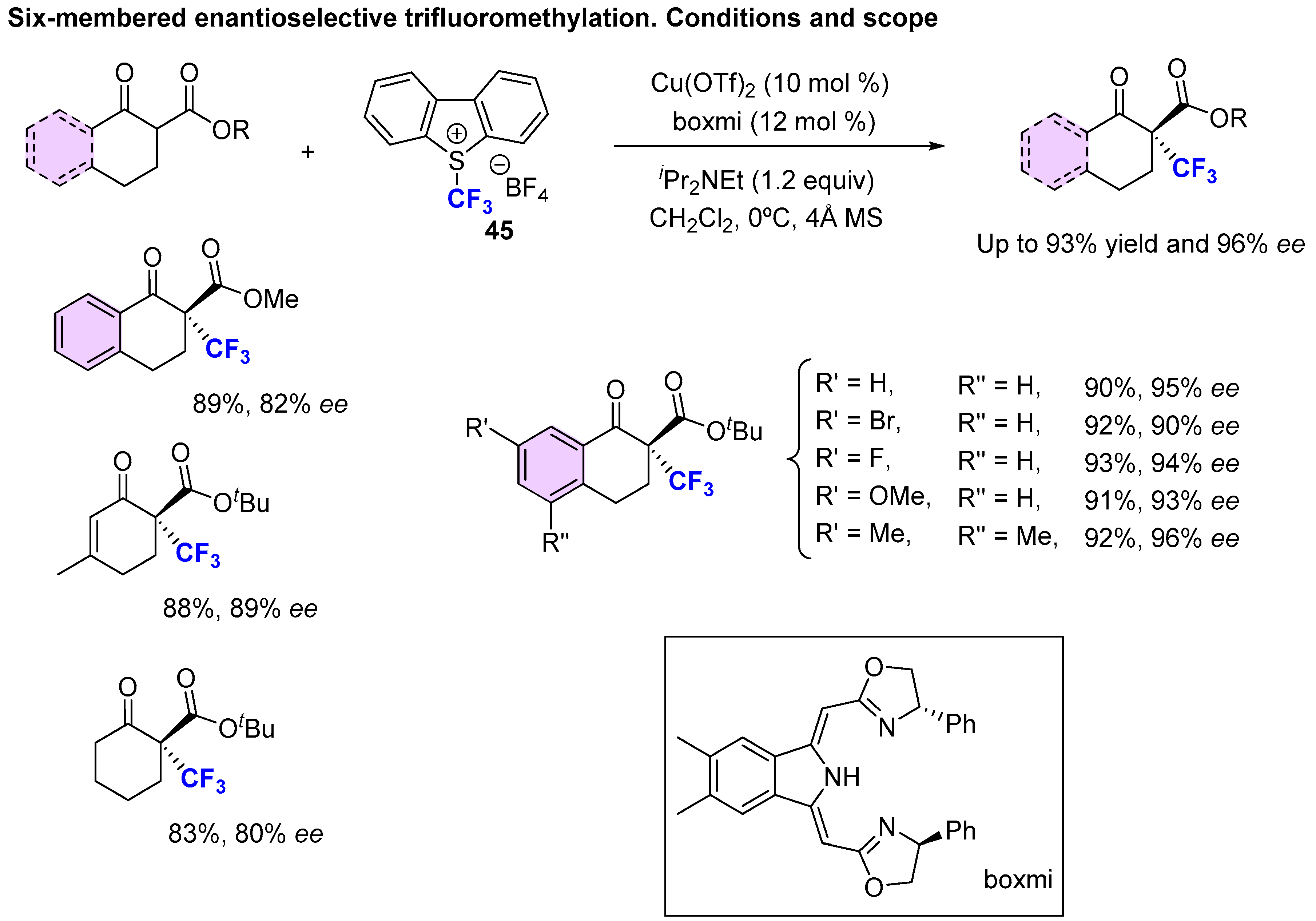
3. Asymmetric Preparation of Quaternary C-CF3 Stereocentres on β-keto Esters
3.1. Metal-Catalyzed Methods
3.2. Organocatalytic Methods
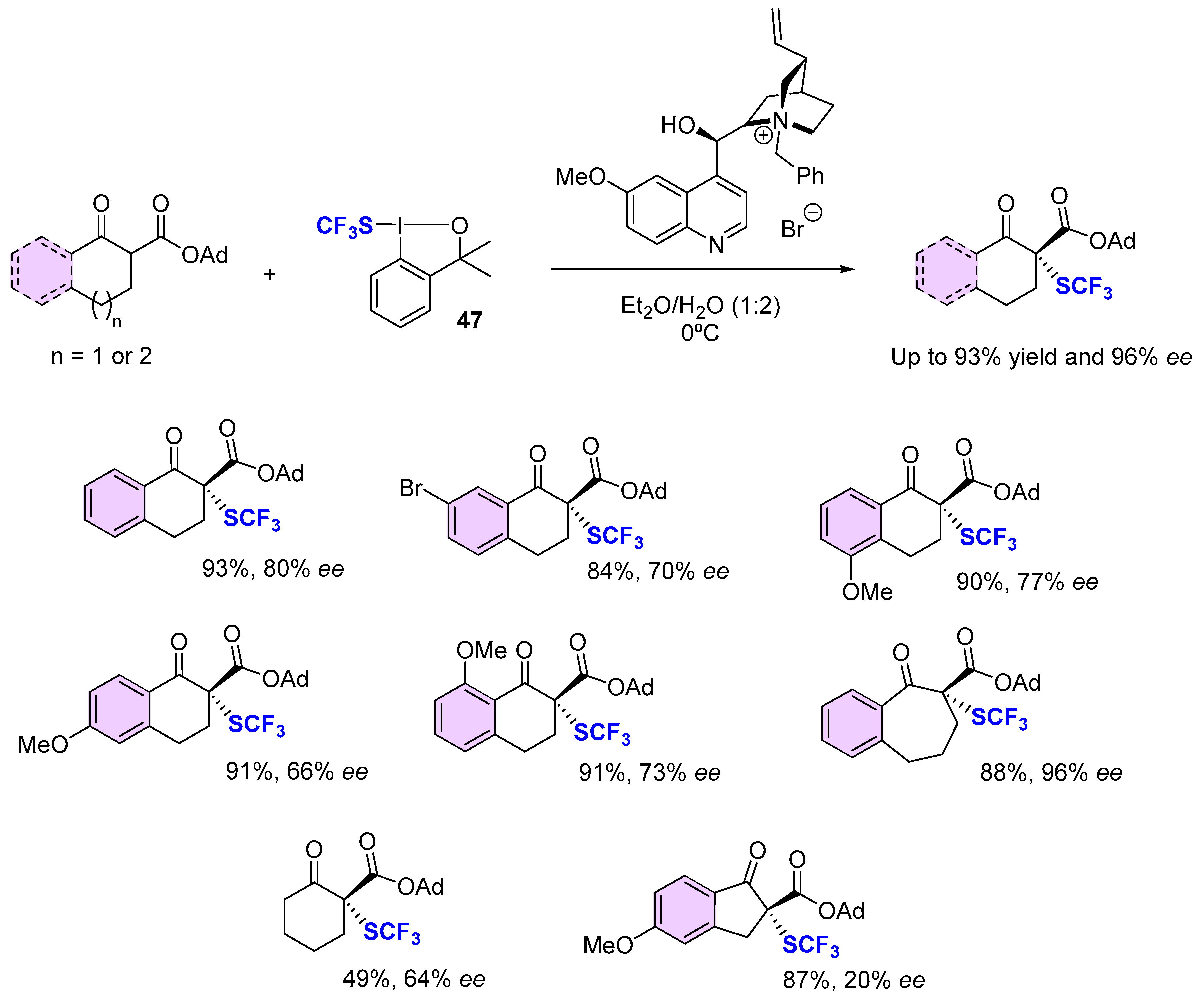
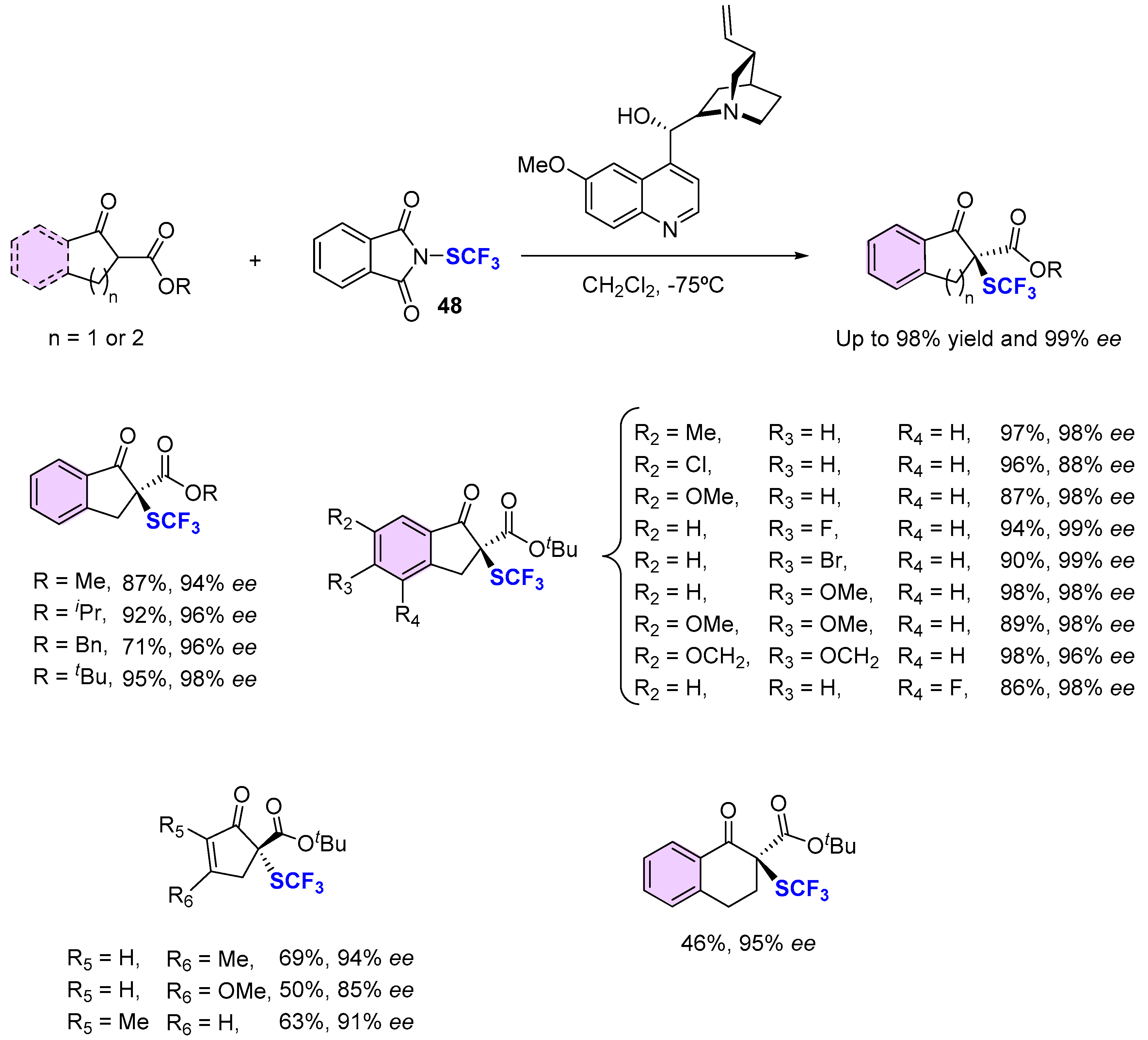
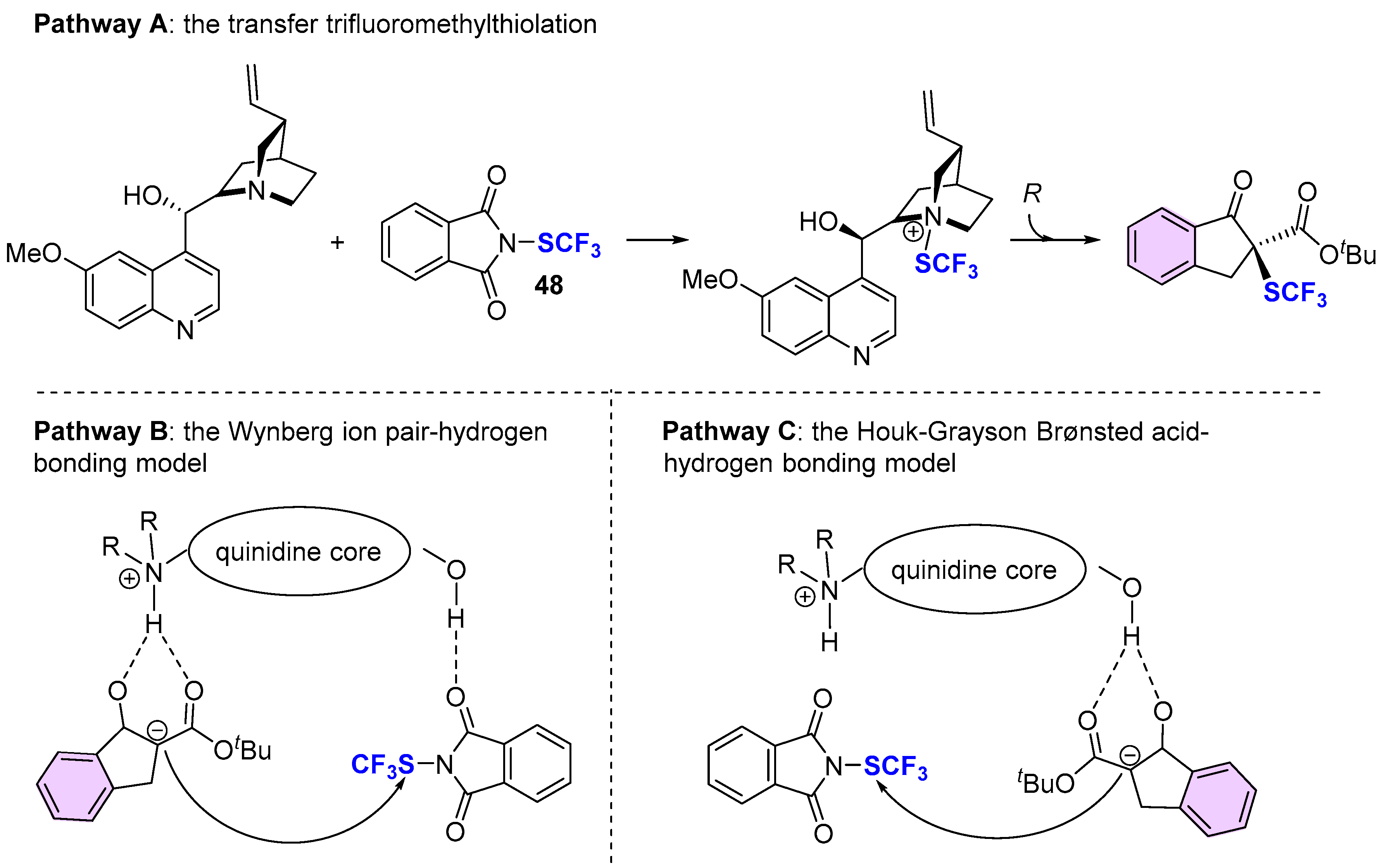
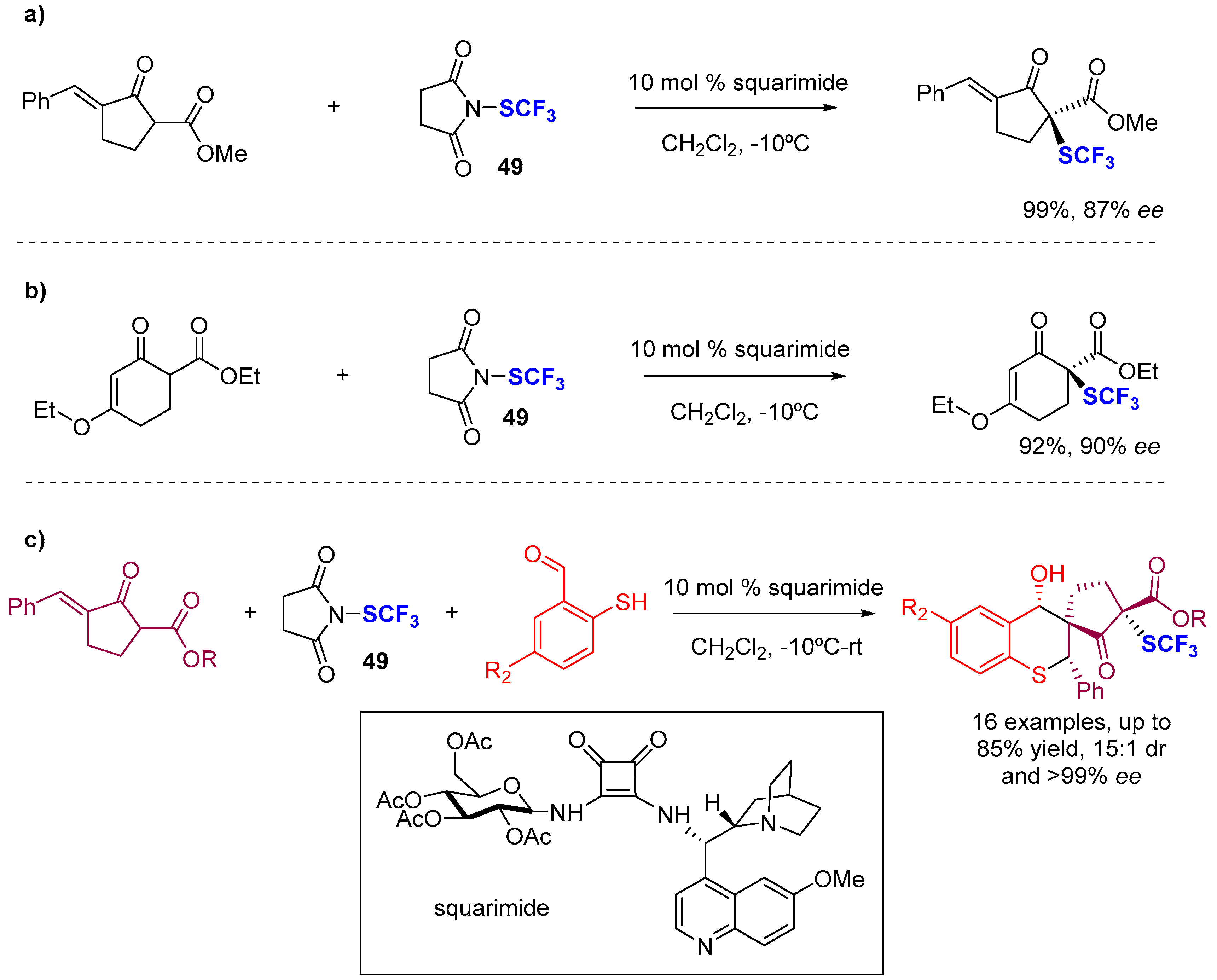
4. Asymmetric Preparation of Quaternary C-SCF3 Stereocentres on β-keto Esters
4.1. Metal-Catalyzed Methods
4.2. Organocatalytic Methods
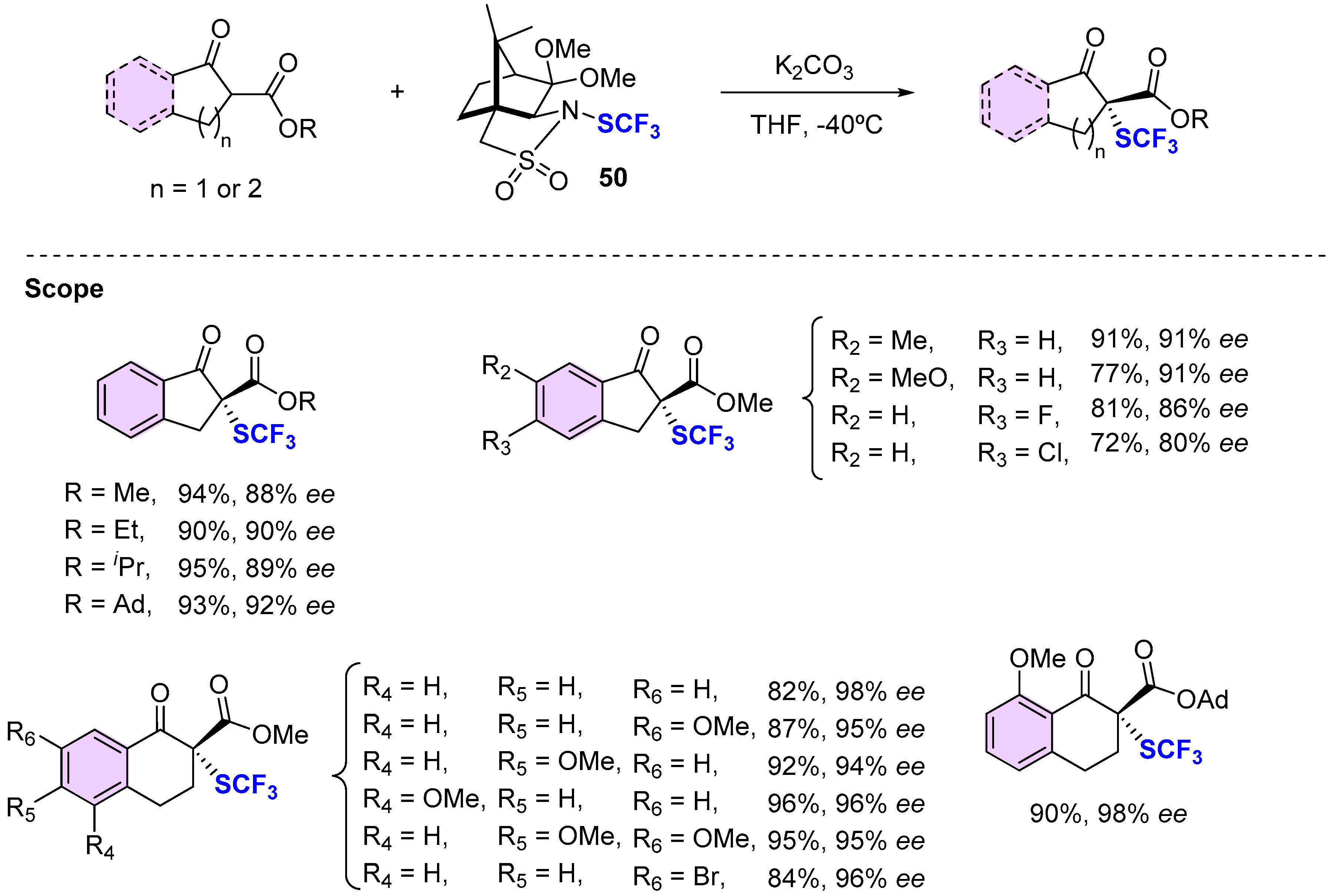
4.3. Methods Involving Chiral -SCF3 Reagents
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Kirsch, P. Modern Fluoroorganic Chemistry; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Gladysz, J.A.; Curran, D.E.; Horváth, I.T. Handbook of Fluorous Chemistry; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- O’Hagan, D. Understanding Organofluorine Chemistry. An Introduction to the C–F Bond. Chem. Soc. Rev. 2008, 37, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in Pharmaceuticals: Looking Beyond Intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Ilardi, E.A.; Vitaku, E.; Njardarson, J.T. Data-Mining for Sulfur and Fluorine: An Evaluation of Pharmaceuticals to Reveal Opportunities for Drug Design and Discovery. J. Med. Chem. 2014, 57, 2832–2842. [Google Scholar] [CrossRef]
- Haufe, G.; Leroux, F. Fluorine in Life Sciences: Pharmaceuticals, Medicinal Diagnostics, and Agrochemicals. Volume Four in Alain Tressaud’s Progress in Fluorine Science; Elsevier: Amsterdam, The Netherlands, 2019. [Google Scholar]
- Meanwell, N.A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef] [PubMed]
- Isanbor, C.; O’Hagan, D. Fluorine in Medicinal Chemistry: A Review of Anti-cancer Agents. J. Fluor. Chem. 2006, 127, 303–319. [Google Scholar] [CrossRef]
- Wang, J.; Sanchez-Roselló, M.; Aceña, J.L.; del Pozo, C.; Sorochinsky, A.E.; Fustero, S.; Soloshonok, V.A.; Liu, H. Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011). Chem. Rev. 2014, 114, 2432–2506. [Google Scholar] [CrossRef]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Application of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef]
- Smart, B.E. Fluorine Substituent Effects (on Bioactivity). J. Fluor. Chem. 2001, 109, 3–11. [Google Scholar] [CrossRef]
