3. Materials and Methods
Reactions were carried out under argon atmosphere and solvents were dried using standard methods and distilled before use. DCM, Pyridine and DMF were dried over calcium hydride and THF over sodium and benzophenone. Unless otherwise specified, materials were purchased from commercial suppliers and used without further purification. TLC was performed using Merck commercial aluminum sheets coated with silica gel 60 F254 (Merck, Darmstadt, Germany). Compounds were detected by charring with 10% H2SO4 in ethanol followed by heating. Purification was performed by flash chromatography on silica gel (60 Å, 180–240 mesh; Merck, Darmstadt, Germany). Preparative HPLC was performed using a HPLC system with a reverse phase C-18 column (250 × 21.2 mm) using a solvent system consisting of H2O and CH3CN (linear gradient from 0:100 to 100:0 in 30 min) at a flow rate of 15 mL.min−1 and UV detection at 254 nm. The purity of final compounds (≥ 95%) was established by analytical HPLC, which was performed on Macherey Nagel C18 100-5 NUCLEOSIL column (25 × 4.6, 5 μm) with UV detection at 214 and 254 nm. NMR spectra were recorded on Bruker spectrometers (Avance II 500 and Avance III HD 4000) (Bruker Biospin, Fällanden, Switzerland). Chemical shifts (δ) are reported in parts per million (ppm) and referenced to the residual proton or carbon resonance of the solvents: CDCl3 (δ 7.26), MeOD (δ 3.31), D2O (δ 4.79) or (CD3)2SO (δ 2.50) for 1H and CDCl3 (δ 77.16), MeOD (δ 49.0) or (CD3)2SO (δ 39.52) for 13C. Signals were assigned using 1D (1H and 13C) and 2D (HSQC, COSY and HMBC) experiments. NMR coupling constants (J) are reported in Hertz (Hz) and splitting patterns are indicated as follows: s (singlet), bs (broad singlet), d (doublet), t (triplet), q (quartet), dd (doublet of doublet), m (multiplet). High-resolution mass spectroscopy (HRMS) was recorded with an ion trap mass analyzer under electrospray ionization (ESI) in the negative or positive ionization detection mode, using Thermo Scientific LTQ Orbitrap XL (Thermo Scientific, Illkirch, France.
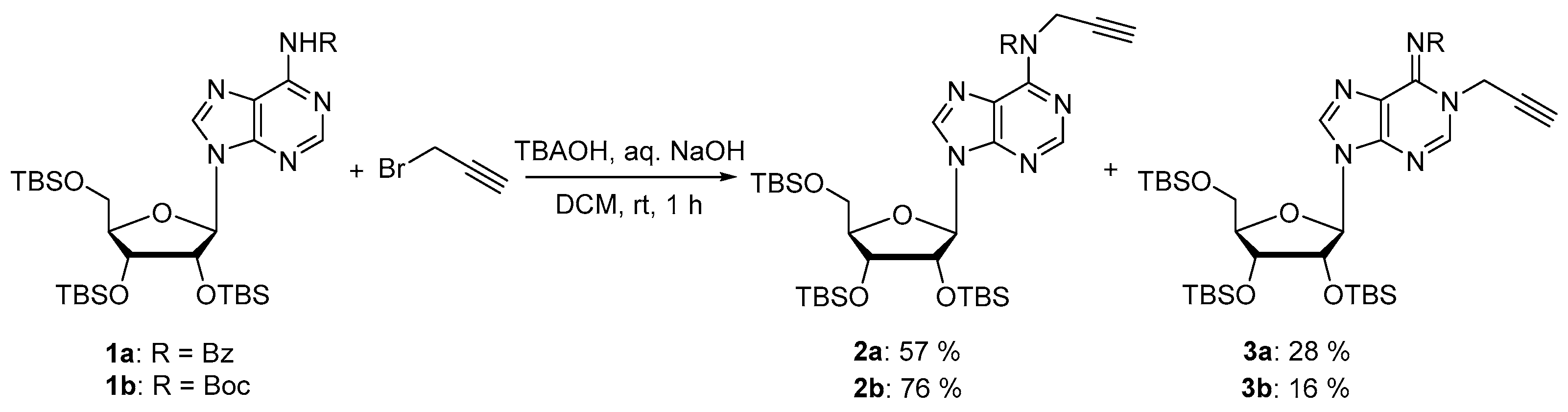
Compounds 2a and 3a: The trisilylated adenosine 1a (1.59 g, 2.22 mmol) and propargyl bromide 80% in toluene (674 μL, 8.88 mmol) were dissolved in DCM (50 mL) and tetrabutylammonium hydroxyde (1.78 g, 2.22 mmol) and 1M aqueous NaOH (22.2 mL) were added to the solution. After vigorous stirring at room temperature for 1 h, the reaction mixture was diluted in DCM and washed with brine, dried over MgSO4, and the solvent was removed under reduced pressure. The residue was purified by silica gel chromatography (eluent: Cyclohexane/EtOAc 9:1 then 7:3) to provide the desired compounds as white foams (946 mg, 57% for 2a and 475 mg, 28% for 3a). 