
Continuous Flow Synthesis of Heterocycles: A Recent Update on the Flow Synthesis of Indoles




Abstract
{kind=link}
{kind=link}
{kind=link}
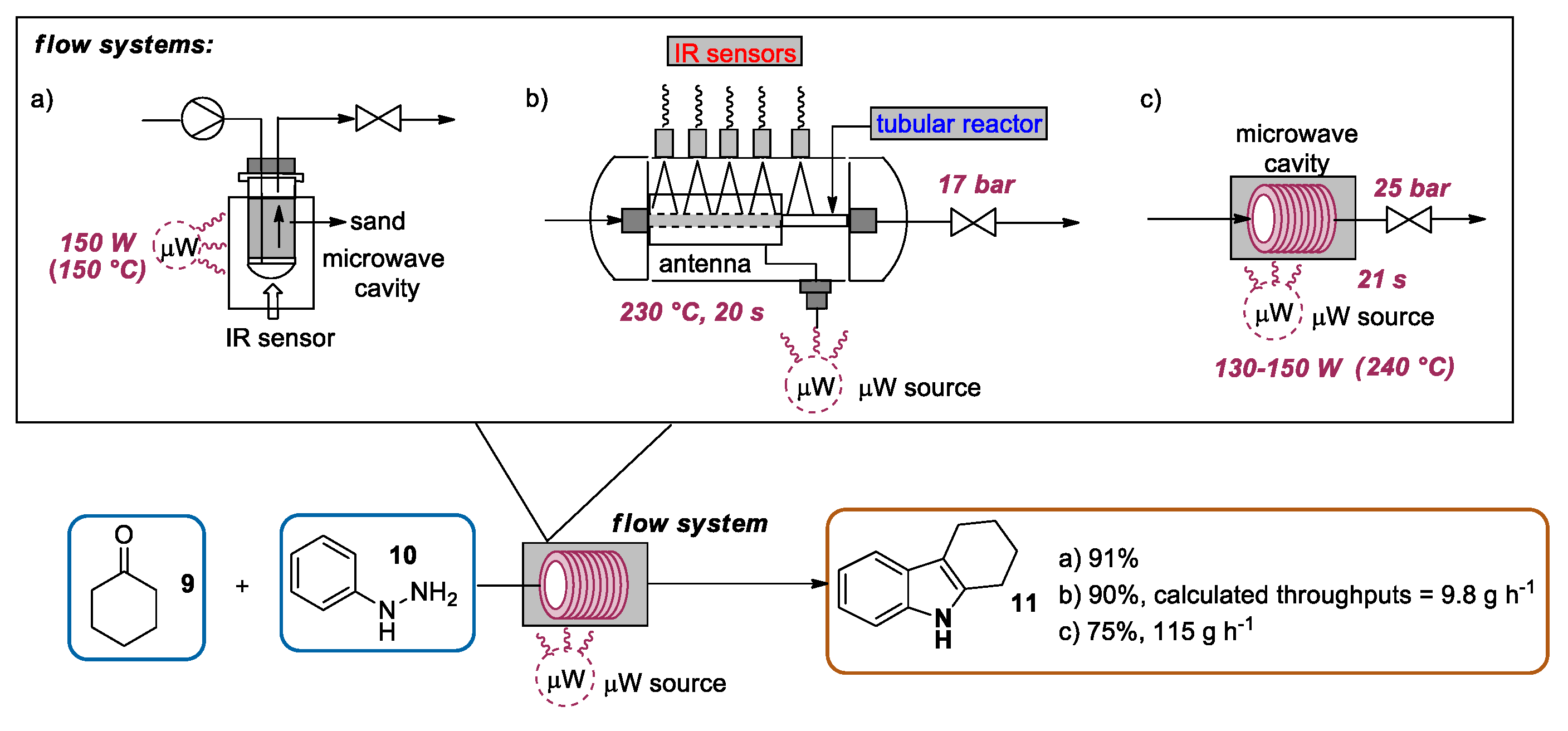{kind=link}
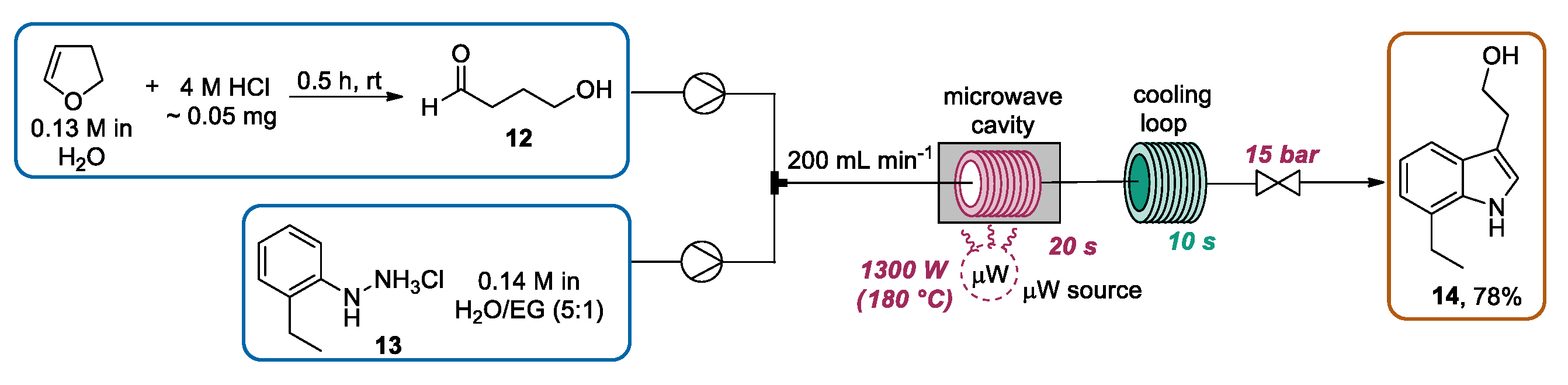{kind=link}
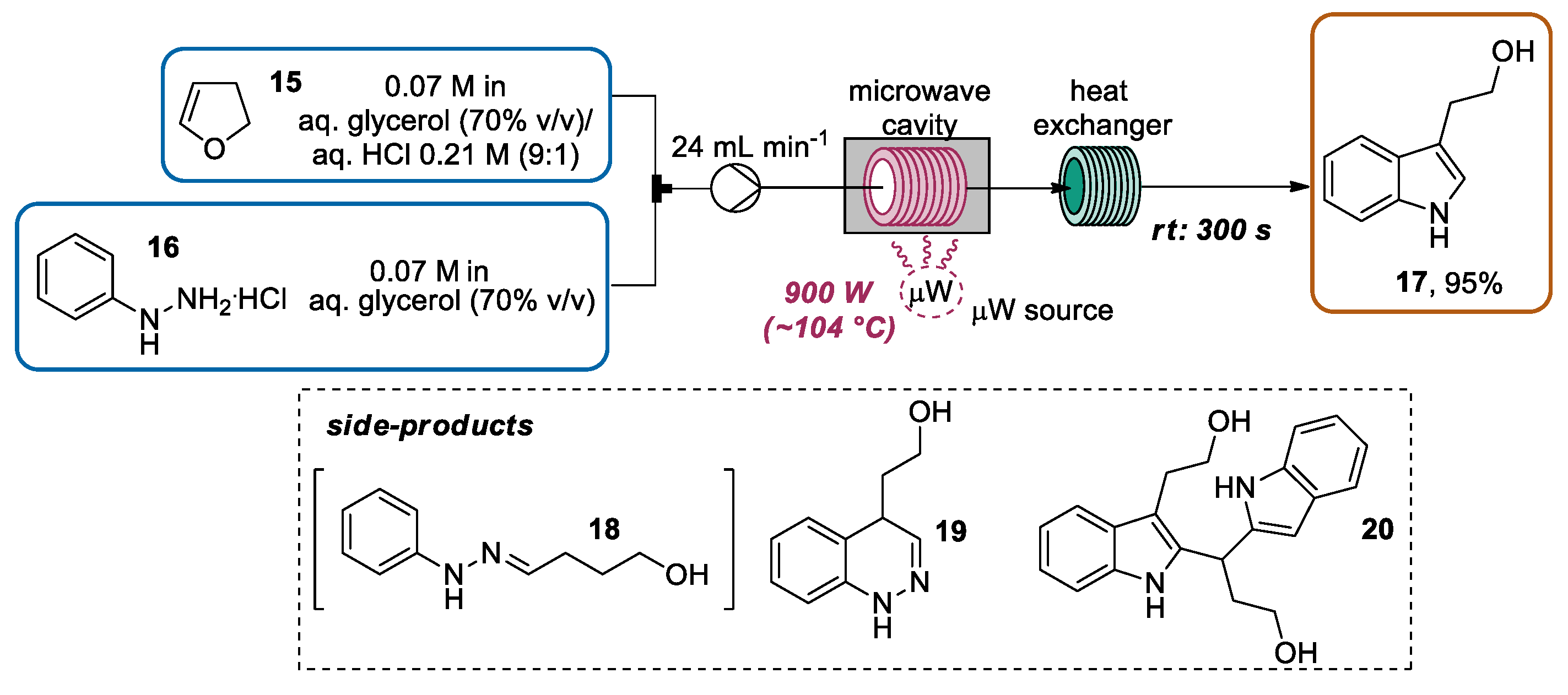{kind=link}
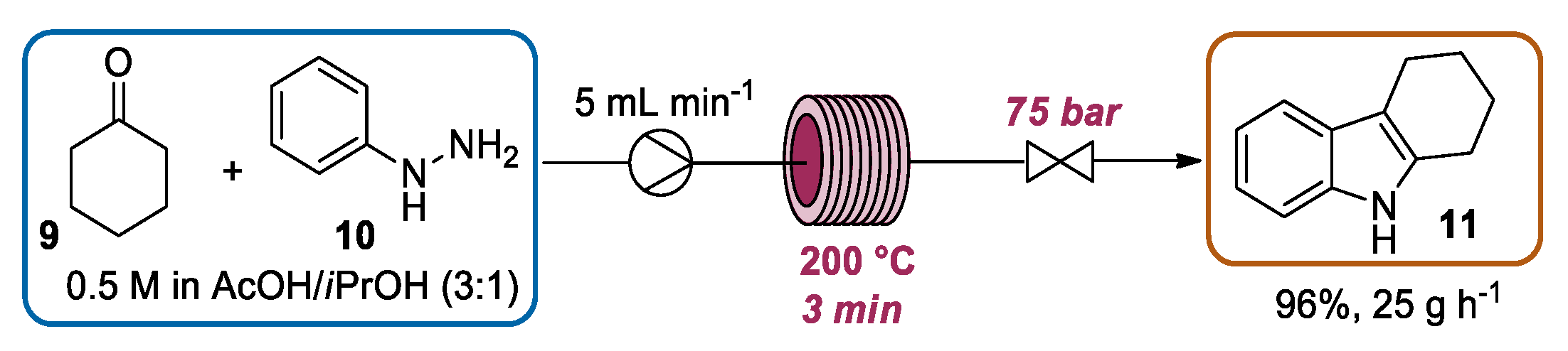{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
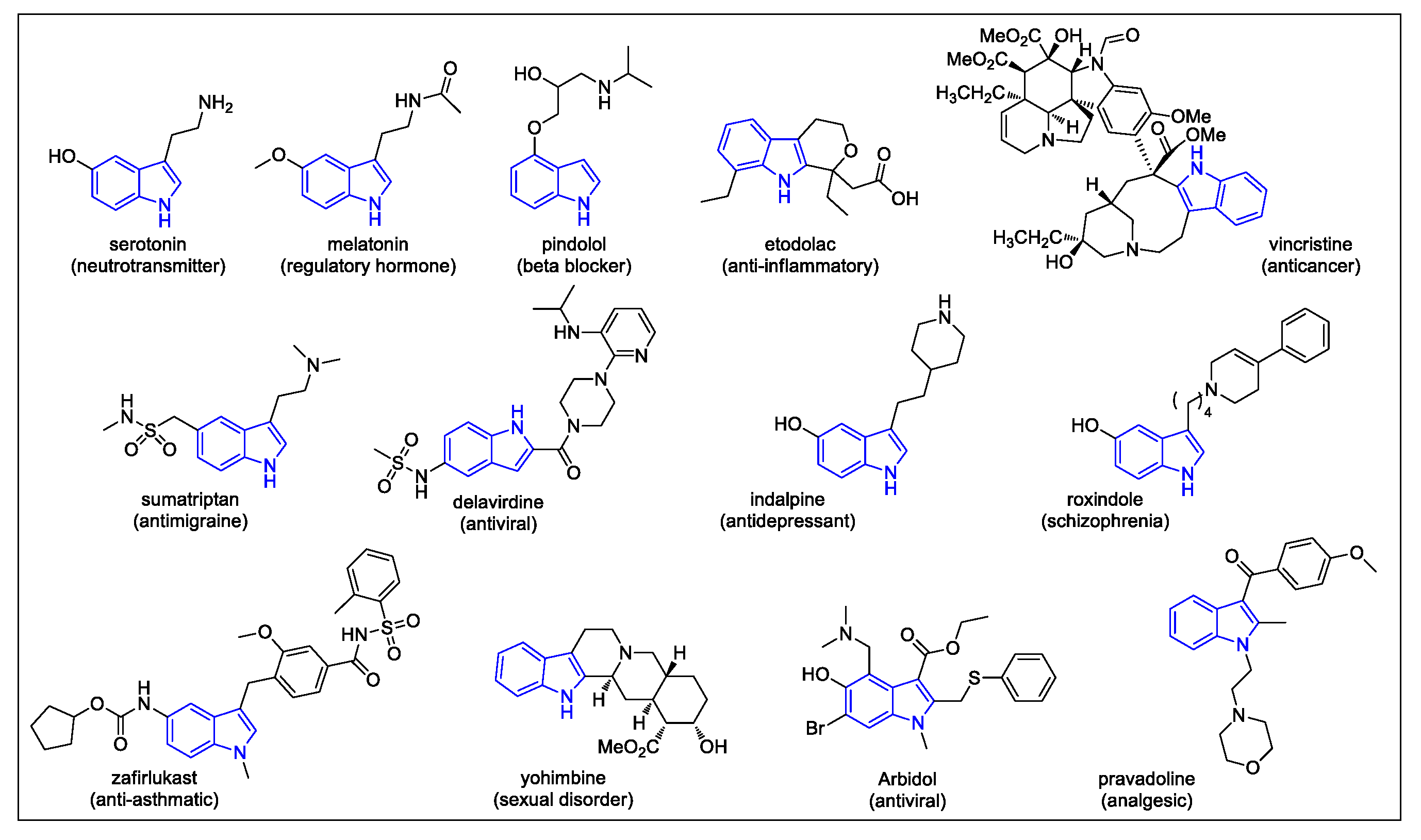
1. Introduction
2. Synthesis of Indole Ring under Continuous Flow Conditions
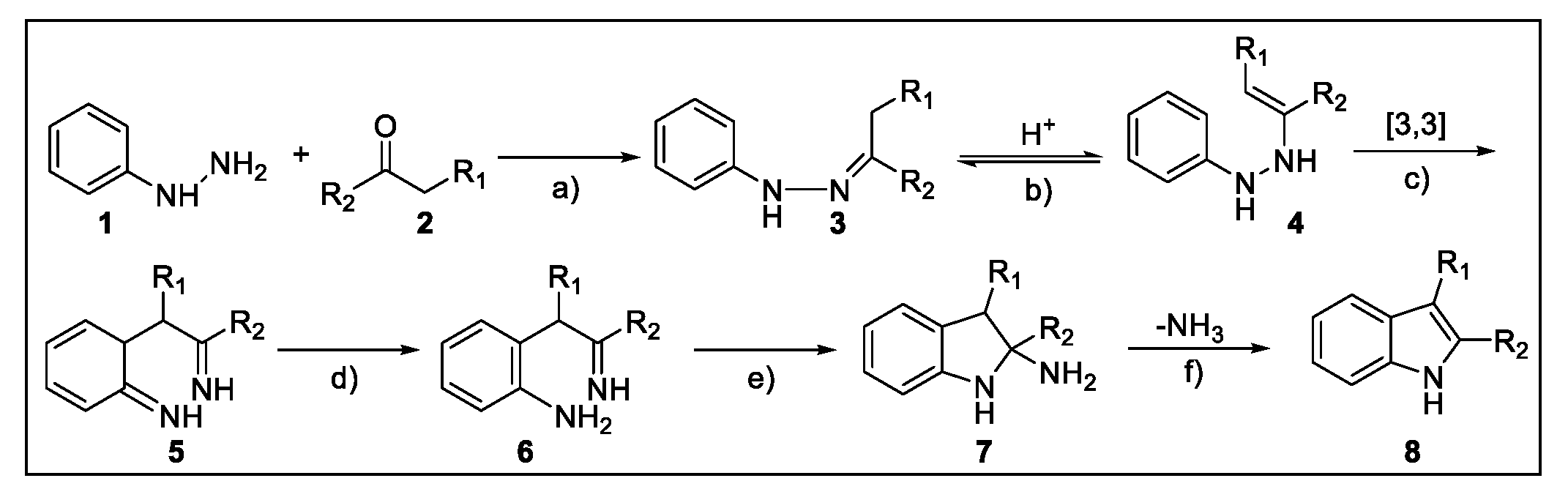
2.1. Fischer Indole Synthesis
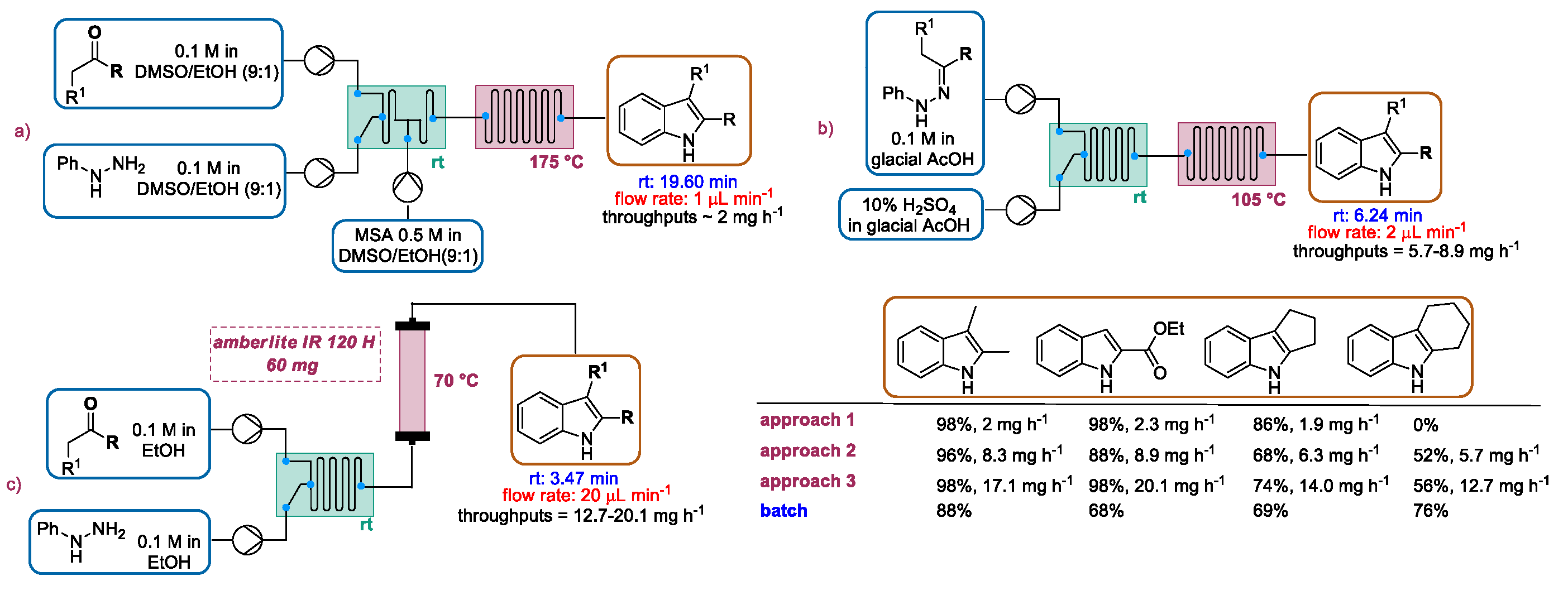
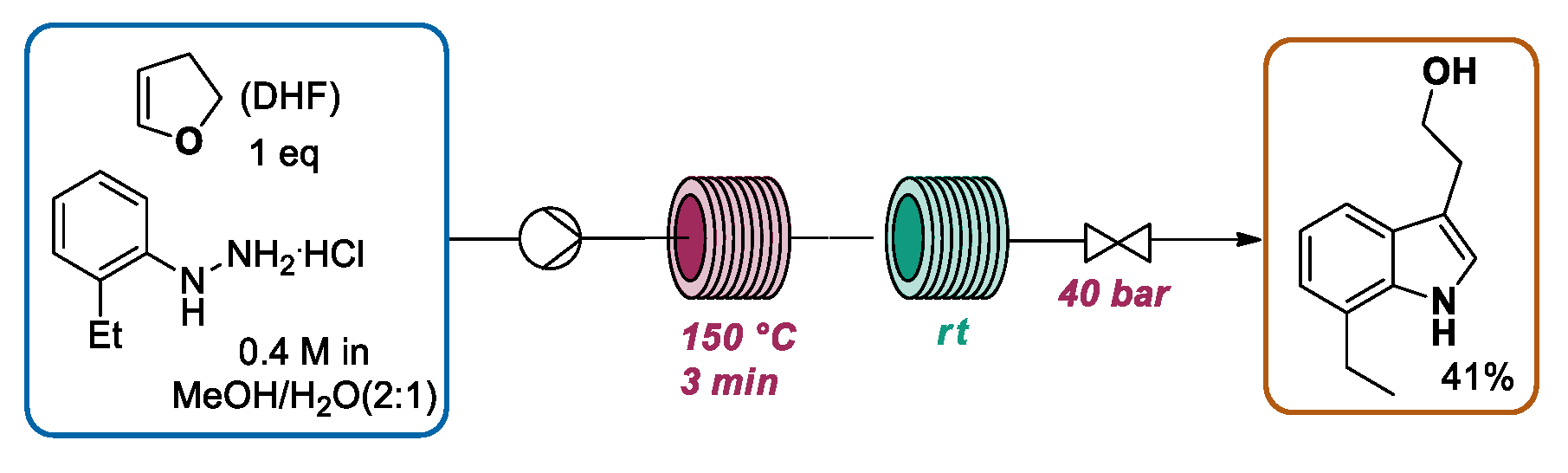
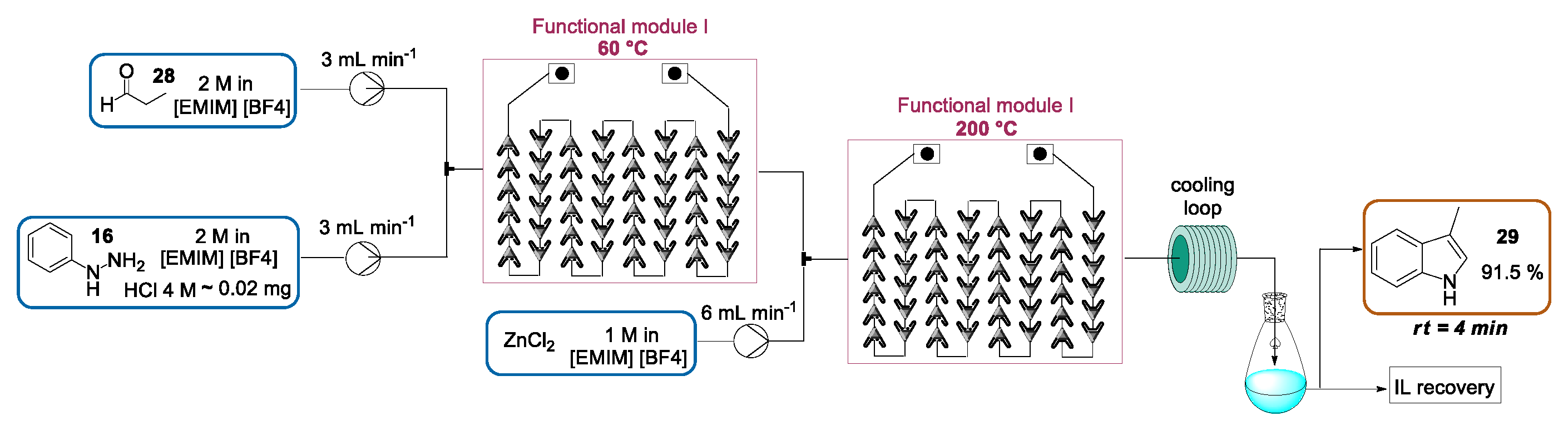
2.1.1. Continuous Flow Microwave-Assisted Fischer Indolization
2.1.2. Continuous Flow Fischer Indole Synthesis Mediated by Conventional Heating
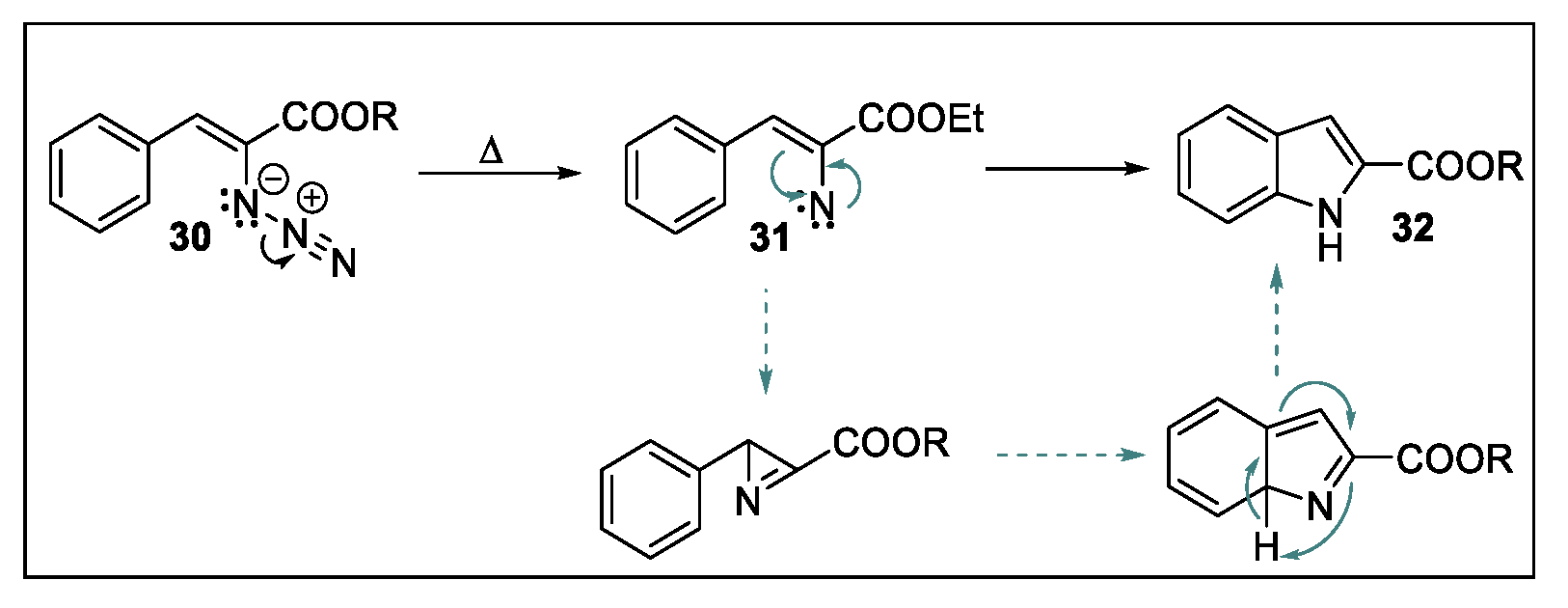
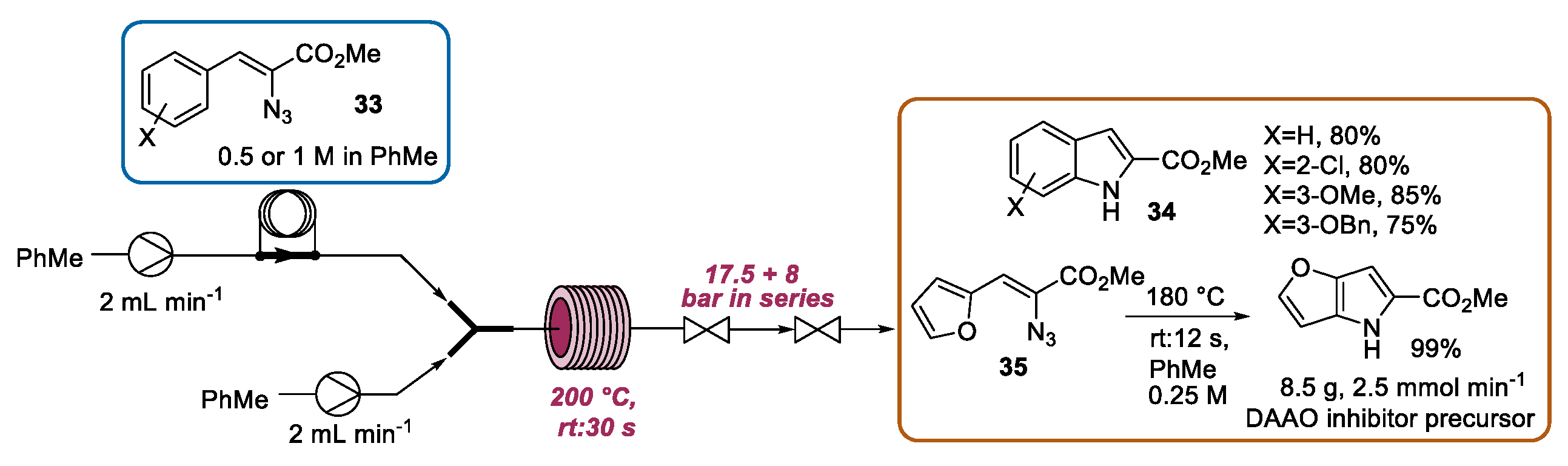
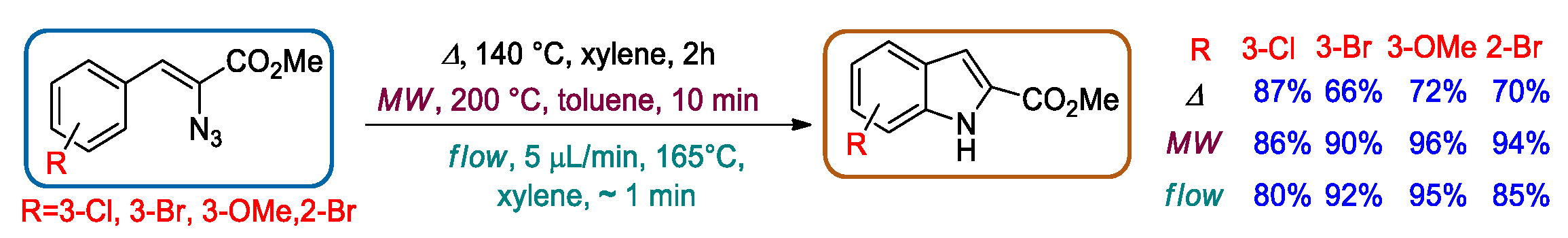
2.2. Hemetsberger-Knittel Indole Synthesis under Continuous Flow Conditions
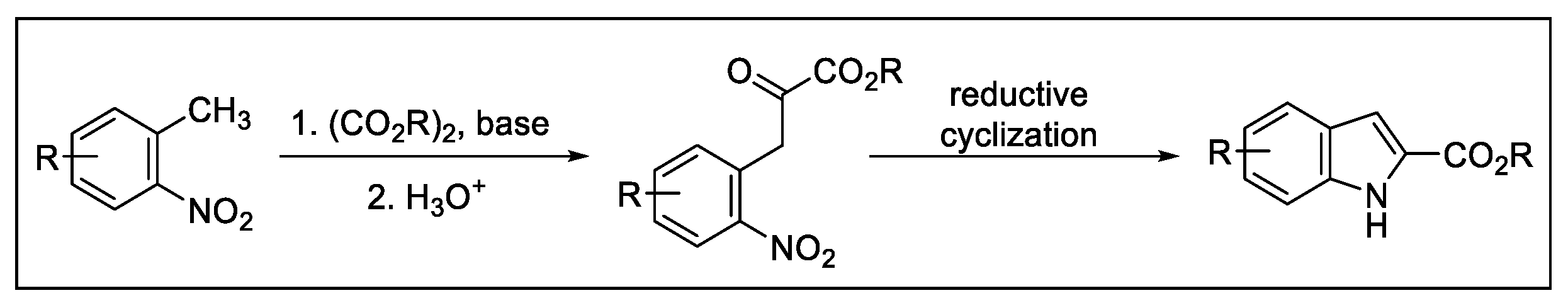
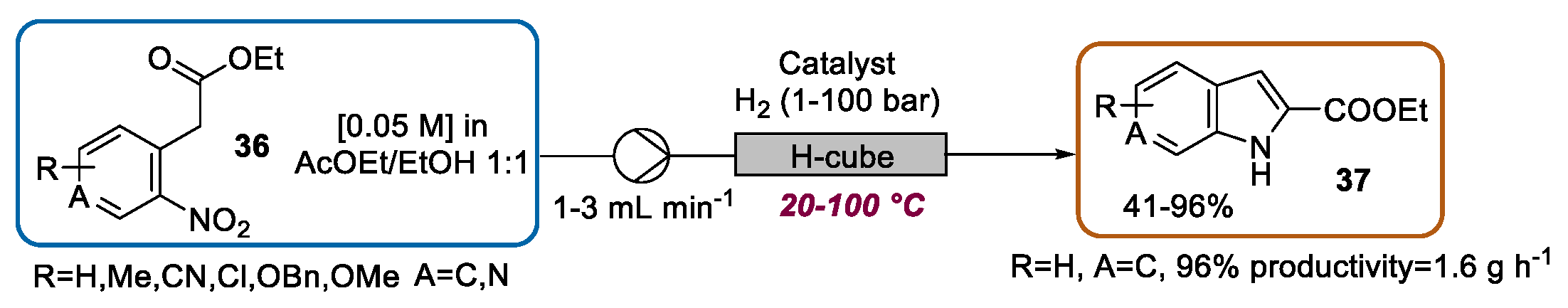
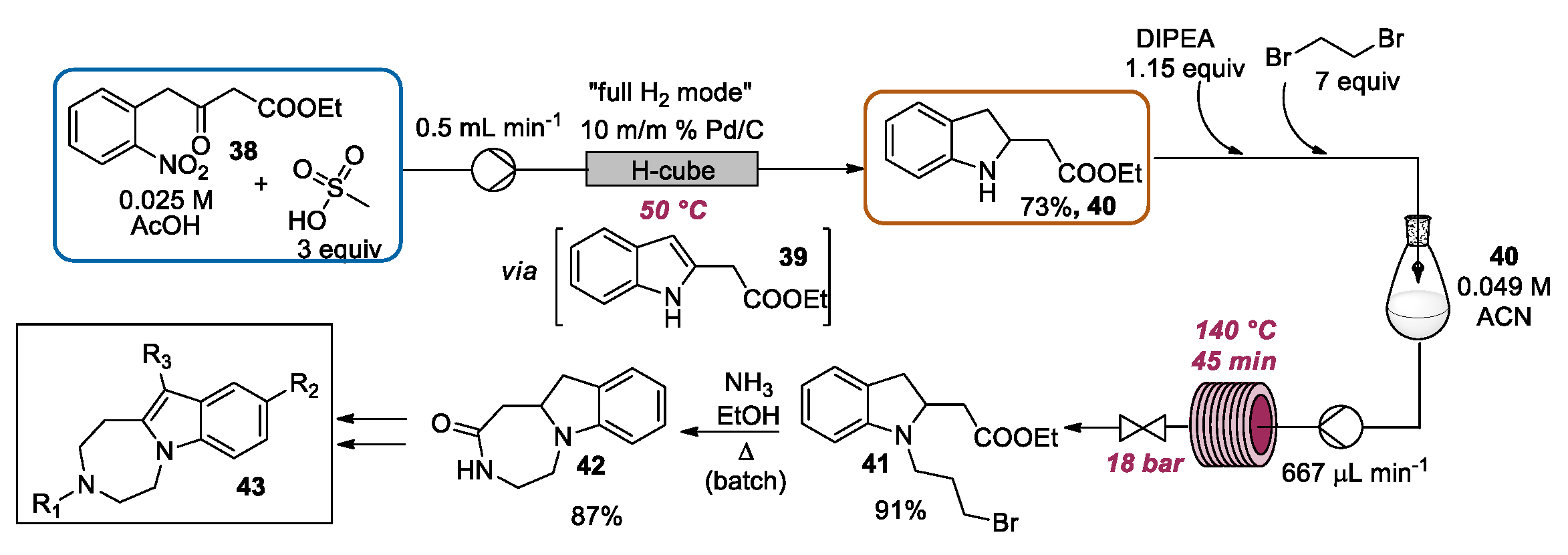
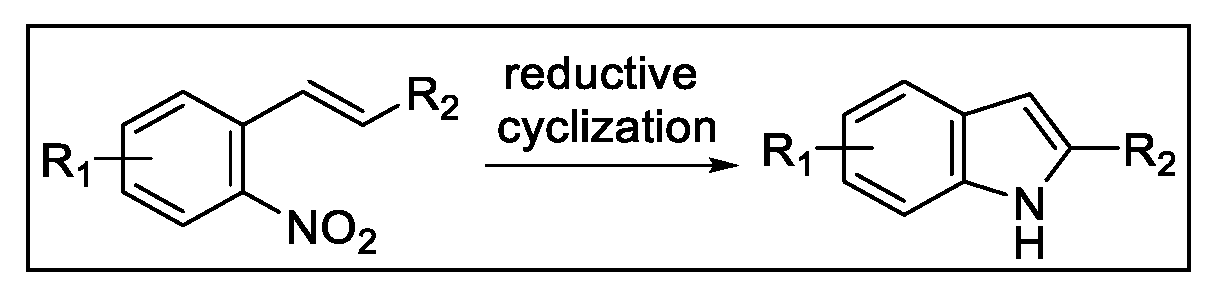
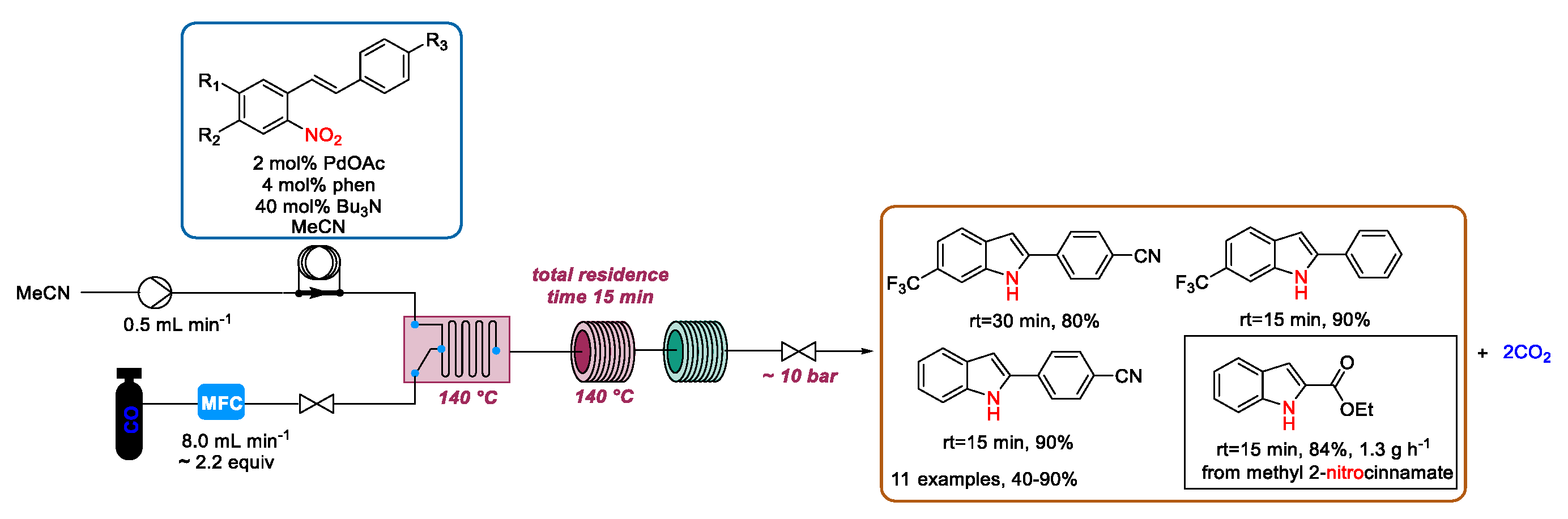
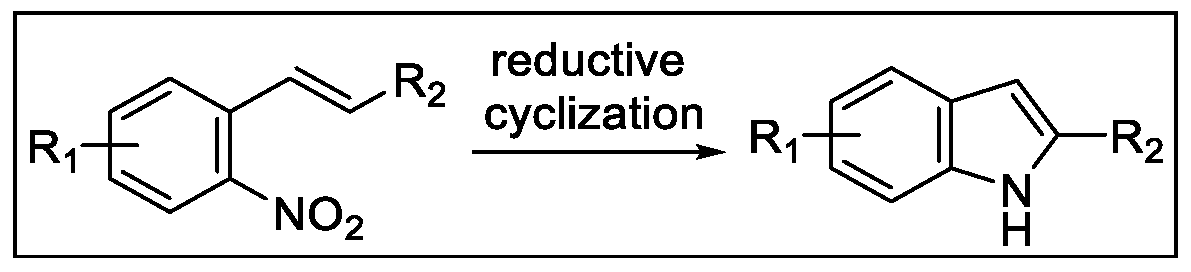
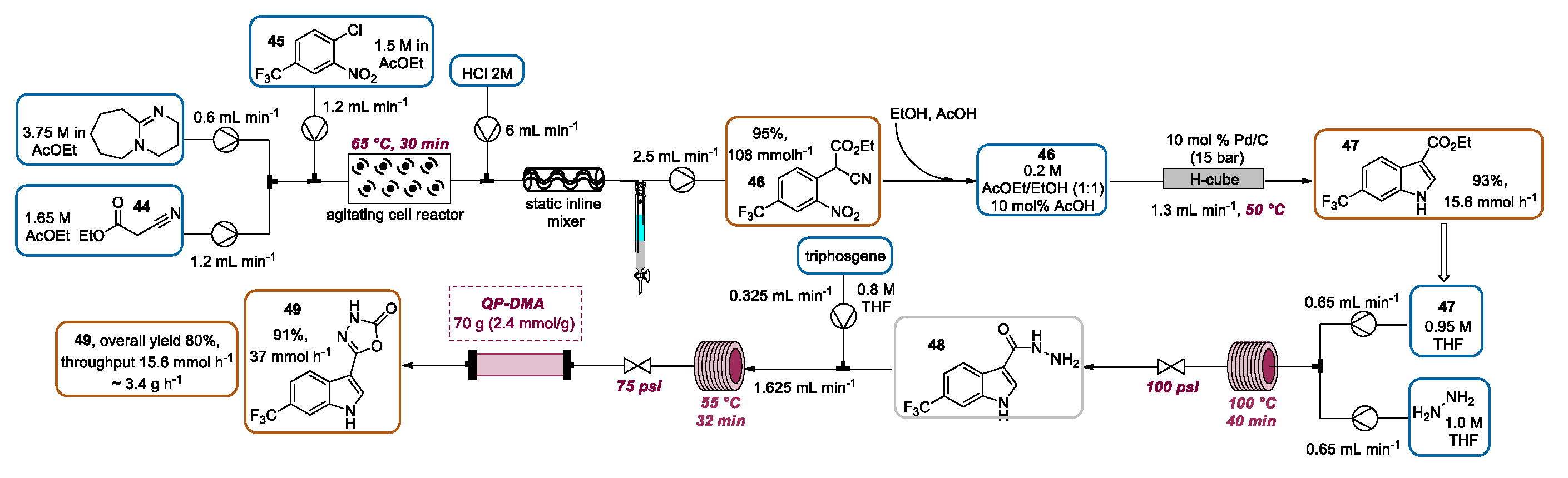
2.3. Indolization by Reductive Cyclization of Nitro Compounds
2.4. Heumann Indole Synthesis
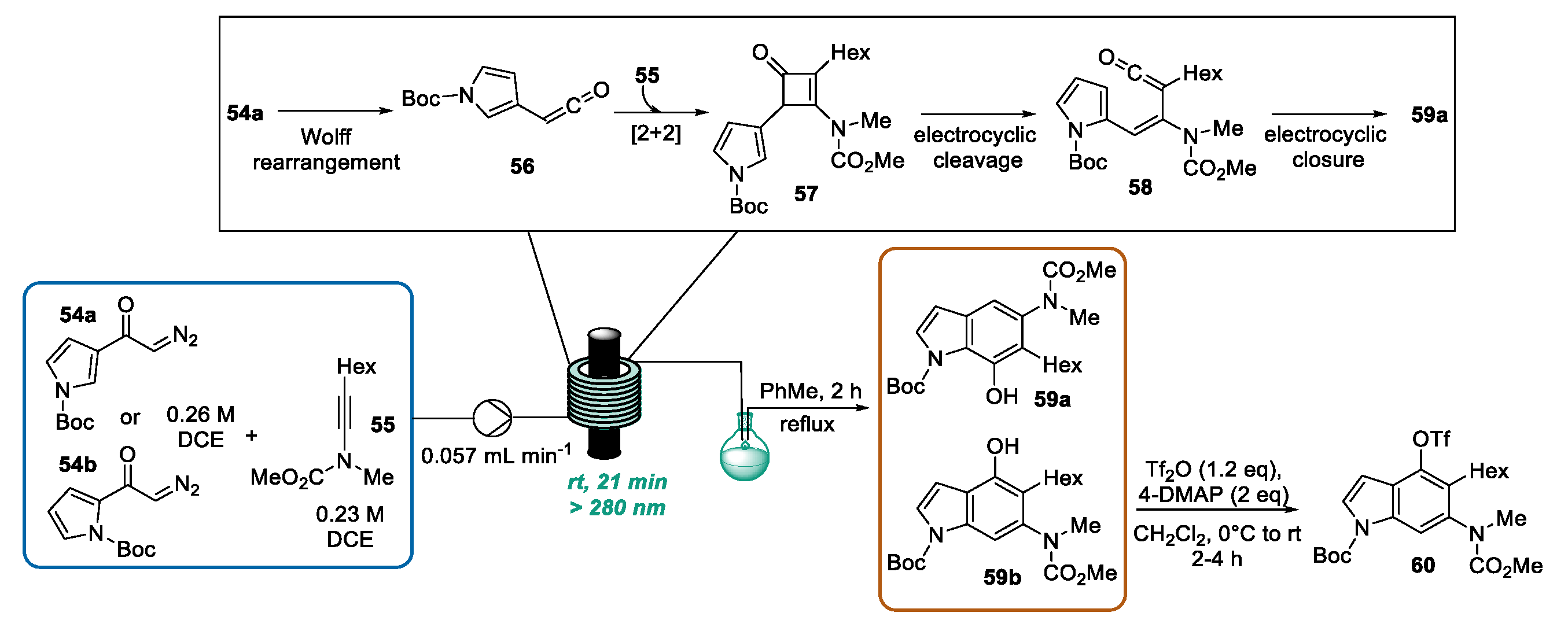
2.5. Indole Synthesis through Photochemical Benzannulation
3. Derivatization of Indole Ring under Continuous Flow Conditions
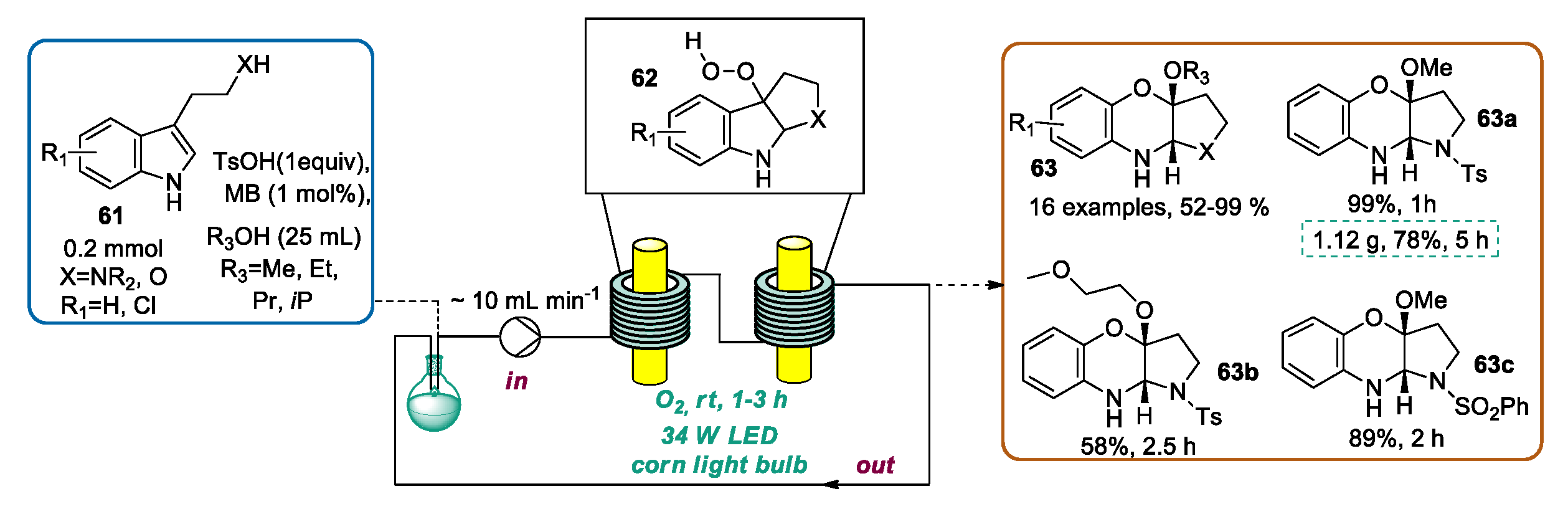
3.1. Indole Ring Derivatization Mediated by Visible Light-Photoredox Catalysis in Flow Mode
3.2. Continuous Flow Functionalization of Indole Nitrogen
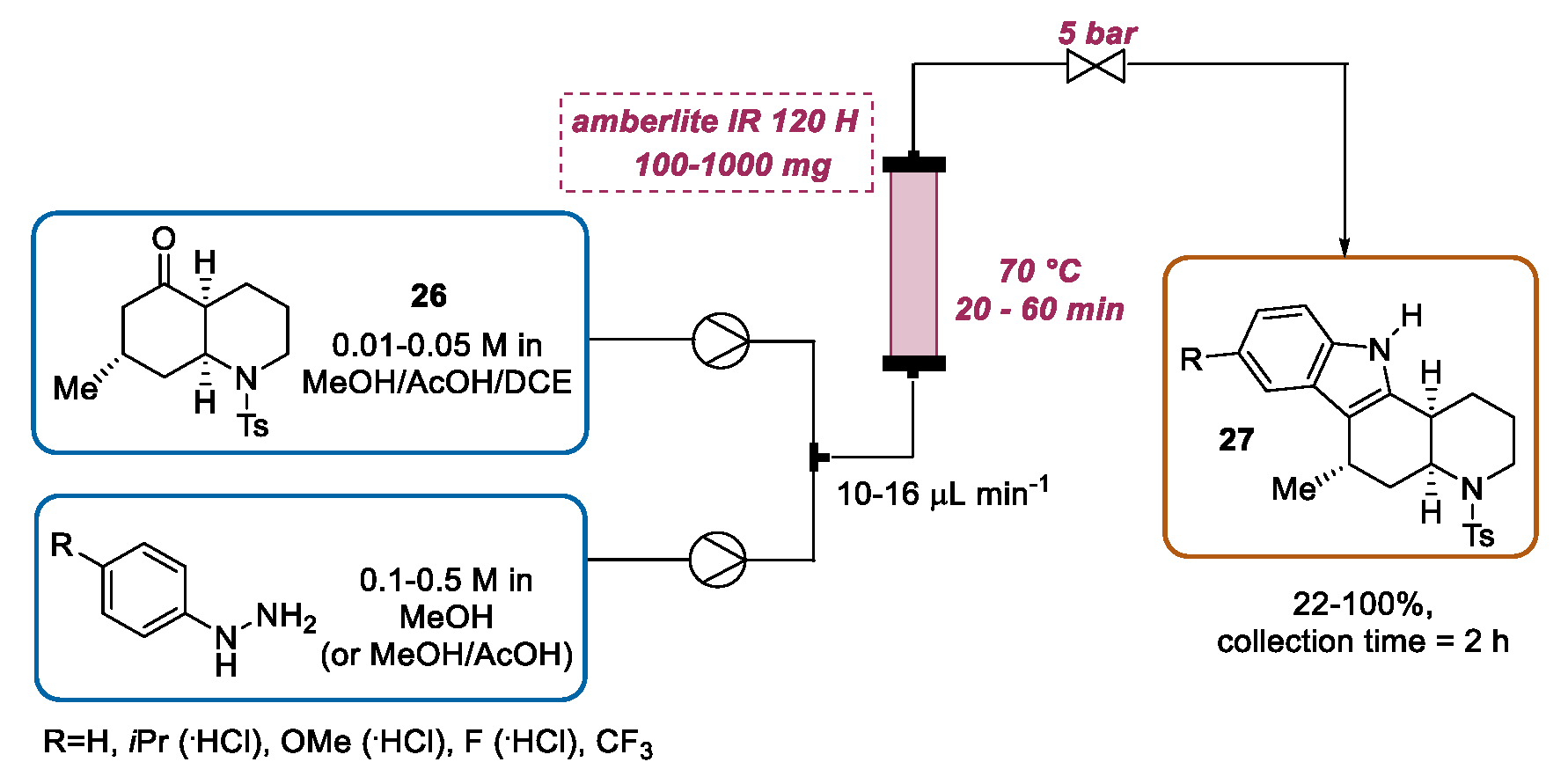
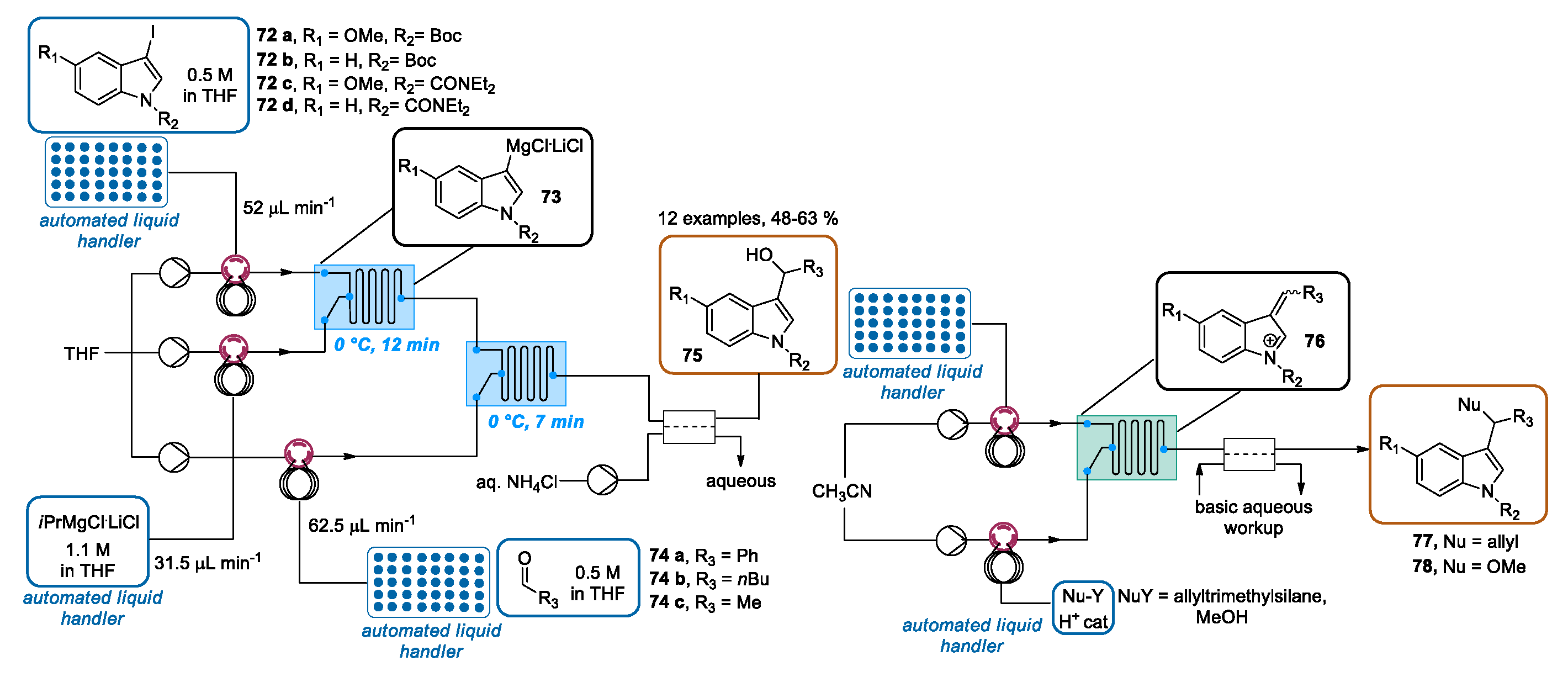
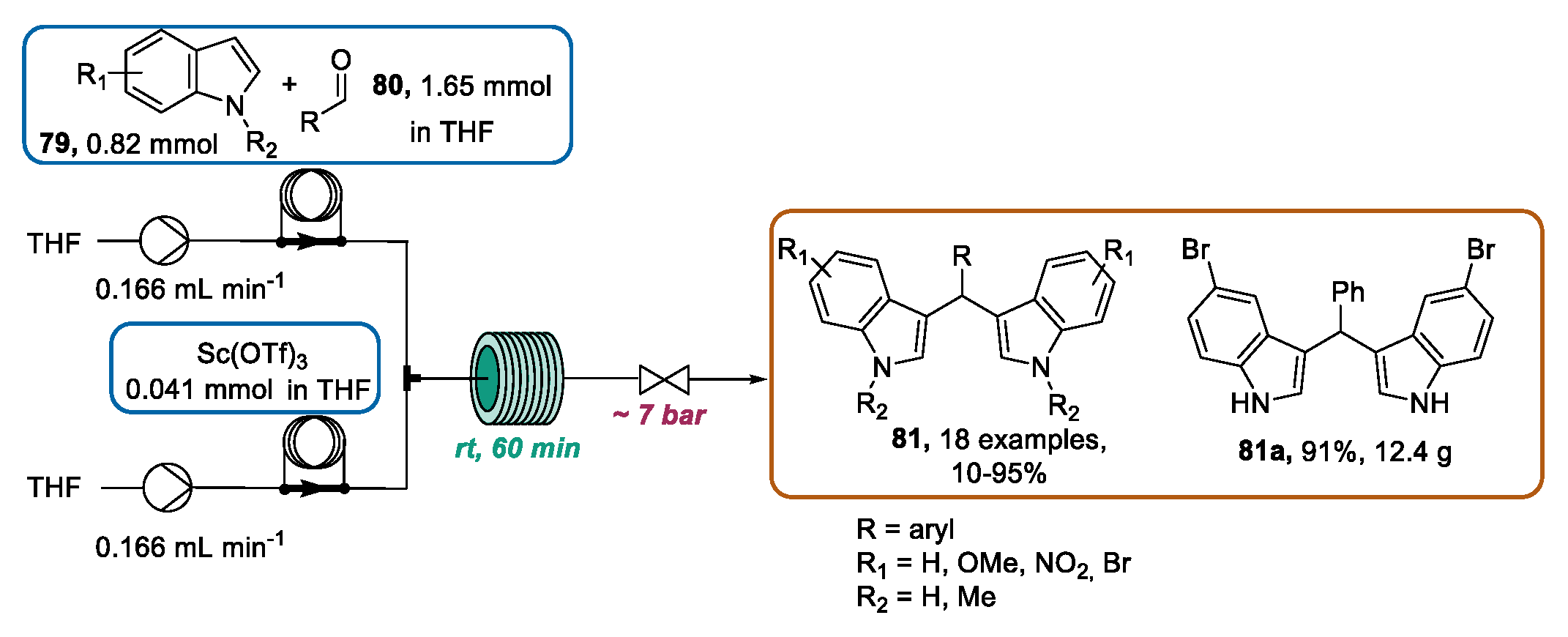
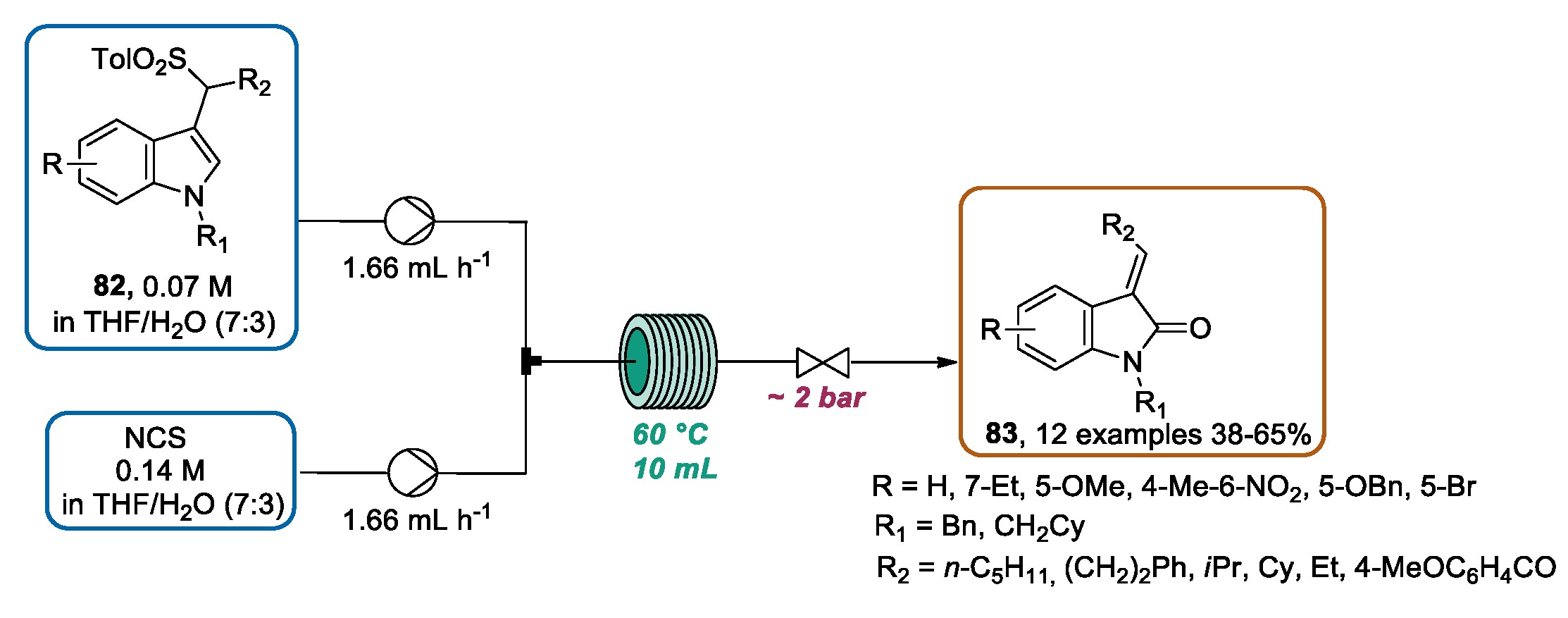
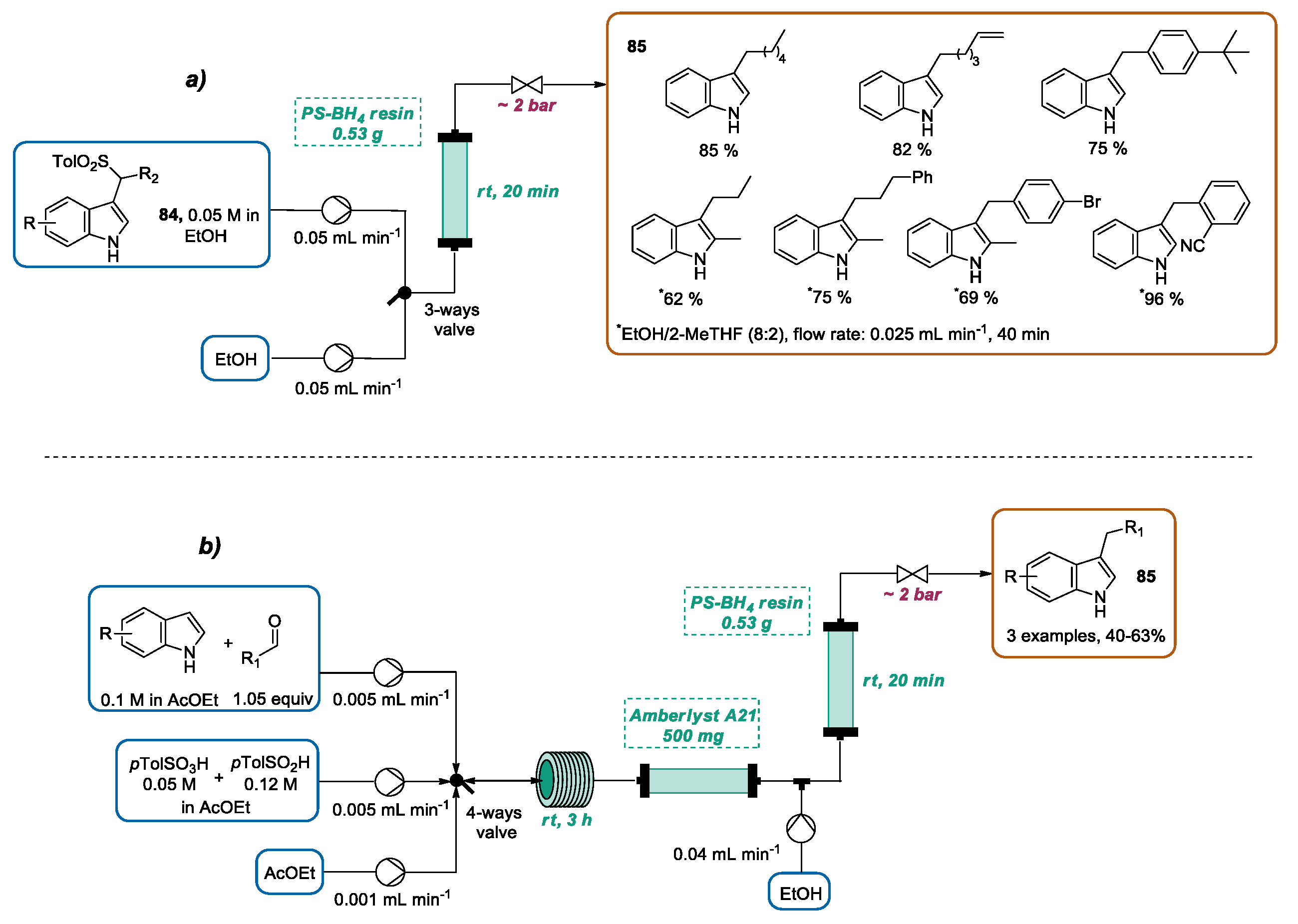
3.3. Continuous Flow Functionalization of C-3 Position of Indole Ring
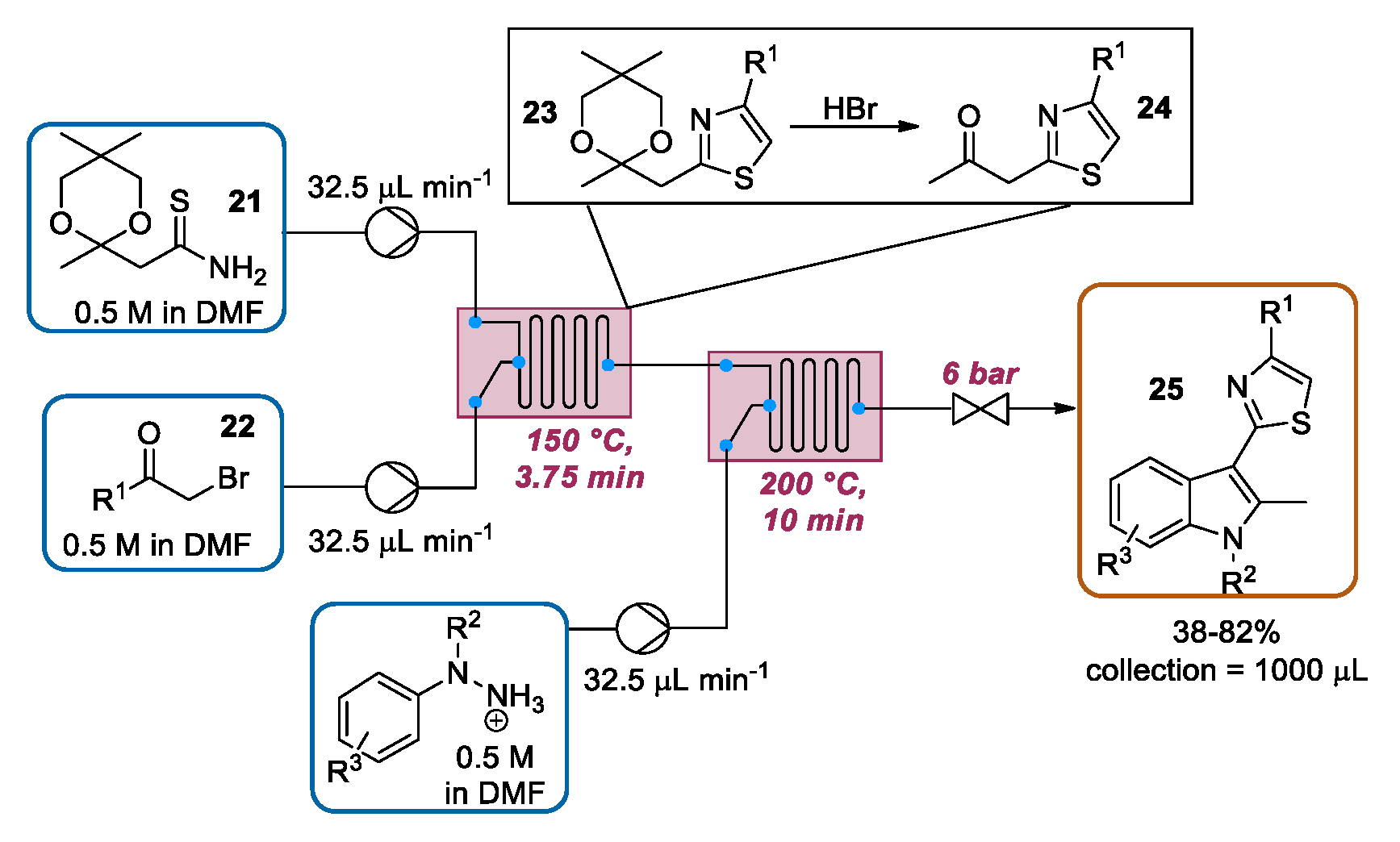
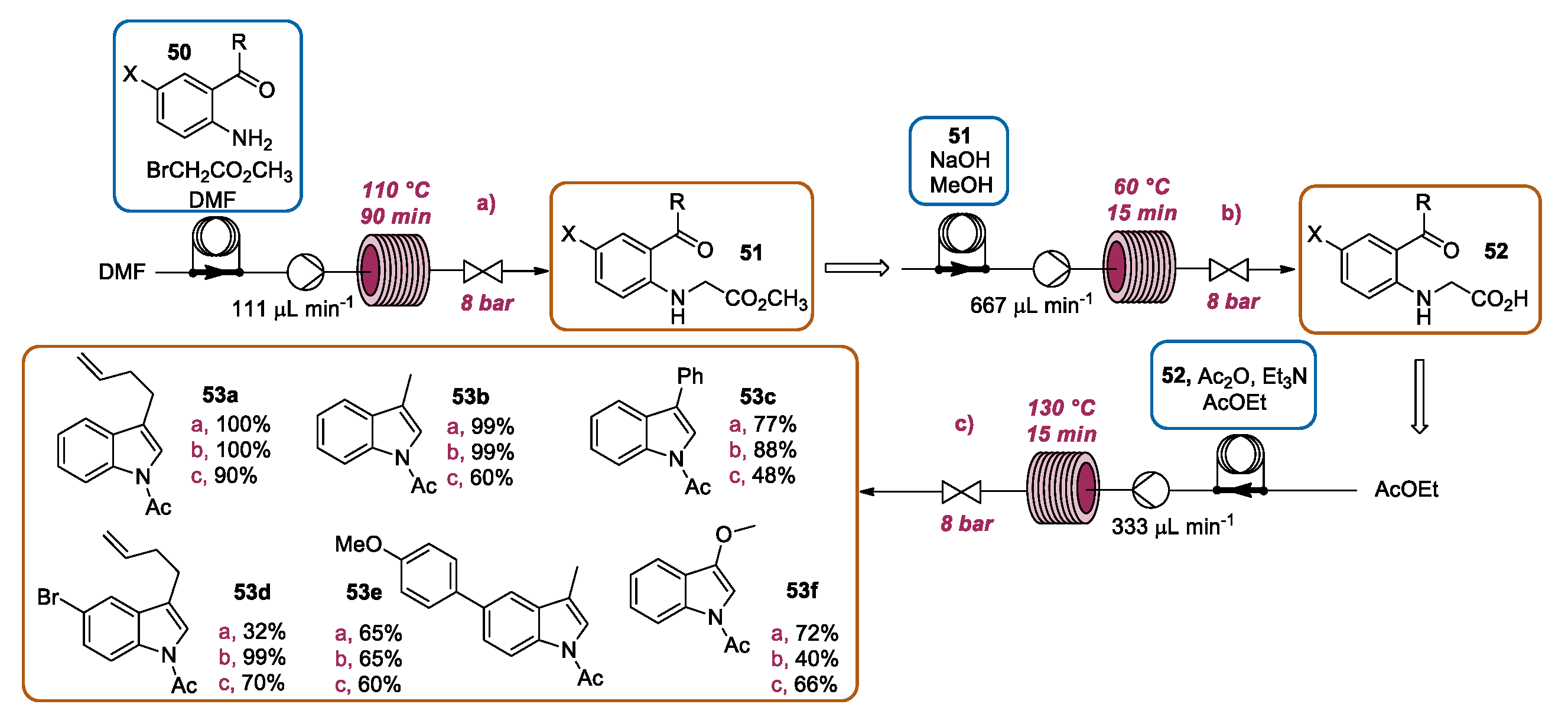
3.4. Continuous Flow Functionalization of C-2 Position of Indole Ring
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Vicente, R. Recent advances in indole syntheses: New routes for a classic target. Org. Biomol. Chem. 2011, 9, 6469–6480. [Google Scholar] [CrossRef] [PubMed]
- Gribble, G.W. Indole Ring Synthesis: From Natural Products to Drug Discovery; Wiley-VCH: Chichester, UK, 2016. [Google Scholar]