- Park, B.K.; Kitteringham, N.R.; O’Neill, P.M. Metabolism of Fluorine-Containing Drugs. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 443–470. [Google Scholar] [CrossRef] [PubMed]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in Medicinal Chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Ojima, I. Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell: Hobpken, NY, USA, 2009. [Google Scholar]
- Littich, R.; Scott, P.J.H. Novel Strategies for Fluorine-18 Radiochemistry. Angew. Chem. Int. Ed. 2012, 51, 1106–1109. [Google Scholar] [CrossRef] [PubMed]
- Tirotta, I.; Dichiarante, V.; Pigliacelli, C.; Cavallo, G.; Terraneo, G.; Bombelli, F.B.; Metrangolo, P.; Resnati, G. 19F Magnetic Resonance Imaging (MRI): From Design of Materials to Clinical Applications. Chem. Rev. 2015, 115, 1106–1129. [Google Scholar] [CrossRef] [PubMed]
- Harsanyi, A.; Sandford, G. Organofluorine Chemistry: Applications, Sources and Sustainability. Green Chem. 2015, 17, 2081–2086. [Google Scholar] [CrossRef]
- Soler, R.; Badetti, E.; Moreno-Mañas, M.; Vallribera, A.; Sebastián, R.M.; Vera, F.; Serrano, J.L.; Sierra, T. Wide Temperature Range Mesomorphic Behaviour of Highly Fluorinated 15-Membered Macrocycles and their Open Trisulphonamide Precursor. Liq. Cryst. 2007, 34, 235–240. [Google Scholar] [CrossRef]
- Ghosh, A.; Nakanishi, T. Frontiers of Solvent-free Functional Molecular Liquids. Chem. Comm. 2017, 53, 10344–10357. [Google Scholar] [CrossRef] [PubMed]
- Montagut, A.M.; Galvez, E.; Shafir, A.; Sebastián, R.M.; Vallribera, A. Triarylmethane Dyes for Artificial Repellent Cotton Fibers. Chem. Eur. J. 2017, 23, 3810–3814. [Google Scholar] [CrossRef]
- Bernini, R.; Cacchi, S.; Fabrizi, G.; Forte, G.; Petrucci, F.; Prastaro, A.; Niembro, S.; Shafir, A.; Vallribera, A. Green Chemistry Perfluoro-tagged Phosphine-free Palladium Nanoparticles Supported on Silica Gel: Application to Alkynylation of Aryl Halides, Suzuki–Miyauracross-coupling, and Heck Reactions under Aerobic Conditions. Green Chem. 2010, 12, 150–158. [Google Scholar] [CrossRef]
- Bernini, R.; Cacchi, S.; Fabrizi, G.; Niembro, S.; Prastaro, A.; Shafir, A.; Vallribera, A. Perfluoro-Tagged Gold Nanoparticles Immobilized on Fluorous Silica Gel: A Reusable Catalyst for the Benign Oxidation and Oxidative Esterification of Alcohols. ChemSusChem 2009, 2, 1036–1040. [Google Scholar] [CrossRef]
- Salabert, J.; Sebastián, R.M.; Vallribera, A. Anthraquinone Dyes for Superhydrophobic Cotton. Chem. Commun. 2015, 51, 14251–14254. [Google Scholar] [CrossRef]