2a: 1H NMR (500 MHz, CDCl3): δ 8.49 (s, 1H, H2), 8.14 (s, 1H, H8), 7.39–7.37 (m, 2H, HBz), 7.18–7.15 (m, 1H, HBz), 7.06–7.03 (m, 2H, HBz), 5.95 (d, J = 5.9 Hz, 1H, H1′), 5.7 (d, J = 4.9 Hz, 2H, CH2N), 4.54–4.52 (m, 1H, H2′), 4.17–4.15 (m, 1H, H3′), 4.03–4.01 (m, 1H, H4′), 3.89 (dd, J = 4.4, 11.4 Hz, 1H, H5′), 3.68 (dd, J = 2.8, 11.2 Hz, 1H, H5′), 2.01 (t, J = 4.7 Hz, 1H, C≡CH), 0.85 (s, 9H, tBuTBS), 0.83 (s, 9H, tBuTBS), 0.64 (s, 9H, tBuTBS), 0.02 (s, 3H, MeTBS), 0.01 (s, 3H, MeTBS), 0.00 (s, 6H, 2 MeTBS), −0.19 (s, 3H, MeTBS), −0.51 (s, 3H, MeTBS). 13C NMR (126 MHz, CDCl3): δ 171.4 (C=O), 153.2 (C6), 152.8 (C4), 151.9 (C2), 142.8 (C8), 135.9 (CqBz), 130.9 (CBz), 128.9 (2C, CBz), 127.9 (2C, CBz), 127.0 (C5), 88.2 (C1′), 86.2 (C4′), 79.0 (C≡CH), 75.8 (C2′), 72.4 (C3′), 71.7 (C≡CH), 62.8 (C5′), 37.5 (CH2N), 26.1 (3C, tBuTBS), 25.9 (3C, tBuTBS), 25.7 (3C, tBuTBS), 18.6 (CqTBS), 18.1 (CqTBS), 17.9 (CqTBS), −4.3 (MeTBS), −4.4 (MeTBS), −4.5 (MeTBS), −5.0 (MeTBS), −5.2 (MeTBS), −5.3 (MeTBS). HRMS (ESI) m/z: calcd for C38H61N5NaO5Si3 [M + Na]+: 774.3878; found: 774.3904. 3a: 1H NMR (500 MHz, CDCl3): δ 8.24 (s, 1H, H2), 8.16–8.15 (m, 2H, HBz), 7.88 (s, 1H, H8), 7.48–7.45 (m, 1H, HBz), 7.40–7.37 (m, 2H, HBz), 5.90 (d, J = 5.0 Hz, 1H, H1′), 4.97 (t, J = 2.8 Hz, 2H, CH2N), 4.48–4.47 (m, 1H, H2′), 4.26–4.24 (m, 1H, H3′), 4.08–4.06 (m, 1H, H4′), 3.88 (dd, J = 4.0, 11.3 Hz, 1H, H5′), 3.73 (dd, J = 3.4, 11.4 Hz, 1H, H5′), 2.59 (t, J = 2.5 Hz, 1H, C≡CH), 0.92 (s, 9H, tBuTBS), 0.89 (s, 9H, tBuTBS), 0.81 (s, 9H, tBuTBS), 0.09 (s, 3H, MeTBS), 0.08 (s, 3H, MeTBS), 0.06 (s, 3H, MeTBS), 0.05 (s, 3H, MeTBS), −0.03 (s, 3H, MeTBS), −0.19 (s, 3H, MeTBS). 13C NMR (126 MHz, CDCl3): δ 177.2 (C=O), 146.1 (C6), 145.4 (C2), 145.1 (C4), 138.8 (C8), 135.8 (CqBz), 132.0 (CBz), 129.9 (2C, CBz), 128.1 (2C, CBz), 122.3 (C5), 88.2 (C1′), 85.4 (C4′), 76.7 (C≡CH), 76.1 (C2′), 75.9 (C≡CH), 72.0 (C3′), 62.7 (C5′), 37.8 (CH2N), 26.1 (3C, tBuTBS), 25.9 (3C, tBuTBS), 25.8 (3C, tBuTBS), 18.6 (CqTBS), 18.2 (CqTBS), 18.0 (CqTBS), −4.2 (MeTBS), −4.4 (MeTBS), −4.5 (MeTBS), −4.8 (MeTBS), −5.2 (MeTBS), −5.3 (MeTBS). HRMS (ESI) m/z: calcd for C38H61N5NaO5Si3 [M + Na]+: 774.3878; found: 774.3911.
Compounds 2b and 3b: To a stirred solution of compound 1b (1.5 g, 2.11 mmol) in DCM (47.5 mL) was added propargyl bromide (80% in toluene) (0.795 mL, 8.44 mmol), tetrabutylammonium hydroxide (1.68 g, 1.68 mmol) and 1M aqueous NaOH (21.1 mL). The reaction mixture was stirred at room temperature for 1 h. The reaction mixture was dissolved in DCM and washed with brine. The organic layer was dried over MgSO4, concentrated in vacuo and purified on silica gel chromatography (eluent: cyclohexane/EtOAc 9:1 then 7:3) to afford the desired compounds as white foams (1.2 g, 76% for 2b and 0.251 g, 16% for 3b). 2b: 1H NMR (500 MHz, CDCl3): δ 8.77 (s, 1H, H2), 8.41 (s, 1H, H8), 6.10 (d, J = 4.9 Hz, 1H, H1′), 4.80 (d, J = 2.4 Hz, 2H, CH2N), 4.64 (t, J = 4.6 Hz, 1H, H2′), 4.33 (t, J = 4.0 Hz, 1H, H3′), 4.14 (q, J = 3.8 Hz, 1H, H4′), 4.04 (dd, J = 11.4, 4.1 Hz, 1H, H5′), 3.80 (dd, J = 11.4, 2.8 Hz, 1H, H5′), 2.16 (t, J = 2.4 Hz, 1H, C≡CH), 1.48 (s, 9H, tBuBoc), 0.96 (s, 9H, tBuTBS), 0.93 (s, 9H, tBuTBS), 0.80 (s, 9H, tBuTBS), 0.15 (s, 3H, MeTBS), 0.14 (s, 3H, MeTBS), 0.10 (s, 3H, MeTBS), 0.09 (s, 3H, MeTBS), −0.04 (s, 3H, MeTBS), −0.22 (s, 3H, MeTBS). 