- Kaushik, N.K.; Kaushik, N.; Attri, P.; Kumar, N.; Kim, C.H.; Verma, A.K.; Choi, E.H. Biomedical importance of indoles. Molecules 2013, 18, 6620–6662. [Google Scholar] [CrossRef] [PubMed]
- Ghinea, I.O.; Dinica, R.D. Breakthoughts in Indole and Indolizine Chemistry, New Synthetic Pathways, New Applications. In Scope of Selective Heterocycles from Organic and Pharmaceutical Perspective; Varala, V., Ed.; Intech Open: Rijeka, Croatia, 2016; Chapter 5; pp. 1043–1561. [Google Scholar]
- Zhang, M.Z.; Chen, Q.; Yang, G.-F. A review on recent developments of indole-containing antiviral agents. Eur. J. Med. Chem. 2015, 89, 421–441. [Google Scholar] [CrossRef] [PubMed]
- Baeyer, A. Ueber die beziehungen der zimmtsäure zu der indigogruppe. Chem. Ber. 1880, 13, 2254–2263. [Google Scholar] [CrossRef]
- Colella, M.; Carlucci, C.; Luisi, R. Supported catalysts for continuous flow synthesis. Top. Curr. Chem. 2018, 376, 46. [Google Scholar] [CrossRef]
- Gutmann, B.; Cantillo, D.; Kappe, C.O. Continuous-flow technology—A tool for the safe manufacturing of active pharmaceutical ingredients. Angew. Chem. Int. Ed. 2015, 54, 6688–6728. [Google Scholar] [CrossRef]
- Fanelli, F.; Parisi, G.; Degennaro, L.; Luisi, R. Contribution of microreactor technology and flow chemistry to the development of green and sustainable synthesis. Beilstein J. Org. Chem. 2017, 13, 520–542. [Google Scholar] [CrossRef]
- Gioiello, A.; Piccinno, A.; Lozza, A.M.; Cerra, B. The medicinal chemistry in the era of machines and automation: Recent advances in continuous flow technology. J. Med. Chem. 2020, 63, 6624–6647. [Google Scholar] [CrossRef]
- Bloemendal, V.R.L.J.; Janssen, M.A.C.H.; van Hest, J.C.M.; Rutjes, F.P.J.T. Continuous one-flow multi-step synthesis of active pharmaceutical ingredients. React. Chem. Eng. 2020. [Google Scholar] [CrossRef]
- Colella, M.; Nagaki, A.; Luisi, R. Flow technology for the genesis and use of (highly) reactive organometallic reagents. Chem. Eur. J. 2020, 26, 19–32. [Google Scholar] [CrossRef]
- Guttman, B.; Kappe, C.O. Forbidden chemistries-paths to a sustainable future engaging continuous processing. J. Flow Chem. 2017, 7, 65–71. [Google Scholar] [CrossRef]
- Fischer, E.; Jourdan, F. Ueber die Hydrazine der Brenztraubensäure. Ber. Dtsch. Chem. Ges. 1883, 16, 2241–2245. [Google Scholar] [CrossRef]
- Heravi, M.M.; Rohani, S.; Zadsirjan, V.; Zahedi, N. Fischer indole synthesis applied to the total synthesis of natural products. RSC Adv. 2017, 7, 52852–52887. [Google Scholar] [CrossRef]
- Humphrey, G.R.; Kuethe, J.T. Practical methodologies for the synthesis of indoles. Chem. Rev. 2006, 106, 2875–2911. [Google Scholar] [CrossRef] [PubMed]