- Schlögla, S.; Kramera, R.; Lenkoa, D.; Schröttnerb, H.; Schallerc, R.; Holznerc, A.; Kern, W. Fluorination of Elastomer Materials. Eur. Polym. J. 2011, 47, 2321–2330. [Google Scholar] [CrossRef]
- Liang, T.; Neumann, C.N.; Ritter, T. Introduction of Fluorine and Fluorine-Containing Functional Groups. Angew. Chem. Int. Ed. 2013, 52, 8214–8264. [Google Scholar] [CrossRef]
- Orsi, D.L.; Altman, R.A. Exploiting the Unusual Effects of Fluorine in Methodology. Chem. Commun. 2017, 53, 7168–7181. [Google Scholar] [CrossRef] [PubMed]
- Ni, C.; Hu, J. The Unique Fluorine Effects in Organic Reactions: Recent Facts and Insights into Fluoroalkylations. Chem. Soc. Rev. 2016, 45, 5441–5454. [Google Scholar] [CrossRef]
- Zhu, Y.; Han, J.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V.A.; Coelho, J.A.S.; Toste, F.D. Modern Approaches for Asymmetric Construction of Carbon−Fluorine Quaternary Stereogenic Centers: Synthetic Challenges and Pharmaceutical Needs. Chem. Rev. 2018, 118, 3887–3964. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wu, T.; Phipps, R.J.; Toste, F.D. Advances in Catalytic Enantioselective Fluorination, Mono-, Di-, and Trifluoromethylation, and Trifluoromethylthiolation Reactions. Chem. Rev. 2015, 115, 826–870. [Google Scholar] [CrossRef]
- Lin, J.-H.; Xiao, J.-C. Recent Advances in Asymmetric Fluorination and Fluoroalkylation Reactions via Organocatalysis. Tetrahedron Lett. 2014, 55, 6147–6155. [Google Scholar] [CrossRef]
- Toulgoat, F.; Alazet, S.; Billard, T. Direct Trifluoromethylthiolation Reactions: The “Renaissance” of an Old Concept. Eur. J. Org. Chem. 2014, 2014, 2415–2428. [Google Scholar] [CrossRef]
- Hintermann, L.; Togni, A. Catalytic Enantioselective Fluorination of β-Ketoesters. Angew. Chem. Int. Ed. 2000, 39, 4359–4362. [Google Scholar]
- Nagib, D.A.; Scott, M.E.; MacMillan, D.W. C Enantioselective α-Trifluoromethylation of Aldehydes via Photoredox Organocatalysis. J. Am. Chem. Soc. 2009, 131, 10875–10877. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.E.; MacMillan, D.W.C. The Productive Merger of Iodonium Salts and Organocatalysis: A Non-photolytic Approach to the Enantioselective α-Trifluoromethylation of Aldehydes. J. Am. Chem. Soc. 2010, 132, 4986–4987. [Google Scholar] [CrossRef]