13C NMR (126 MHz, CDCl3): δ 152.7 (C6), 152.5 (C4), 152.4 (C=O), 151.8 (C2), 142.2 (C8), 127.60 (C5), 88.7 (C1′), 85.5 (C4′), 82.9 (CqBoc), 79.9 (C≡CH), 76.1 (C2′), 72.0 (C3′), 71.2 (C≡CH), 62.6 (C5′), 37.3 (CH2N), 28.1 (3C, tBuBoc), 26.3 (3C, tBuTBS), 26.0 (3C, tBuTBS), 25.8 (3C, tBuTBS), 18.7 (CqTBS), 18.2 (CqTBS), 18.0 (CqTBS), −4.2 (MeTBS), −4.5 (MeTBS), −4.6 (MeTBS), −4.8 (MeTBS), −5.2 (MeTBS), −5.2 (MeTBS). HRMS (ESI) m/z: calcd for C36H66N5O6Si3 [M + H]+: 748,4315; found 748.4304. 3b: 1H NMR (500 MHz, CDCl3): δ 8.22 (s, 1H, H2), 8.12 (s, 1H, H8), 5.91 (d, J = 3.3 Hz, 1H, H1′), 4.83 (d, J = 2.7 Hz, 2H, CH2N), 4.36–4.30 (m, 2H, H2′, H3′), 4.11–4.08 (m, 1H, H4′), 3.99 (dd, J = 11.6, 3.4 Hz, 1H, H5′), 3.78 (dd, J = 11.6, 2.5 Hz, 1H, H5′), 2.56 (t, J = 2.6 Hz, 1H, C≡CH), 1.60 (s, 9H, tBuBoc), 0.94 (s, 9H, tBuTBS), 0.92 (s, 9H, tBuTBS), 0.86 (s, 9H, tBuTBS), 0.12 (s, 3H, MeTBS), 0.11 (s, 3H, MeTBS), 0.10 (s, 3H, MeTBS), 0.08 (s, 3H, MeTBS), 0.02 (s, 3H, MeTBS), −0.04 (s, 3H, MeTBS). 13C NMR (126 MHz, CDCl3): δ 161.0 (C=O), 147.1 (C6), 145.1 (C2), 144.2 (C4), 138.2 (C8), 122.0 (C5), 88.7 (C1′), 84.5 (C4′), 80.9 (CqBoc), 76.8 (C≡CH), 76.7 (C2′), 76.4 (C≡CH), 70.9 (C3′), 61.9 (C5′), 37.0 (CH2N), 28.3 (3C, tBuBoc), 26.3 (3C, tBuTBS), 26.0 (3C, tBuTBS), 25.9 (3C, tBuTBS), 18.7 (CqTBS), 18.2 (CqTBS), 18.1 (CqTBS), −4.1 (MeTBS), −4.6 (2C, MeTBS), −4.7 (MeTBS), −5.2 (MeTBS), −5.3 (MeTBS). HRMS (ESI) m/z: calcd for C36H66N5O6Si3 [M + H]+: 748.4315; found 748.4309.
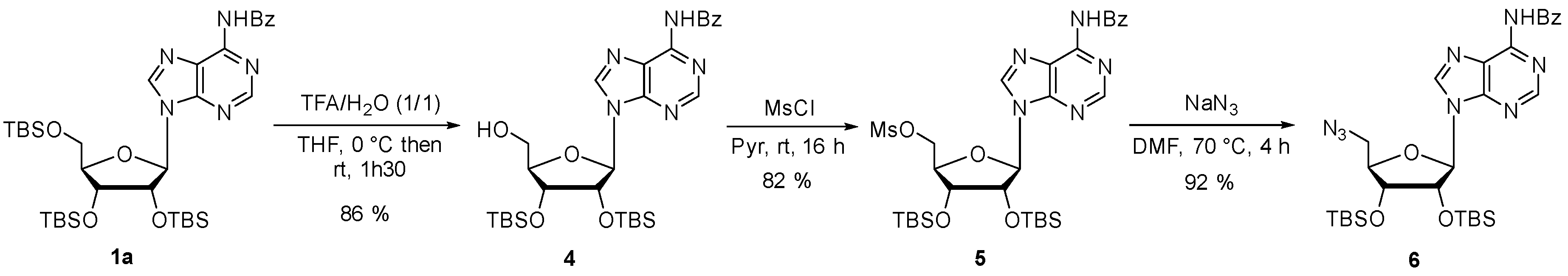
Compound 4: To a stirred solution of compound 1a (1.42 g, 1.97 mmol) in THF (20 mL) at 0 °C, was added dropwise an aqueous solution of TFA (1:1, 7.55 mL, 98.72 mmol). The solution was stirred at room temperature for 1.5 h. The reaction mixture was neutralized with saturated aqueous NaHCO3 solution and dissolved in EtOAc. The organic layer was washed with brine, dried over MgSO4, filtered and concentrated in vacuo. The residue was purified on silica gel chromatography (eluent: Cyclohexane/EtOAc 6:4) to afford compound 4 as a white foam (1.02 g, 86%). 1H NMR (500 MHz, CDCl3): δ 8.83 (s, 1H, H2), 8.13 (s, 1H, H8), 8.07–8.03 (m, 2H, HBz), 7.65–7.60 (m, 1H, HBz), 7.56–7.51 (m, 2H, HBz), 5.88 (d, J = 7.6 Hz, 1H, H1′), 5.03 (dd, J = 7.6, 4.5 Hz, 1H, H2′), 4.35 (d, J = 4.5 Hz, 1H, H3′), 4.20 (d, J = 1.7 Hz, 1H, H4′), 3.98 (dd, J = 13.1, 1.8 Hz, 1H, H5′), 3.74 (dd, J = 13.0, 1.6 Hz, 1H, H5′), 0.96 (s, 9H, tBuTBS), 0.75 (s, 9H, tBuTBS), 0.14 (s, 3H, MeTBS), 0.13 (s, 3H, MeTBS), −0.12 (s, 3H, MeTBS), −0.60 (s, 3H, MeTBS). 13C NMR (126 MHz, CDCl3): δ 164.4 (C=O), 152.4 (C6), 150.6 (C2), 150.5 (C4), 143.4 (C8), 133.7 (CqBz), 133.1 (CBz), 129.1 (2C, CBz), 127.9 (2C, CBz), 124.5 (C5), 91.4 (C1′), 89.7 (C4′), 74.2 (C2′), 74.0 (C3′), 63.1 (C5′), 25.9 (3C, tBuTBS), 25.8 (3C, tBuTBS), 18.2 (CqTBS), 17.9 (CqTBS), −4.4 (MeTBS), −4.4 (MeTBS), −4.5 (MeTBS), −5.7 (MeTBS). HRMS (ESI) m/z: calcd for C29H46N5O5Si2 [M + H]+: 600.3032; found: 600.3022.