- Hughes, D.L. Progress in the Fischer indole reaction. A review. Org. Prep. Proced. Int. 1993, 25, 607–632. [Google Scholar] [CrossRef]
- Przheval’ski, L.; Kostromina, Y.; Grandberg, I.I. New data on the mechanism of the Fischer indole synthesis (review). Chem. Heterocycl. Compd. (Engl. Transl.) 1988, 24, 709–721. [Google Scholar] [CrossRef]
- Bagley, M.C.; Jenkins, R.L.; Lubinu, M.C.; Mason, C.; Wood, R. A Simple Continuous Flow Microwave Reactor. J. Org. Chem. 2005, 70, 7003–7006. [Google Scholar] [CrossRef]
- Razzaq, T.; Glasnov, T.N.; Kappe, C.O. Continuous-flow microreactor chemistry under high-temperature/pressure conditions. Eur. J. Org. Chem. 2009, 9, 1321–1325. [Google Scholar] [CrossRef]
- Gedye, R.; Smith, F.; Westaway, K.; Ali, H.; Baldisera, L.; Laberge, L.; Rousell, J. The use of microwave ovens for rapid organic synthesis. Tetrahedron Lett. 1986, 27, 279–282. [Google Scholar] [CrossRef]
- Giguere, R.J.; Bray, T.L.; Duncan, S.M.; Majetich, G. Application of commercial microwave-ovens to organic-synthesis. Tetrahedron Lett. 1986, 27, 4945–4948. [Google Scholar] [CrossRef]
- Öhrngren, P.; Fardost, A.; Russo, F.; Schanche, J.; Fagrell, M.; Larhed, M. Evaluation of a nonresonant microwave applicator for continuous-flow chemistry applications. Org. Process. Res. Dev. 2012, 16, 1053–1063. [Google Scholar] [CrossRef]
- Ferguson, J.D. CEM Corporation. Mol. Diver. 2003, 7, 281–286. Available online: http://www.cemsynthesis.com (accessed on 15 July 2020). [CrossRef] [PubMed]
- Favretto, L. Milestone Inc. Mol. Divers. 2003, 7, 287–291. Available online: http://www.milestonesci.com (accessed on 15 July 2020). [CrossRef] [PubMed]
- Schanche, J.S. Microwave synthesis solutions from personal chemistry. Mol. Divers. 2003, 7, 293–300. [Google Scholar] [CrossRef]
- Kappe, C.O.; Stadler, A. Microwaves in Organic and Medicinal Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005; Volume 25. [Google Scholar]
- Kappe, C.O. Microwave dielectric heating in synthetic organic chemistry. Chem. Soc. Rev. 2008, 37, 1127–1139. [Google Scholar] [CrossRef]
- Kappe, C.O. Controlled microwave heating in modern organic synthesis. Angew. Chem. Int. Ed. 2004, 43, 6250–6284. [Google Scholar] [CrossRef]
- Adam, D. Microwave chemistry: Out of the kitchen. Nature 2003, 421, 571–572. [Google Scholar] [CrossRef]
- Loupy, A. Microwaves in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Arvela, R.K.; Leadbeater, N.E.; Collins, M.J. Automated batch scale-up of microwave-promoted Suzuki and Heck coupling reactions in water using ultra-low metal catalyst concentrations. Tetrahedron 2005, 61, 9349–9355. [Google Scholar] [CrossRef]