- Comelles, J.; Pericas, A.; Moreno-Mañas, M.; Vallribera, A.; Drudis-Solé, G.; Lledós, A.; Parella, T.; Roglans, A.; García-Granda, S.; Roces-Fernández, L. Highly Enantioselective Electrophilic Amination and Michael Addition of Cyclic β-Ketoesters Induced by Lanthanides and (S,S)-Ip-pybox: The Mechanism. J. Org. Chem. 2007, 72, 2077–2087. [Google Scholar] [CrossRef]
- Pericas, A.; Shafir, A.; Vallribera, A. Asymmetric Synthesis of l-Carbidopa Based on a Highly Enantioselective α-Amination. Org. Lett. 2013, 15, 1448–1451. [Google Scholar] [CrossRef] [PubMed]
- Pericas, A.; Jiménez, R.; Granados, A.; Shafir, A.; Vallribera, A.; Roglans, A.; Molins, E. Lanthanides–pybox: An Excellent Combination for Highly Enantioselective Electrophilic α-Amination of Acyclic β-Keto Esters. Isolation of Ternary Pybox/Ln/β-Keto Ester Complexes. ChemistrySelect 2016, 1, 4305–4312. [Google Scholar] [CrossRef]
- Deng, Q.-H.; Wadepohl, H.; Gade, L.H. Highly Enantioselective Copper-Catalyzed Electrophilic Trifluoromethylation of β-Ketoesters. J. Am. Chem. Soc. 2012, 134, 10769–10772. [Google Scholar] [CrossRef] [PubMed]
- Differding, E.; Ofner, H. N-Fluorobenzenesulfonimide: A Practical Reagent for Electrophilic Fluorinations. Synlett 1991, 1991, 187–189. [Google Scholar] [CrossRef]
- Nyffeler, P.T.; Gonzalez, S.; Burkart, M.D.; Vicent, S.P.; Wong, C.-H. Selectfluor: Mechanistic Insight and Applications. Angew. Chem. Int. Ed. 2005, 44, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yuan, H.; Lu, H.; Zheng, W.-H. Development of Planar Chiral Iodoarenes Based on [2.2]Paracyclophane and Their Application in Catalytic Enantioselective Fluorination of β-ketoesters. Org. Lett. 2018, 20, 2555–2558. [Google Scholar] [CrossRef]
- Pluta, R.; Krach, P.E.; Cavallo, L.; Falivene, L.; Rueping, M. Metal-free Catalytic Fluorination of Keto Esters Using a Combination of Hydrogen Fluoride (HF) and Oxidant: Experiment and Computation. ACS Catal. 2018, 8, 2582–2588. [Google Scholar] [CrossRef]
- Lectard, S.; Hamashima, Y.; Sodeoka, M. Recent Advances in Catalytic Enantioselective Fluorination Reactions. Adv. Synth. Catal. 2010, 352, 2708–2732. [Google Scholar] [CrossRef]
- Bertogg, A.; Hintermann, L.; Huber, D.P.; Perseghini, M.; Sanna, M.; Togni, A. Substrate Range of the Titanium TADDOLate Catalyzed Asymmetric Fluorination of Activated Carbonyl Compounds. Helv. Chim. Acta 2012, 95, 353–382. [Google Scholar] [CrossRef]