Compound 5: Methanesulfonyl chloride (0.26 mL, 3.33 mmol) in pyridine (10 mL) was added dropwise to a solution of compound 4 (1 g, 1.66 mmol) in pyridine (7 mL) at 0 °C. The reaction mixture was stirred at room temperature for 16 h. The reaction was quenched by addition of water and then diluted in DCM. The organic layer was washed with a saturated solution of NaHCO3 and brine. The combined organic layer was dried over MgSO4 and concentrated to dryness. The residue was purified on silica gel chromatography (eluent: Cyclohexane/EtOAc 6:4) and compound 5 was isolated as a white foam (0.92 g, 82%). 1H NMR (500 MHz, CDCl3): δ 8.82 (s, 1H, H2), 8.20 (s, 1H, H8), 8.04 (d, J = 7.3 Hz, 2H, HBz), 7.62 (t, J = 7.4 Hz, 1H, HBz), 7.54 (t, J = 7.6 Hz, 2H, HBz), 6.00 (d, J = 4.8 Hz, 1H, H1′), 4.99 (t, J = 4.4 Hz, 1H, H2′), 4.62 (dd, J = 11.2, 4.1 Hz, 1H, H5′), 4.50 (dd, J = 11.2, 4.6 Hz, 1H, H5′), 4.40 – 4.35 (m, 2H, H3′, H4′), 3.03 (s, 3H, CH3), 0.95 (s, 9H, tBuTBS), 0.82 (s, 9H, tBuTBS), 0.15 (s, 3H, MeTBS), 0.13 (s, 3H, MeTBS), 0.00 (s, 3H, MeTBS), −0.20 (s, 3H, MeTBS). 13C NMR (126 MHz, CDCl3): δ 168.3 (C=O), 152.8 (C6), 151.5 (C2), 149.9 (C4), 142.5 (C8), 134.8 (CqBz), 132.9 (CBz), 129.0 (2C, CBz), 127.9 (2C, CBz),123.9 (C5), 89.9 (C1′), 82.4 (C4′), 74.3 (C2′), 72.1 (C3′), 67.9 (C5′), 37.8 (CH3), 25.9 (3C, tBuTBS), 25.8 (3C, tBuTBS), 18.2 (CqTBS), 18.00 (CqTBS), −4.3 (MeTBS), −4.5 (MeTBS), −4.7 (MeTBS), −4.8 (MeTBS). HRMS (ESI) m/z: calcd for C30H48N5O7SSi2 [M + H]+: 678.2807; found: 678.2788.
Compound 6: To a stirred solution of compound 5 (920 mg, 1.35 mmol) in DMF (7 mL) was added sodium azide (264 mg, 4.06 mmol) and the mixture was heated at 70 °C for 4 h. The reaction mixture was cooled to room temperature and was dissolved in EtOAc and washed with brine. The organic layer was dried over MgSO4, concentrated in vacuo and purified on silica gel chromatography (eluent: Cyclohexane/EtOAc 8:2) to afford compound 6 as a white foam (770 mg, 92%). 1H NMR (500 MHz, CDCl3): δ 8.80 (s, 1H, H2), 8.28 (s, 1H, H8), 8.03 (d, J = 7.3, 2H, HBz), 7.60 (t, J = 7.3, 1H, HBz), 7.52 (t, J = 7.8, 2H, HBz), 5.98 (d, J = 4.0 Hz, 1H, H1′), 4.88 (t, J = 4.1 Hz, 1H, H2′), 4.31 (t, J = 4.6 Hz, 1H, H3′), 4.23 (q, J = 4.6 Hz, 1H, H4′), 3.80–3.67 (m, 2H, H5′), 0.94 (s, 9H, tBuTBS), 0.84 (s, 9H, tBuTBS), 0.12 (s, 3H, MeTBS), 0.11 (s, 3H, MeTBS), −0.01 (s, 3H, MeTBS), −0.12 (s, 3H, MeTBS). 13C NMR (126 MHz, CDCl3): δ 164.6 (C=O), 152.8 (C6), 151.5 (C2), 149.8 (C4), 142.4 (C8), 133.8 (CqBz), 132.9 (CBz), 129.0 (2C, CBz), 127.9 (2C, CBz), 123.90 (C5), 90.1 (C1′), 82.8 (C4′), 74.8 (C2′), 72.3 (C3′), 51.6 (C5′), 25.9 (3C, tBuTBS), 25.8 (3C, tBuTBS), 18.2 (CqTBS), 18.0 (CqTBS), −4.2 (MeTBS), −4.5 (MeTBS), −4.6 (MeTBS), −4.7 (MeTBS). HRMS (ESI) m/z: calcd for C29H45N8O4Si2 [M + H]+: 625.3096; found 625.3088.