- Wathey, B.; Tierney, J.; Lidström, P.; Westman, J. The impact of microwave-assisted organic chemistry on drug discovery. Drug Discov. Today 2002, 7, 373–380. [Google Scholar] [CrossRef]
- De la Hoz, A.; Díaz-Ortiz, Á.; Moreno, A. Selectivity in organic Synthesis under microwave irradiation. Curr. Org. Chem. 2004, 8, 903–918. [Google Scholar] [CrossRef]
- Bowman, M.D.; Schmink, J.R.; McGowan, C.M.; Kormos, C.M.; Leadbeater, N.E. Scale-up of microwave-promoted reactions to the multigram level using a sealed-vessel microwave apparatus. Org. Process Res. Dev. 2008, 12, 1078–1088. [Google Scholar] [CrossRef]
- Dallinger, D.; Lehmann, H.; Moseley, J.D.; Stadler, A.; Kappe, C.O. Scale-up of microwave-assisted reactions in a multimode bench-top reactor. Org. Process Res. Dev. 2011, 15, 841–854. [Google Scholar] [CrossRef]
- Schön, M.; Schnürch, M.; Mihovilovic, M.D. Application of continuous flow and alternative energy devices for 5-hydroxymethylfurfural production. Mol. Divers. 2011, 15, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Yokozawa, S.; Ohneda, N.; Muramatsu, K.; Okamoto, T.; Odajima, H.; Ikawa, T.; Sugiyama, J.; Fujita, M.; Sawairi, T.; Egami, H.; et al. Development of a highly efficient single-mode microwave applicator with a resonant cavity and its application to continuous flow syntheses. RSC Adv. 2015, 5, 10204–10210. [Google Scholar] [CrossRef]
- Cablewski, T.; Faux, A.F.; Strauss, C.R. Development and application of a continuous microwave reactor for organic synthesis. J. Org. Chem. 1994, 59, 3408–3412. [Google Scholar] [CrossRef]
- Strauss, C.R.; Trainor, R.W. Developments in microwave-assisted organic chemistry. Aust. J. Chem. 1995, 48, 1665–1692. [Google Scholar] [CrossRef]
- Tfelt-Hansen, P.; De Vries, P.; Saxena, P.R. Triptans in migraine: A comparative review of pharmacology, pharmacokinetics and efficacy. Drugs 2000, 60, 1259–1287. [Google Scholar] [CrossRef]
- Garden, S.J.; da Silva, R.B.; Pinto, A.C. A versatile synthetic methodology for the synthesis of tryptophols. Tetrahedron 2002, 58, 8399–8412. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Lieberman, D.R.; Larsen, R.D.; Reamer, R.A.; Verhoeven, T.R.; Reider, P.J.; Cottrel, I.F.; Houghton, P.G. Synthesis of the 5-HT1D receptor agonist MK-0462 via a Pd-catalyzed coupling reaction. Tetrahedron Lett. 1994, 35, 6981–6984. [Google Scholar] [CrossRef]
- Brodfuehrer, P.R.; Chen, B.-C.; Sattelberg, T.R.; Smith, P.R.; Reddy, J.P.; Stark, D.R.; Quinlan, S.L.; Reid, J.G. An efficient Fischer indole synthesis of avitriptan, a potent 5-HT1D receptor agonist. J. Org. Chem. 1997, 62, 9192–9202. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Senanayake, C.H.; Bill, T.J.; Larsen, R.D.; Verhoeven, T.R.; Reider, P.J. Improved Fischer indole reaction for the preparation of N, N-dimethyltryptamines: Synthesis of L-695,894, a potent 5-HT1D receptor agonist. J. Org. Chem. 1994, 59, 3738–3741. [Google Scholar] [CrossRef]