- Balaraman, K.; Vasanthan, R.; Kesavan, V. Enantioselective Fluorination of β-Ketoesters Using Tartrate Derived Bidentate Bioxazoline-Cu(II) Complexes. Tetrahedron-Asymmetry 2013, 24, 919–924. [Google Scholar] [CrossRef]
- Ma, J.-A.; Cahard, D. Copper(II) Triflate-bis(Oxazoline)-Catalyzed Enantioselective Electrophilic Fluorination of β-Ketoesters. Tetrahedron-Asymmetry 2004, 15, 1007–1011. [Google Scholar] [CrossRef]
- Zheng, L.-S.; Wei, Y.-L.; Jiang, K.-Z.; Deng, Y.; Zheng, Z.-J.; Xua, L.-W. Enantioselective Fluorination of β-Ketoamides Catalyzed by Ar-BINMOL-derived Salan Copper Complex. Adv. Synth. Catal. 2014, 356, 3769–3776. [Google Scholar] [CrossRef]
- Peng, J.; Du, D.-M. Efficient Enantioselective Fluorination of β-Ketoesters/amides Catalyzed by Diphenylamine-linked bis(thiazoline) and Cu(OTf)2 Complexes. RSC Adv. 2014, 4, 2061–2067. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.; Jiang, Y.; Zhang, C.; Shao, J.; Xu, D. Fast, Solvent-free and Highly Enantioselective Fluorination of β-Keto esters Catalyzed by Chiral Copper Complexes in a Ball Mill. Green Chem. 2017, 19, 1674–1677. [Google Scholar] [CrossRef]
- Gu, X.; Zhang, Y.; Xu, Z.-J.; Che, C.-M. Iron(III)–salan Complexes Catalysed Highly Enantioselective Fluorination and Hydroxylation of β-Keto esters and N-Boc oxindoles. Chem. Commun. 2014, 50, 7870–7873. [Google Scholar] [CrossRef]
- Hayamizu, K.; Teramaya, N.; Hashizume, D.; Dodo, K.; Sodeoka, M. Unique Features of Chiral Palladium Enolates Derived from β-Ketoamide: Structure and Catalytic Asymmetric Michael and Fluorination Reactions. Tetrahedron 2015, 71, 6594–6601. [Google Scholar] [CrossRef]
- Niu, T.; Han, X.; Huang, D.; Wang, K.-H.; Su, Y.; Hu, Y.; Fu, Y. Enantioselective Fluorination of β-Ketoesters Catalysed by Complexes of New Mono-oxazoline Ligands. J. Fluor. Chem. 2015, 175, 6–11. [Google Scholar] [CrossRef]
- Granados, A.; Sarró, P.; Vallribera, A. Catalytic Asymmetric Fluorination of Alkyl 1-Indanone-2-carboxylates Ruled by Pybox-Eu(III) Combination. Molecules 2019, 24, 1141. [Google Scholar] [CrossRef] [PubMed]