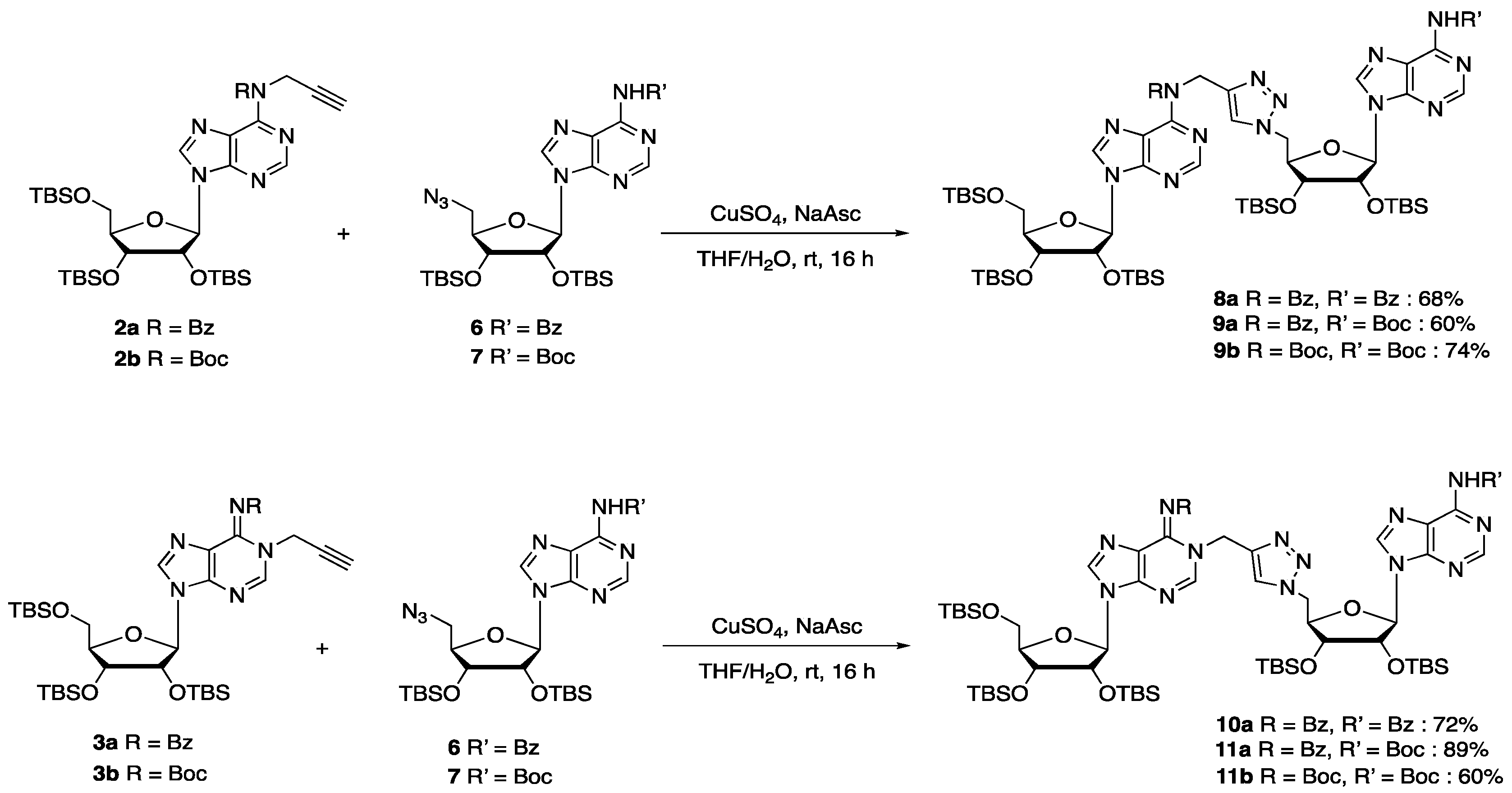
General procedure A for CuAAC reaction: To a solution of alkyne (1 eq) in THF (13 mL/mmol), were successively added azido compound 6 or 7 (1.2 eq), CuSO4 (0.3 eq, in water 3 mL/mmol) and sodium ascorbate (0.6 eq, in water 3 mL/mmol). The heterogeneous mixture was stirred at room temperature for 16 h. EtOAc was added and the organic layer was washed with brine, dried over MgSO4, and concentrated in vacuo. The crude was purified by flash chromatography to afford the desired compounds.
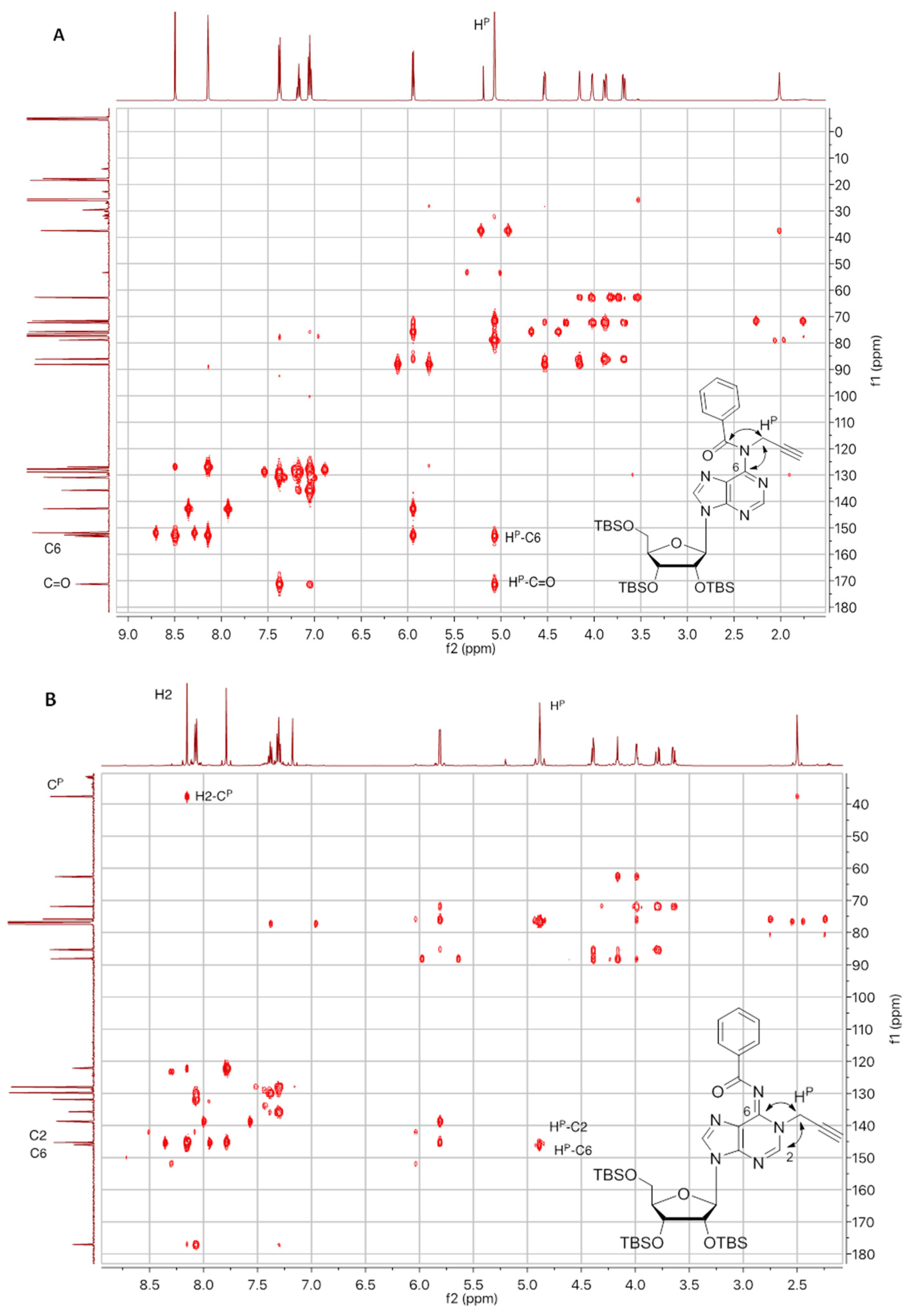
Compound 8a: Following the general procedure A for CuAAC, starting from alkyne 2a (200 mg, 0.26 mmol) and azido compound 6 (194 mg, 0.31 mmol) and using Cyclohexane/EtOAc 6:4 as eluent for flash chromatography purification, compound 8a was obtained as a white foam (245 mg, 68%). 1H NMR (500 MHz, CDCl3): δ 8.84 (s, 1H, H2a or H2b), 8.52 (s, 1H, H2a or H2b), 8.21 (s, 1H, H8a or H8b), 8.02 (bs, 3H, H8a or H8b and HBz), 7.81 (s, 1H, HTriazole), 7.60 (bs, 1H, HBz), 7.53 (bs, 2H, HBz), 7.42 (d, J = 7.1 Hz, 2H, HBz), 7.22 (t, J = 7.4 Hz, 1H, HBz), 7.10 (t, J = 7.6 Hz, 2H, HBz), 5.99 (d, J = 5.6 Hz, 1H, H1′a), 5.90 (bs, 1H, H1′b), 5.64 (s, 2H, CH2N), 5.07 (bs, 1H, H2′b), 4.89–4.81 (m, 1H, H5′b), 4.65–4.58 (m, 2H, H5′b and H2′a), 4.43 (bs, 