- Xu, J.; Yu, J.; Jin, Y.; Li, J.; Yu, Z.; Lv, Y. A continuous flow microwave-assisted Fischer indole synthesis of 7-Ethyltryptophol. Chem. Eng. Process. 2017, 121, 144–148. [Google Scholar] [CrossRef]
- Demerson, C.A.; Humber, L.G.; Philipp, A.H. Etodolic acid and related compounds. Chemistry and antiinflammatory actions of some potent di- and trisubstituted 1,3,4,9-tetrahydropyrano[3,4-b]indole-1-acetic acids. J. Med. Chem. 1976, 19, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Gopalsamy, A.; Lim, K.; Ciszewski, G.; Park, K.; Ellingboe, J.W.; Bloom, J.; Insaf, S.; Upeslacis, J.; Mansour, T.S.; Krishnamurthy, G.; et al. Discovery of pyrano[3,4-b]indoles as potent and selective HCV NS5B polymerase inhibitors. J. Med. Chem. 2004, 47, 6603–6608. [Google Scholar] [CrossRef]
- Humber, L.G.; Ferdinandi, E.; Demerson, C.A.; Ahmed, S.; Shah, U.; Mobilio, D.; Sabatucci, J.; De Lange, B.; Labbadia, F.; Hughes, P.; et al. Etodolac, a novel antiinflammatory agent. The syntheses and biological evaluation of its metabolites. J. Med. Chem. 1988, 31, 1712–1719. [Google Scholar] [CrossRef]
- Dubhashe, Y.R.; Sawant, V.M.; Gaikar, V.G. Process intensification of continuous flow synthesis of tryptophol. Ind. Eng. Chem. Res. 2018, 57, 2787–2796. [Google Scholar] [CrossRef]
- Wahab, B.; Ellames, G.; Passey, S.; Watts, P. Synthesis of substituted indoles using continuous flow micro reactors. Tetrahedron 2010, 66, 3861–3865. [Google Scholar] [CrossRef]
- Gutmann, B.; Gottsponer, M.; Elsner, P.; Cantillo, D.; Roberge, D.M.; Kappe, C.O. On the Fischer indole synthesis of 7-Ethyltryptophol-mechanistic and process intensification studies under continuous flow conditions. Org. Process Res. Dev. 2013, 17, 294–302. [Google Scholar] [CrossRef]
- Lv, Y.; Yu, Z.; Su, W. A Continuous kilogram-scale process for the manufacture of 7-Ethyltryptophol. Org. Process Res. Dev. 2011, 15, 471–475. [Google Scholar] [CrossRef]
- Bosch, C.; López-Lledó, P.; Bonjoch, J.; Bradshaw, B.; Nieuwland, P.J.; Blanco-Ania, D.; Rutjes, F.P.J.T. Fischer indole reaction in batch and flow employing a sulfonic acid resin: Synthesis of pyrido[2,3-a]carbazoles. J. Flow Chem. 2016, 6, 240–243. [Google Scholar] [CrossRef]
- Yu, J.; Xu, J.; Yu, Z.; Jin, Y.; Li, J.; Lv, Y. A continuous-flow Fischer indole synthesis of 3-methylindole in an Ionic Liquid. J. Flow Chem. 2017, 7, 33–36. [Google Scholar] [CrossRef]