- Blasius, C.K.; Ren, B.-T.; Liu, Y.-K.; Li, B.; Michalsky, I.; Wadepohl, H.; Dend, Q.-H.; Gade, L.H. Expanding the Boxmi Ligand Family: Synthesis and Application of NON and NSN Ligands. J. Org. Chem. 2020, 85, 6719–6731. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Park, E.J. Catalytic Enantioselective Fluorination of β-Keto Esters by Phase-Transfer Catalysis Using Chiral Quaternary Ammonium Salts. Org. Lett. 2002, 4, 545–547. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lan, Q.; Shirakawa, S.; Maruoka, K. Chiral Bifunctional Phase Transfer Catalysts for Asymmetric Fluorination of β-Keto esters. Chem. Commun. 2010, 46, 321–323. [Google Scholar] [CrossRef] [PubMed]
- Tanzer, E.-M.; Schweizer, W.B.; Ebert, M.-O.; Gilmour, R. Designing Fluorinated Cinchona Alkaloids for Enantioselective Catalysis: Controlling Internal Rotation by a Fluorine-Ammonium Ion Gauche Effect (ΦNCCF). Chem. Eur. J. 2012, 18, 2006–2013. [Google Scholar] [CrossRef]
- Shang, J.-Y.; Li, L.; Lu, Y.; Yang, K.-F.; Xu, L.-X. Enantioselective Fluorination Reaction of β-Keto Ester- Catalyzed Chiral Primary Amine-Based Multifunctional Catalyst Systems. Synth. Commun. 2014, 44, 101–104. [Google Scholar] [CrossRef]
- Xu, J.; Hu, Y.; Huang, D.; Wang, K.-H.; Xu, C. Thiourea-Catalyzed Enantioselective Fluorination of β-Keto Esters. Adv. Synth. Catal. 2012, 354, 515–526. [Google Scholar] [CrossRef]
- Novacek, J.; Waser, M. Synthesis and Applications of (Thio)Urea-Containing Chiral Quaternary Ammonium Salt Catalysts. Eur. J. Org. Chem. 2014, 2014, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Miyake, A.; Akiyama, T. Enantioselective Fluorination of β-Ketoesters Catalyzed by Chiral Sodium Phosphate: Remarkable Enhancement of Reactivity by Simultaneous Utilizationof Metal Enolate and Metal Phosphate. Chem. Lett. 2014, 43, 137–139. [Google Scholar] [CrossRef]
- Lehmann, F. Chemical Constitution and Activity. Aromatic Fluorine Compounds. Arch. Exp. Pathol. Pharmakol. 1928, 130, 250. [Google Scholar] [CrossRef]
- Yagupol’skii, L.M.; Kondratenko, N.V.; Timofeeva, G.N. Fluoro(trifluoromethyl)aryl- and (trifluoromethyl)diarylsulfonium Salts. Zhurnal Org. Khimii 1984, 20, 115–118. [Google Scholar]
- Umemoto, T.; Ishihara, S. Power-variable Electrophilic Trifluoromethylating Agents. S-, Se-, and Te-(trifluoromethyl)dibenzothio-, -Seleno-, and -Tellurophenium Salt System. J. Am. Chem. Soc. 1993, 115, 2156–2164. [Google Scholar] [CrossRef]
- Ma, J.-A.; Cahard, D. Mild Electrophilic Trifluoromethylation of β-Ketoesters and Silyl Enol Ethers with 5-Trifluoro Methyldibenzothiophenium Tetrafluoroborate. J. Org. Chem. 2003, 68, 8726. [Google Scholar] [CrossRef] [PubMed]