1H, H3′b), 4.36 (bs, 1H, H4′b), 4.26–4.24 (m, 1H, H3′a), 4.11–4.08 (m, 1H, H4′a), 3.97 (dd, J = 11.3, 4.3 Hz, 1H, H5′a), 3.76 (dd, J = 11.3, 2.9 Hz, 1H, H5′a), 0.94 (s, 9H, tBuTBS), 0.92 (s, 9H, tBuTBS), 0.89 (s, 9H, tBuTBS), 0.76 (s, 9H, tBuTBS), 0.74 (s, 9H, tBuTBS), 0.11 (s, 3H, MeTBS), 0.11 (s, 3H, MeTBS), 0.09 (s, 3H, MeTBS), 0.09 (s, 3H, MeTBS), 0.04 (s, 3H, MeTBS), −0.05 (s, 3H, MeTBS), −0.08 (s, 3H, MeTBS), −0.11 (s, 3H, MeTBS), −0.37 (s, 3H, MeTBS), −0.42 (s, 3H, MeTBS). 13C NMR (126 MHz, CDCl3): δ 172.2 (2C, C=O), 164.5 (Cq), 153.7 (Cq), 152.8 (Cq), 152.0 (C2a or C2b), 151.5 (Cq), 150.1 (C2a or C2b), 144.7 (CqTriazole), 143.2 (C8a or C8b), 143.0 (C8a or C8b), 136.0 (CqBz) 133.9 (CqBz) 132.9 (CBz), 130.9 (CBz), 129.1 (2C, CBz), 129.0 (2C, CBz), 127.9 (4C, CBz), 127.30 (Cq), 124.9 (CHTriazole), 124.2 (Cq), 90.1 (C1′b), 88.4 (C1′a), 86.0 (C4′a), 84.1 (C4′b), 75.8 (C2′a), 73.4 (C3′b), 73.3 (C2′a), 72.3 (C3′a), 62.8 (C5′a), 51.7 (C5′b), 44.0 (CH2N), 26.2 (3C, tBuTBS), 26.0 (3C, tBuTBS), 25.9 (3C, tBuTBS), 25.8 (6C, tBuTBS), 18.6 (CqTBS), 18.2 (CqTBS), 18.1 (CqTBS), 17.9 (CqTBS), 17.8 (CqTBS), −4.3 (MeTBS), −4.5 (3C, MeTBS), −4.6 (MeTBS), −4.7 (MeTBS), −4.9 (MeTBS), −5.0 (MeTBS), −5.2 (MeTBS), −5.3 (MeTBS). HRMS (ESI) m/z: calcd for C67H106N13O9Si5 [M + H]+: 1376.7077; found 1376.7056.