- Hemetsberger, H.; Knittel, D.; Weidmann, H. Synthese von α-Azidozimtsäureestern. Monatshefte Chem. 1969, 100, 1599–1603. [Google Scholar] [CrossRef]
- Cui, G.; Lai, F.; Wang, X.; Chen, X.; Xu, B. For some biological properties of indole-2-carboxylic acid derivatives see: Design, Synthesis and Biological Evaluation of Indole-2-Carboxylic Acid Derivatives as IDO1/TDO Dual Inhibitors. Eur. J. Med. Chem. 2020, 188, 111985–112005. [Google Scholar] [CrossRef]
- Lindley, J.M.; McRobbie, I.M.; Meth-Cohn, O.; Suschitzky, H. Competitive cyclisation of singlet and triplet nitrenes. Part III. The effect of temperature on tile reactivity of thermally and photochemically derived arylnitrenes. Tetrahedron Lett. 1976, 17, 4513–4516. [Google Scholar] [CrossRef]
- Knittel, D. Verbesserte synthese von α-azidozimtsäure-estern und 2H-azirinen. Synthesis 1985, 1985, 186–188. [Google Scholar] [CrossRef]
- O’Brien, A.G.; Lévesque, F.; Seeberger, P.H. Continuous flow thermolysis of azidoacrylates for the synthesis of heterocycles and pharmaceutical intermediates. Chem. Commun. 2011, 47, 2688–2690. [Google Scholar] [CrossRef]
- Ranasinghe, N.; Jones, G.B. Extending the versatility of the Hemetsberger–Knittel indole synthesis through microwave and flow chemistry. Bioorganic Med. Chem. Lett. 2013, 23, 1740–1742. [Google Scholar] [CrossRef]
- Söderberg, B.C.G. For a review, see: Synthesis of heterocycles via intramolecular annulation of nitrene intermediates. Curr. Org. Chem. 2000, 4, 727–764. [Google Scholar] [CrossRef]
- Colombo, E.; Ratel, P.; Mounier, L.; Guillier, F. Reissert indole synthesis using continuous-flow hydrogenation. J. Flow Chem. 2011, 1, 68–73. [Google Scholar] [CrossRef]
- Suzuki, H.; Gyoutoku, H.; Yokoo, H.; Shinba, M.; Sato, Y.; Yamada, H.; Murakami, Y. Unexpected formation of quinolone derivatives in Reissert indole synthesis. Synlett 2000, 8, 1196–1198. [Google Scholar]
- Sall, D.J.; Arfsten, A.E.; Bastian, J.A.; Denney, M.L.; Harms, C.S.; McCowan, J.R.; Morin, J.M., Jr.; Rose, J.W.; Scarborough, R.M.; Smyth, M.S.; et al. Use of conformationally restricted benzamidines as arginine surrogates in the design of platelet GPIIb-IIIa receptor antagonists. J. Med. Chem. 1997, 40, 2843–2857. [Google Scholar] [CrossRef]
- Chapman, N.; Conway, B.; O’Grady, F.; Wall, M.D. A Convenient method to aniline compounds using microwave-assisted transfer hydrogenation. Synlett 2006, 7, 1043–1046. [Google Scholar] [CrossRef]
- The Design Expert® Software, Versione 7.0 (DX7) Is Available from Stat-Ease, Inc. (Minneapolis, USA). Available online: http:www.statease.com (accessed on 15 July 2020).