- Granados, A.; Rivilla, I.; Cossío, F.P.; Vallribera, A. Lanthanum-Catalyzed Enantioselective Trifluoromethylation by Using an Electrophilic Hypervalent Iodine Reagent. Chem. Eur. J. 2019, 25, 8214–8218. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.A.; Cahard, D. Strategies for Nucleophilic, Electrophilic, and Radical Trifluoromethylations. J. Fluor. Chem. 2007, 128, 975–996. [Google Scholar] [CrossRef]
- Noritake, S.; Shibata, N.; Nomura, Y.; Huang, Y.; Matsnev, A.; Nakamura, S.; Toru, T.; Cahard, D. Enantioselective Electrophilic Trifluoromethylation of β-Keto esters with Umemoto Reagents Induced by Chiral Nonracemic Guanidines. Org. Biomol. Chem. 2009, 7, 3599–3604. [Google Scholar] [CrossRef]
- Woźniak, L.; Murphy, J.J.; Melchiorre, P. Photo-organocatalytic Enantioselective Perfluoroalkylation of β-Ketoesters. J. Am. Chem. Soc. 2015, 137, 5678–5681. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhang, W.; Li, Y.-H.; Xue, X.-S.; Li, X.; Cheng, J.-P. Origin of Stereoselectivity of the Photoinduced Asymmetric Phase-Transfer-Catalyzed Perfluoroalkylation of β-Ketoesters. J. Org. Chem. 2017, 82, 9321–9327. [Google Scholar] [CrossRef]
- Toulgoat, F.; Billard, T. Toward CF3S Group: From Trifluoromethylation of Sulfides to Direct Trifluoromethylthiolation. In Modern Synthesis Processes and Reactivity of Fluorinated Compounds; Groults, H., Leroux, F., Tressaud, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 141–179. [Google Scholar]
- Deng, Q.-H.; Rettenmeier, C.; Wadepohl, H.; Gade, L.H. Copper–Boxmi Complexes as Highly Enantioselective Catalysts for Electrophilic Trifluoromethylthiolations. Chem. Eur. J. 2014, 20, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.-H.; Bleith, T.; Wadepohl, H.; Gade, L.H. Enantioselective Iron-Catalyzed Azidation of β-Keto Esters and Oxindoles. J. Am. Chem. Soc. 2013, 135, 5356–5359. [Google Scholar] [CrossRef]
- Shao, X.; Wang, X.; Yang, T.; Lu, L.; Shen, Q. An Electrophilic Iodine Reagent for Trifluoromethylthiolation. Angew. Chem. Int. Ed. 2013, 52, 3457–3460. [Google Scholar] [CrossRef]
- Wang, X.; Yang, T.; Cheng, X.; Shen, Q. Enantioselective Electrophilic Trifluoromethylthiolation of β-Ketoesters: A Case of Reactivity and Selectivity Bias for Organocatalysis. Angew. Chem. Int. Ed. 2013, 52, 12860–12864. [Google Scholar] [CrossRef]