Compound 9a: Following the general procedure A for CuAAC, starting from alkyne 2a (578 mg, 0.77 mmol) and azido compound 7 (277 mg, 0.38 mmol) and using Cyclohexane/EtOAc 7:3 as eluent for flash chromatography purification, compound 9a was obtained as a white foam (384 mg, 60%). 1H NMR (500 MHz, CDCl3): δ 8.75 (s, 1H, H8 or H2), 8.51 (s, 1H, H8 or H2), 8.19 (s, 1H, H8 or H2), 8.04 (bs, 1H, NH), 7.92 (bs, 1H, H8 or H2), 7.80 (s, 1H, HTriazole), 7.42–7.40 (m, 2H, HBz), 7.23–7.20 (m, 1H, HBz), 7.11–7.08 (m, 2H, HBz), 5.98 (d, J = 5.6 Hz, 1H, H1′a), 5.86 (d, J = 6.2 Hz, 1H, H1′b), 5.63 (s, 2H, CH2N), 5.04–5.02 (m, 1H, H2′b), 4.84 (dd, J = 6.3, 14.2 Hz, 1H, H5′b), 4.61–4.57 (m, 2H, H5′b and H2′a), 4.39–4.38 (m, 1H, H3′b), 4.35–4.32 (m, 1H, H4′b), 4.25–4.23 (m, 1H, H3′a), 4.10–4.08 (m, 1H, H4′a), 3.96 (dd, J = 4.4, 11.3 Hz, 1H, H5′a), 3.75 (dd, J = 3.0, 11.4 Hz, 1H, H5′a), 1.56 (s, 9H, tBuBoc), 0.93 (s, 9H, tBuTBS), 0.91 (s, 9H, tBuTBS), 0.87 (s, 9H, tBuTBS), 0.73 (s, 18H, tBuTBS), 0.11 (s, 3H, MeTBS), 0.10 (s, 3H, MeTBS), 0.08 (s, 6H, MeTBS), 0.02 (s, 3H, MeTBS), −0.08 (s, 3H, MeTBS), −0.11 (s, 6H, MeTBS), −0.43 (s, 6H, MeTBS). 13C NMR (126 MHz, CDCl3): δ 172.1 (C=O), 153.7 (C2 or C8), 153.1 (Cq), 152.8 (Cq), 152.0 (C2 or C8), 150.6 (Cq), 150.4 (Cq), 149.6 (Cq), 144.7 (Cq), 142.9 (C2 or C8), 142.6 (C2 or C8), 135.9 (Cq), 130.9 (CBz), 129.0 (2C, CBz), 127.9 (2C, CBz), 127.3 (CqBz), 124.9 (CHTriazole), 122.9 (Cq), 89.8 (C1′b), 88.3 (C1′a), 86.0 (C4′a), 84.2 (C4′b), 82.4 (CqBoc), 75.8 (C2′a), 73.4 (C3′b), 73.1 (C2′b), 72.2 (C3′a), 62.7 (C5′a), 51.7 (C5′b), 44.0 (CH2N), 28.3 (3C, tBuBoc), 26.2 (3C, tBuTBS), 25.9 (3C, tBuTBS), 25.8 (3C, tBuTBS), 25.7 (6C, tBuTBS), 18.6 (CqTBS), 18.2 (CqTBS), 18.1 (CqTBS), 17.9 (2C, CqTBS), −4.3 (MeTBS), −4.5 (3C, 3 MeTBS), −4.6 (MeTBS), −4.8 (MeTBS), −5.0 (MeTBS), −5.1 (MeTBS), −5.2 (MeTBS), −5.3 (MeTBS). HRMS (ESI) m/z: calcd for C65H108N13O10Si5 [M − H]−: 1370.7188; found: 1370.7132.
Compound 9b: Following the general procedure A for CuAAC, starting from alkyne 2b (50 mg, 0.067 mmol) and azido compound 7 (49 mg, 0.08 mmol) and using Cyclohexane/EtOAc 7:3 as eluent for flash chromatography purification, compound 9b was obtained as a white foam (68 mg, 74%). 1H NMR (500 MHz, CDCl3): δ 8.74 (s, 1H, H2b), 8.70 (s, 1H, H2a), 8.38 (s, 1H, H8a), 8.00 (s, 1H, NH), 7.90 (s, 1H, H8b), 7.70 (s, 1H, HTriazole), 6.07 (d, J = 4.7 Hz, 1H, H1′a), 5.86 (d, J = 6.2 Hz, 1H, H1′b), 5.30 (s, 2H, CH2N), 5.10 (dd, J = 6.2, 4.3 Hz, 1H, H2′b), 4.89 (dd, J = 14.3, 6.3 Hz, 1H, H5′b), 4.65–4.58 (m, 2H, H5′b, H2′a), 4.41 (dd, J = 4.3, 2.4 Hz, 1H, H3′b), 4.36–4.31 (m, 2H, H4′b, H3′a), 4.15–4.12 (m, 1H, H4′a), 4.03 (dd, J = 11.4, 4.1 Hz, 1H, H5′a), 3.79 (dd, J = 11.4, 2.9 Hz, 1H, H5′a), 1.57 (s, 9H, tBuBoc), 1.38 (s, 9H, tBuBoc), 0.95 (s, 9H, tBuTBS), 0.93 (s, 9H, tBuTBS), 0.88 (s, 9H, tBuTBS), 0.80 (s, 9H, tBuTBS), 0.74 (s, 9H, tBuTBS), 0.13 (s, 3H, MeTBS), 0.12 (s, 3H, MeTBS), 0.10 (s, 3H, MeTBS), 0.09 (s, 3H, MeTBS), 0.04 (s, 3H, MeTBS), −0.04 (s, 3H, MeTBS), −0.06 (s, 3H, MeTBS), −0.10 (s, 3H, MeTBS), −0.21 (s, 3H, MeTBS), −0.42 (s, 3H, MeTBS). 13C NMR (126 MHz, CDCl3): δ 153.4 (C2b), 153.1 (Cq), 152.9 (Cq), 152.5 (Cq), 151.9 (C2a), 150.6 (Cq), 150.4 (C=O), 149.6 (C=O), 145.8 (CqTriazole), 142.6 (C8b), 142.2 (C8a), 127.7 (Cq), 124.1 (CHTriazole), 122.9 (Cq), 90.1 (C1′b), 88.8 (C1′a), 85.3 (C4′a), 84.4 (C4′b), 82.5 (2C, CqBoc), 75.9 (C2′a), 73.5 (C3′b), 73.1 (C2′b), 71.8 (C3′a), 62.5 (C5′a), 51.8 (C5′b), 43.4 (CH2N), 28.3 (3C, tBuBoc), 28.0 (3C, tBuBoc), 26.2 (3C, tBuTBS), 26.0 (3C, tBuTBS), 25.9 (3C, tBuTBS), 25.8 (3C, tBuTBS), 25.7 (3C, tBuTBS), 18.7 (CqTBS), 18.2 (CqTBS), 18.1 (CqTBS), 18.0 (CqTBS), 17.9 (CqTBS), −4.2 (MeTBS), −4.4 (MeTBS), −4.5 (MeTBS), −4.5 (MeTBS), −4.6 (MeTBS), −4.7 (MeTBS), −4.8 (MeTBS), −5.1 (MeTBS), −5.2 (MeTBS), −5.3 (MeTBS). HRMS (ESI) m/z: calcd for C63H114N13O11Si5 [M + H]+: 1368.7601; found 1368.7601.