- Örkényi, R.; Beke, G.; Riethmüller, E.; Szakács, Z.; Kóti, J.; Faigl, F.; Éles, J.; Greiner, I. Environmentally friendly synthesis of indoline derivatives using flow-chemistry techniques. Eur. J. Org. Chem. 2017, 6525–6532. [Google Scholar]
- Cadogan, J.I.G.; Cameron-Wood, M. Reduction of nitro-compounds by triethyl phosphite: A new cyclisation reaction. Proc. Chem. Soc. 1962, 361. [Google Scholar]
- Cadogan, J.I.G.; Mackie, R.K.; Todd, M.J. Reductive cyclisation of nitro-compounds by triethyl phosphite: New syntheses of phenothiazines and anthranils. J. Chem. Soc. Chem. Commun. 1966, 491. [Google Scholar] [CrossRef]
- Sundberg, R.J. Deoxygenation of Nitro Groups by Trivalent Phosphorus. Indoles from o-Nitrostyrenes. J. Org. Chem. 1965, 30, 3604–3610. [Google Scholar] [CrossRef]
- Sundberg, R.J.; Tamazaki, T. Rearrangements and ring expansions during the deoxygenation of beta, beta-disubstituted o-nitrostyrenes. J. Org. Chem. 1967, 32, 290–294. [Google Scholar] [CrossRef]
- Pizzotti, M.; Cenini, S.; Quici, S.; Tollari, S. Role of alkali halides in the synthesis of nitrogen containing heterocycles by reductive carbonylation of aromatic nitro-derivatives catalysed by Ru3(CO)12. J. Chem. Soc. Perkin Trans. 1994, 2, 913–917. [Google Scholar] [CrossRef]
- Crotti, C.; Cenini, S.; Bassoli, A.; Rindone, B.; Demartin, F.J. Synthesis of carbazole by Ru3(CO)12-catalyzed reductive carbonylation of 2-nitrobiphenyl: The crystal and molecular structure of Ru3(μ3-NC6H4-o-C6H5)2(CO)9. Mol. Catal. 1991, 70, 175–187. [Google Scholar] [CrossRef]
- Crotti, C.; Cenini, S.; Todeschini, R.; Tollari, S. Chemometric optimization of the ruthenium carbonyl catalysed cyclization of 2-nitrostilbene to 2-phenylindole. J. Chem. Soc. Faraday Trans. 1991, 87, 2811–2820. [Google Scholar] [CrossRef]
- Akazome, M.; Kondo, T.; Watanabe, Y. Palladium complex-catalyzed reductive N-heterocyclization of nitroarenes: Novel synthesis of indole and 2H-indazole derivatives. J. Org. Chem. 1994, 59, 3375–3380. [Google Scholar] [CrossRef]
- Tollari, S.; Penoni, A.; Cenini, S.J. The unprecedented detection of the intermediate formation of N-hydroxy derivatives during the carbonylation of 2′-nitrochalcones and 2-nitrostyrenes catalysed by palladium. Mol. Catal. 2000, 152, 47–54. [Google Scholar] [CrossRef]
- Ragaini, F.; Sportiello, P.; Cenini, S.J. Investigation of the possible role of arylamine formation in the ortho-substituted nitroarenes reductive cyclization reactions to afford heterocycles. Organomet. Chem. 1999, 577, 283–291. [Google Scholar] [CrossRef]
- Tollari, S.; Cenini, S.; Crotti, C.; Gianella, E. Synthesis of heterocycles via palladium-catalyzed carbonylation of ortho-substituted organic nitro compounds in relatively mild conditions. J. Mol. Catal. 1994, 87, 203–214. [Google Scholar] [CrossRef]
- Söderberg, B.C.; Shriver, J.A. Palladium-catalyzed synthesis of indoles by reductive N-heteroannulation of 2-nitrostyrenes. J. Org. Chem. 1997, 62, 5838–5845. [Google Scholar] [CrossRef]
- Söderberg, B.C.; Rector, S.R.; O’Neil, S.N. Palladium-catalyzed synthesis of fused indoles. Tetrahedron Lett. 1999, 40, 3657–3660. [Google Scholar] [CrossRef]
- Scott, T.L.; Söderberg, B.C.G. Novel palladium-catalyzed synthesis of 1,2-dihydro-4(3H)-carbazolones. Tetrahedron Lett. 2002, 43, 1621–1624. [Google Scholar] [CrossRef]
- Dantale, S.W.; Söderberg, B.C.G. A novel palladium-catalyzed synthesis of β-carbolines: Application in total synthesis of naturally occurring alkaloids. Tetrahedron 2003, 59, 5507–5514. [Google Scholar] [CrossRef]
- Söderberg, B.C.G.; Chisnell, A.C.; O’Neil, S.N.; Shriver, J.A. Synthesis of indoles isolated from Tricholoma species. J. Org. Chem. 1999, 64, 9731–9734. [Google Scholar] [CrossRef]
- Glotz, G.; Gutmann, B.; Hanselmann, P.; Kulesza, A.; Roberge, D.; Kappe, C.O. Continuous flow synthesis of indoles by Pd-catalyzed deoxygenation of 2-nitrostilbenes with carbon monoxide. RSC Adv. 2017, 7, 10469–10478. [Google Scholar] [CrossRef]
- Baumann, M.; Baxendale, I.R.; Deplante, F. A concise flow synthesis of indole-3-carboxylic ester and its derivatisation to an auxin mimic. Beilstein J. Org. Chem. 2017, 13, 2549–2560. [Google Scholar] [CrossRef]
- Steingruber, E. Indigo and Indigo Colorants. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000. [Google Scholar]
- Greshock, T.J.; Funk, R.L. Synthesis of indoles via 6π- electrocyclic ring closures of trienecarbamates. J. Am. Chem. Soc. 2006, 128, 4946–4947. [Google Scholar] [CrossRef]
- Yan, L.; Lai, F.; Chen, X.; Xiao, Z. Discovery of novel indirubin-3’-monoxime derivatives as potent inhibitors against CDK2 and CDK9. Bioorganic Med. Chem. Lett. 2015, 25, 2447–2451. [Google Scholar] [CrossRef]
- Klimovich, I.V.; Leshanskaya, L.I.; Troyanov, S.I.; Anokhin, D.V.; Novikov, D.V.; Piryazev, A.A.; Ivanov, D.A.; Dremova, N.N.; Troshin, P.A. Design of indigo derivatives as environment-friendly organic semiconductors for sustainable organic electronics. J. Mater. Chem. 2014, 2, 7621–7631. [Google Scholar] [CrossRef]
- Jung, D.-W.; Hong, Y.J.; Kim, S.-Y.; Kim, W.-H.; Seo, S.; Lee, J.-E.; Shen, H.; Kim, Y.-C.; Williams, D.R. 5-Nitro-5’hydroxyindirubin-3’oxime Is a Novel Inducer of Somatic Cell Transdifferentiation. Arch. Pharm. 2014, 347, 806–818. [Google Scholar] [CrossRef]
- Nirogi, R.V.S.; Deshpande, A.D.; Kambhampati, R.; Badange, R.K.; Kota, L.; Daulatabad, A.V.; Shinde, A.K.; Ahmad, I.; Kandikere, V.; Jayarajan, P.; et al. Indole-3-piperazinyl derivatives: Novel chemical class of 5-HT6 receptor antagonists. Bioorganic Med. Chem. Lett. 2011, 21, 346–349. [Google Scholar] [CrossRef]
- Friedländer, P.; Bruckner, S.; Deutsch, G. Über brom-und methoxyderivate des indigos. Justus Liebigs Ann. Chem. 1912, 388, 23–49. [Google Scholar] [CrossRef]