- Bootwicha, T.; Liu, X.; Pluta, R.; Atodiresei, I.; Rueping, M. N-Trifluoromethylthiophthalimide: A Stable Electrophilic SCF3-Reagent and its Application in the Catalytic Asymmetric Trifluoromethylsulfenylation. Angew. Chem. Int. Ed. 2013, 52, 12856–12859. [Google Scholar] [CrossRef]
- Li, M.; Xue, X.-S.; Cheng, J.-P. Mechanism and Origins of Stereoinduction in Natural Cinchona Alkaloid Catalyzed Asymmetric Electrophilic Trifluoromethylthiolation of β-Keto Esters with N-Trifluoromethylthiophthalimide as Electrophilic SCF3 Source. ACS Catal. 2017, 7, 7977–7986. [Google Scholar] [CrossRef]
- Grayson, M.N.; Houk, K.N.J. Cinchona Alkaloid-Catalyzed Asymmetric Conjugate Additions: The Bifunctional Brønsted Acid–Hydrogen Bonding Model. J. Am. Chem. Soc. 2016, 138, 1170–1173. [Google Scholar] [CrossRef] [PubMed]
- Hiemstra, H.; Wynberg, H. Addition of Aromatic Thiols to Conjugated Cycloalkenones, Catalyzed by Chiral/3-HydroxyAmines. A Mechanistic Study on Homogeneous Catalytic Asymmetric Synthesis. J. Am. Chem. Soc. 1981, 103, 417–430. [Google Scholar] [CrossRef]
- Zhao, B.-L.; Du, D.-M. Enantioselective Squaramide-Catalyzed Trifluoromethylthiolation− Sulfur−Michael/Aldol Cascade Reaction: One-Pot Synthesis of CF3S-Containing Spiro Cyclopentanone−Thiochromanes. Org. Lett. 2017, 19, 1036–1039. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Leng, X.; Wan, X.; Shen, Q. (1S)-(−)-N-Trifluoromethylthio-2,10-camphorsultam and its Derivatives: Easily Available, Optically Pure Reagents for Asymmetric Trifluoromethylthiolation. Org. Chem. Front. 2017, 4, 1051–1057. [Google Scholar] [CrossRef]

























© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Granados, A.; Vallribera, A. Asymmetric Preparation of α-Quaternary Fluorinated β-keto Esters. Review. Molecules 2020, 25, 3264. https://doi.org/10.3390/molecules25143264
Granados A, Vallribera A. Asymmetric Preparation of α-Quaternary Fluorinated β-keto Esters. Review. Molecules. 2020; 25(14):3264. https://doi.org/10.3390/molecules25143264
Chicago/Turabian StyleGranados, Albert, and Adelina Vallribera. 2020. "Asymmetric Preparation of α-Quaternary Fluorinated β-keto Esters. Review" Molecules 25, no. 14: 3264. https://doi.org/10.3390/molecules25143264
APA StyleGranados, A., & Vallribera, A. (2020). Asymmetric Preparation of α-Quaternary Fluorinated β-keto Esters. Review. Molecules, 25(14), 3264. https://doi.org/10.3390/molecules25143264