- Tighineanu, E.; Chiraleu, F.; Råieanu, D. Double cyclisation of phenylglycine-o-carboxylic acids—I: New stable mesoionic oxazolones. Tetrahedron 1980, 36, 1385–1397. [Google Scholar] [CrossRef]
- Cambié, D.; Bottecchia, C.; Straathof, N.J.W.; Hessel, V.; Noël, T. Applications of continuous-flow photochemistry in organic synthesis, material science, and water treatment. Chem. Rev. 2016, 116, 10276–10341. [Google Scholar] [CrossRef]
- Su, Y.; Straathof, N.J.; Hessel, V.; Noël, T. Photochemical transformations accelerated in continuous-flow reactors: Basic concepts and applications. Chem. Eur. J. 2014, 20, 10562–10589. [Google Scholar] [CrossRef]
- Willumstad, T.P.; Haze, O.; Mak, X.Y.; Lam, T.Y.; Wang, Y.P.; Danheiser, R.L. Batch and flow photochemical benzannulations based on the reaction of ynamides and diazo ketones. Application to the synthesis of polycyclic aromatic and heteroaromatic compounds. J. Org. Chem. 2013, 78, 11450–11469. [Google Scholar] [CrossRef]
- Wu, G.; Lv, T.; Mo, W.; Yang, X.; Gao, Y.; Chen, H. One-pot synthesis of tricyclo-1,4-benzoxazines via visible-light photoredox catalysis in continuous flow. Tetrahedron Lett. 2017, 58, 1395–1398. [Google Scholar] [CrossRef]
- Wu, G.; Lv, T.; Mo, W.; Yang, X.; Gao, Y.; Chen, H. For a detailed description of the reactor, see: Acetic acid accelerated visible-light photoredox catalyzed N-demethylation of N, N-dimethylaminophenyl derivatives. Adv. Synth. Catal. 2017, 359, 687–692. [Google Scholar] [CrossRef]
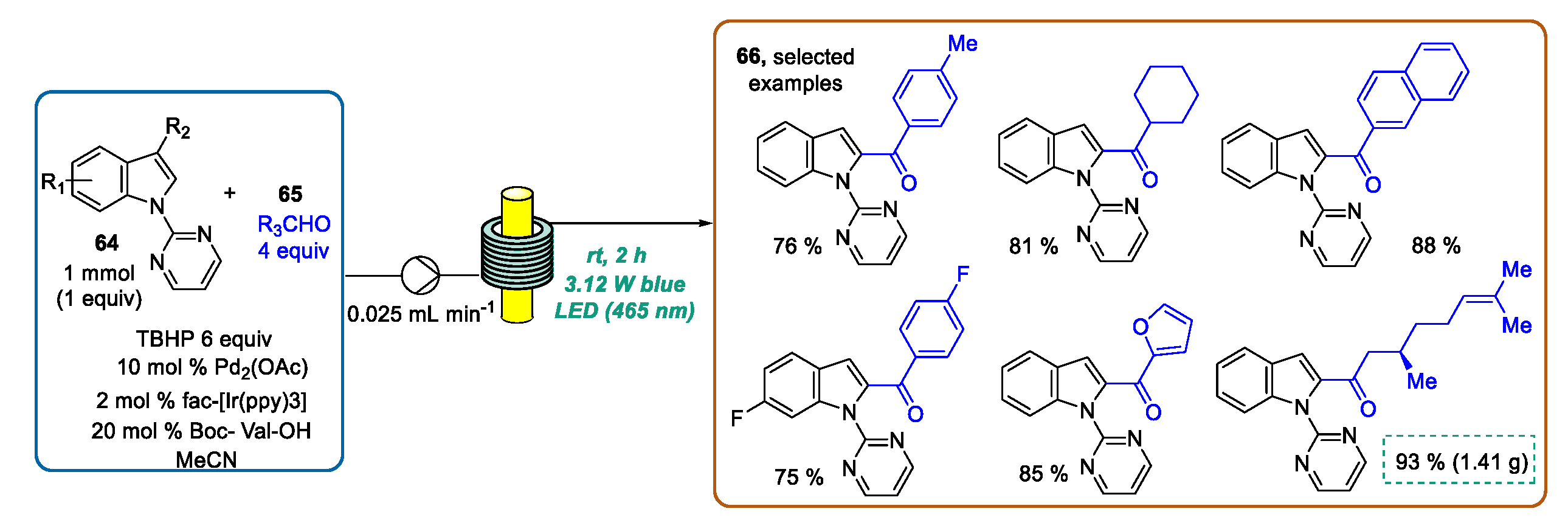
- Sharma, U.K.; Gemoets, H.P.L.; Schröder, F.; Noël, T.; Van der Eycken, E.V. Merger of Visible-Light Photoredox Catalysis and C–H Activation for the RoomTemperature C-2 Acylation of Indoles in Batch and Flow. ACS Catal. 2017, 7, 3818–3823. [Google Scholar] [CrossRef]
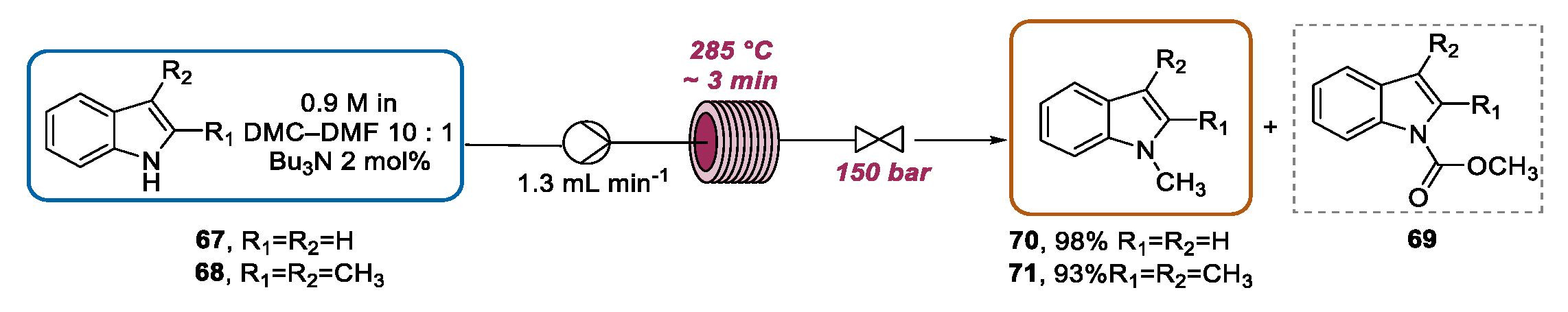
- Glasnov, T.N.; Holbrey, J.D.; Kappe, C.O.; Seddon, K.R.; Yan, T. Methylation using dimethylcarbonate catalysed by ionic liquids under continuous flow conditions. Green Chem. 2012, 14, 3071–3076. [Google Scholar] [CrossRef]
- Holbrey, J.D.; Rogers, R.D.; Shukla, S.S.; Wilfred, C.D. Optimised microwave-assisted synthesis of methylcarbonate salts: A convenient methodology to prepare intermediates for ionic liquid libraries. Green Chem. 2010, 12, 407–413. [Google Scholar] [CrossRef]
- Tricotet, T.; O’Shea, D.F. Automated generation and reactions of 3-hydroxymethylindoles in continuous-flow microreactors. Chem. Eur. J. 2010, 16, 6678–6686. [Google Scholar] [CrossRef]
- Mohapatra, S.S.; Wilson, Z.E.; Roy, S.; Ley, S.V. Utilization of flow chemistry in catalysis: New avenues for the selective synthesis of bis(indolyl)methanes. Tetrahedron 2017, 73, 1812–1819. [Google Scholar] [CrossRef]
- Sujatha, K.; Perumal, P.T.; Muralidharan, D.; Rajendran, M. Synthesis, analgesic and anti-inflammatory activities of bis(indolyl)methanes. Indian J. Chem. B 2009, 48, 267–272. [Google Scholar]
- Shiri, M.; Zolfigol, M.A.; Kruger, H.G.; Tanbakouchian, Z. Bis- and trisindolylmethanes (BIMs and TIMs). Chem. Rev. 2009, 110, 2250–2293. [Google Scholar] [CrossRef]
- Sato, S.; Sato, T. A mild and environmentally friendly scandium(III) trifluoromethanesulfonate-catalyzed synthesis of bis(3’-indolyl)alkanes and bis(3’-indolyl)-1-deoxyalditols. Carbohydr. Res. 2005, 340, 2251–2255. [Google Scholar] [CrossRef]
- Petrini, M.; Chiurchiù, E.; Rossi, F.V.; Palmieri, A. Oxidative conversion of sulfonyl indoles into 3-alkylidene-2-oxindoles under flow chemical conditions. Synthesis 2018, 50, 371–376. [Google Scholar]
- Chiurchiù, E.; Palmieri, A.; Petrini, M. 3-Alkylated indoles by reduction of sulfonyl indoles under flow chemical conditions. Arkivoc 2019, 2019, 69–79. [Google Scholar] [CrossRef]
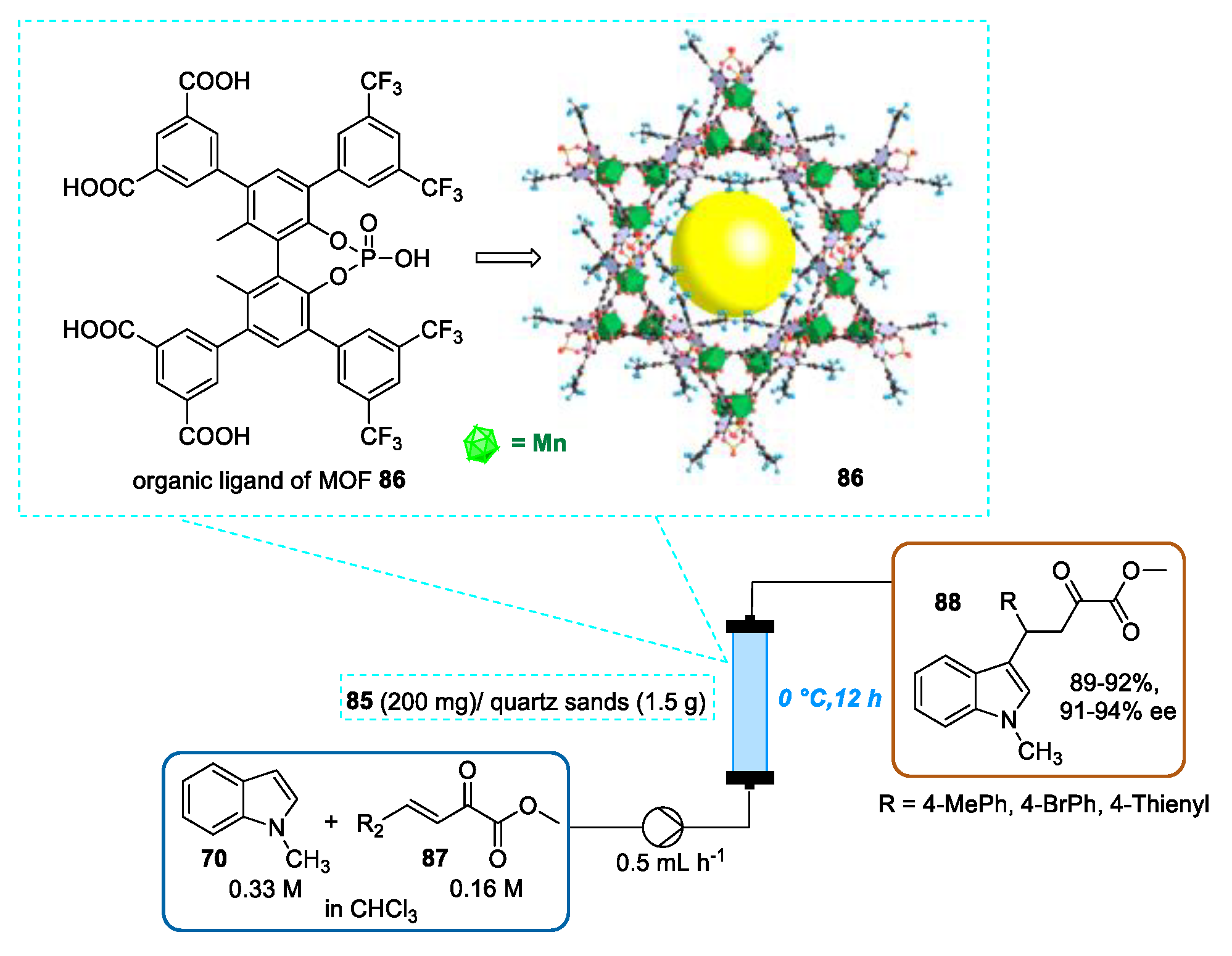
- Chen, X.; Jiang, H.; Hou, B.; Gong, W.; Liu, Y.; Cui, Y. Boosting Chemical Stability, Catalytic Activity, and Enantioselectivity of Metal—Organic Frameworks for Batch and Flow Reactions. J. Am. Chem. Soc. 2017, 139, 13476–13482. [Google Scholar] [CrossRef]
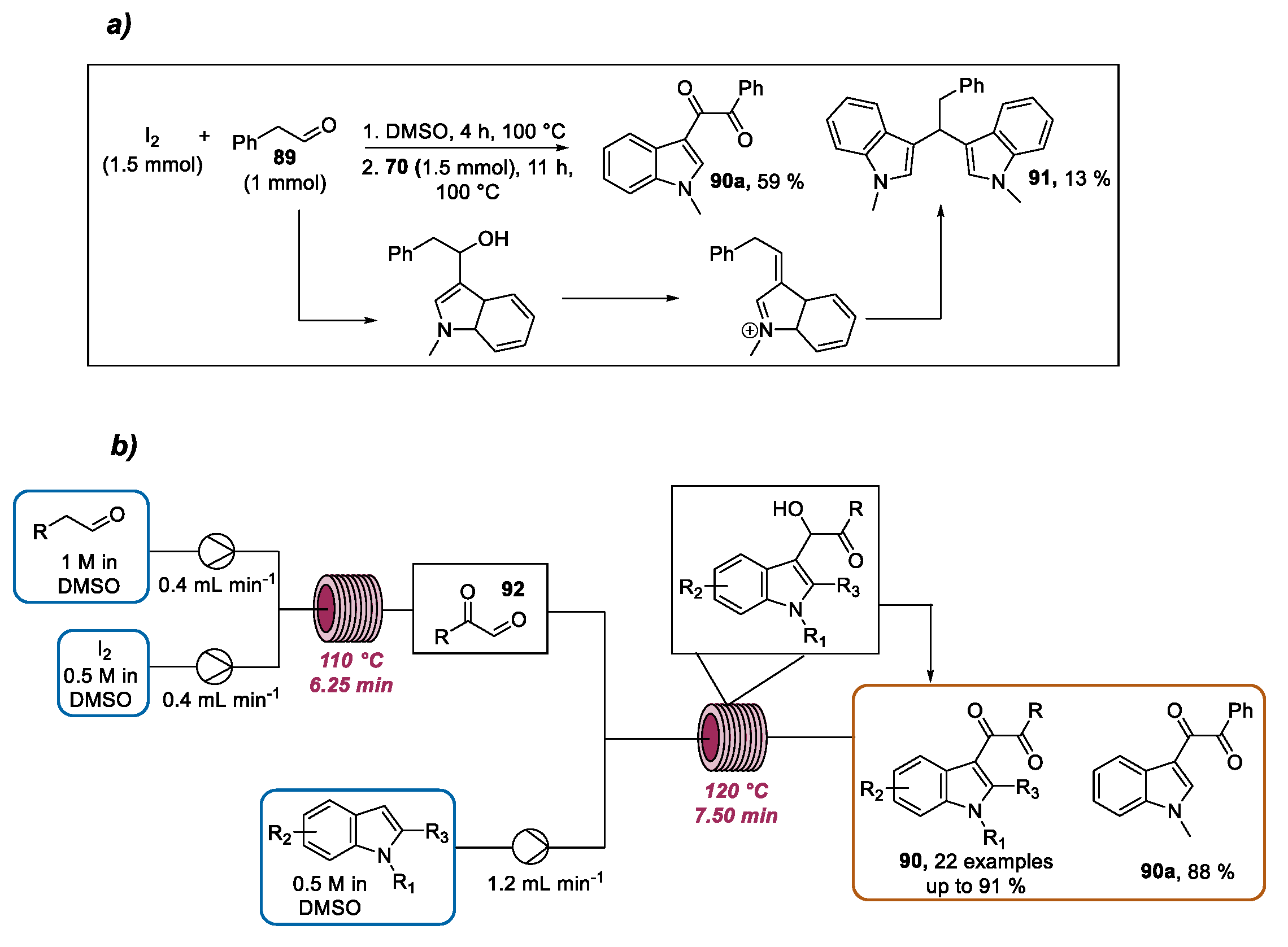
- Guo, S.; Hua, J.; Dai, Z.; Yang, Z.; Fang, Z.; Guo, K. Two-step continuous synthesis of dicarbonyl indoles via I2/DMSO-promoted oxidative coupling: A green and practical approach to valuable diketones from aryl acetaldehydes. ACS Sustain. Chem. Eng. 2018, 6, 7979–7988. [Google Scholar] [CrossRef]
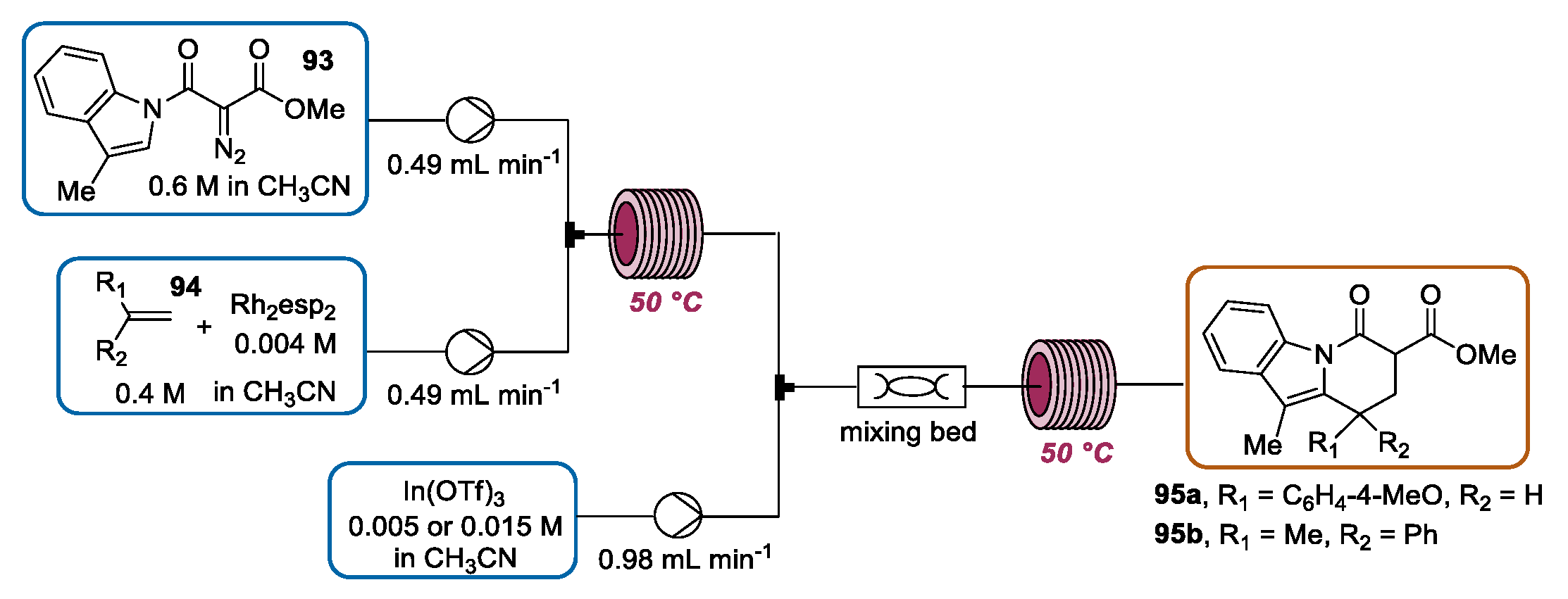
- Aponte-Guzman, J.; Shenje, R.; Huang, Y.; Woodham, W.H.; Saunders, S.R.; Mostaghimi, S.M.; Flack, K.R.; Pollet, P.; Eckert, C.A.; Liotta, C.L.; et al. A Tandem, Bicatalytic Continuous Flow Cyclopropanation-Homo-Nazarov-Type Cyclization. Ind. Eng. Chem. Res. 2015, 54, 9550–9558. [Google Scholar] [CrossRef]

































© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Colella, M.; Degennaro, L.; Luisi, R. Continuous Flow Synthesis of Heterocycles: A Recent Update on the Flow Synthesis of Indoles. Molecules 2020, 25, 3242. https://doi.org/10.3390/molecules25143242
Colella M, Degennaro L, Luisi R. Continuous Flow Synthesis of Heterocycles: A Recent Update on the Flow Synthesis of Indoles. Molecules. 2020; 25(14):3242. https://doi.org/10.3390/molecules25143242
Chicago/Turabian StyleColella, Marco, Leonardo Degennaro, and Renzo Luisi. 2020. "Continuous Flow Synthesis of Heterocycles: A Recent Update on the Flow Synthesis of Indoles" Molecules 25, no. 14: 3242. https://doi.org/10.3390/molecules25143242
APA StyleColella, M., Degennaro, L., & Luisi, R. (2020). Continuous Flow Synthesis of Heterocycles: A Recent Update on the Flow Synthesis of Indoles. Molecules, 25(14), 3242. https://doi.org/10.3390/molecules25143242