Compound 10a: Following the general procedure A for CuAAC, starting from alkyne 3a (200 mg, 0.26 mmol) and azido compound 6 (194 mg, 0.31 mmol) and using Cyclohexane/EtOAc 5:5 as eluent for flash chromatography purification, compound 10a was obtained as a white foam (258 mg, 72%). 1H NMR (500 MHz, CDCl3): δ 8.70 (s, 1H, H2b), 8.26 (s, 1H, H2a), 8.05 (d, J = 7.4 Hz, 2H, HBz), 7.95 (d, J = 7.2 Hz, 2H, HBz), 7.79 (s, 2H, H8a and H8b), 7.74 (s, 1H, HTriazole), 7.62 (t, J = 7.3 Hz, 1H, HBz), 7.54 (t, J = 7.5 Hz, 2H, HBz), 7.40 (t, J = 7.3 Hz, 1H, HBz), 7.31 (t, J = 7.6 Hz, 2H, HBz), 5.85–5.80 (m, 2H, H1′a and H1′b), 5.43–5.31 (m, 2H, CH2N), 5.13–5.07 (m, 1H, H2′b), 4.85 (dd, J = 14.2, 4.8 Hz, 1H, H5′b), 4.73 (dd, J = 14.2, 7.2 Hz, 1H, H5′b), 4.50 (t, J = 3.8 Hz, 1H, H3′b), 4.44 (t, J = 4.4 Hz, 1H, H2′a), 4.40–4.36 (m, 1H, H4′b), 4.23 (t, J = 4.3 Hz, 1H, H3′a), 4.05 (q, J = 3.9 Hz, 1H, H4′a), 3.86 (dd, J = 11.4, 4.1 Hz, 1H, H5′a), 3.69 (dd, J = 11.4, 3.4 Hz, 1H, H5′a), 0.91 (s, 9H, tBuTBS), 0.90 (s, 9H, tBuTBS), 0.86 (s, 9H, tBuTBS), 0.80 (s, 9H, tBuTBS), 0.79 (s, 9H, tBuTBS), 0.10 (s, 3H, MeTBS), 0.07 (s, 3H, MeTBS), 0.06 (s, 3H, MeTBS), 0.05 (s, 3H, MeTBS), 0.04 (s, 3H, MeTBS), 0.03 (s, 3H, MeTBS), −0.04 (s, 3H, MeTBS), −0.06 (s, 3H, MeTBS), −0.19 (s, 3H, MeTBS), −0.34 (s, 3H, MeTBS). 13C NMR (126 MHz, CDCl3): δ 177.1 (C=O), 164.7 (C=O), 152.7 (C2b), 151.2 (Cq), 150.1 (Cq), 146.7 (C2a), 146.6 (Cq), 145.2 (Cq), 143.1 (C8b), 141.9 (CqTriazole), 138.8 (C8a), 135.8 (CqBz), 133.8 (CqBz), 133.0 (CBz), 131.9 (CBz), 129.8 (2C, CBz), 129.0 (2C, CBz), 128.1 (4C, CBz), 125.6 (CHTriazole), 124.2 (Cq), 122.5 (Cq), 90.5 (C1′b), 88.5 (C1′a), 85.1 (C4′a), 83.5 (C4′b), 76.0 (C2′a), 73.5 (C3′b), 73.4 (C2′b), 71.7 (C3′a), 62.6 (C5′a), 51.8 (C5′b), 43.7 (CH2N), 26.2 (3C, tBuTBS), 26.0 (3C, tBuTBS), 25.9 (3C, tBuTBS), 25.8 (3C, tBuTBS), 25.80 (3C, tBuTBS), 18.6 (CqTBS), 18.2 (CqTBS), 18.1 (CqTBS), 18.0 (2C, CqTBS), −4.2 (MeTBS), −4.3 (MeTBS), −4.4 (MeTBS), −4.5 (MeTBS), −4.6 (2C MeTBS), −4.7 (MeTBS), −4.9 (MeTBS), −5.2 (MeTBS), −5.3 (MeTBS). HRMS (ESI) m/z: calcd for C67H106N13O9Si5 [M + H]+: 1376.7077; found 1376.7060.
Compound 11a: Following the general procedure A for CuAAC, starting from alkyne 3a (45 mg, 0.059 mmol) and azido compound 7 (19 mg, 0.03 mmol) and using Cyclohexane/EtOAc 7:3 as eluent for flash chromatography purification, compound 11a was obtained as a white foam (37 mg, 89%). 1H NMR (500 MHz, CDCl3): δ 8.67 (s, 1H, H2b), 8.30 (s, 1H, H2a), 7.97 (m, 2H, HBz), 7.85 (s, 1H, H8a), 7.78 (s, 1H, H8b), 7.77 (s, 1H, HTriazole), 7.46–7.43 (m, 1H, HBz), 7.36–7.32 (m, 2H, HBz), 5.85 (d, J = 4.5 Hz, 1H, H1′a), 5.78 (d, J = 5.4 Hz, 1H, H1′b), 5.44 (d, J = 14.6 Hz, 1H, CH2N), 5.36 (d, J = 14.6 Hz, 1H, CH2N), 5.12 (m, 1H, H2′b), 4.87 (dd, J = 5.2, 14.3 Hz, 1H, H5′b), 4.70 (dd, J = 7.3, 14.2 Hz, 1H, H5′b), 4.48–4.47 (m, 1H, H3′b), 4.46–4.44 (m, 1H, H2′a), 4.37–4.34 (m, 1H, H4′b), 4.24–4.23 (m, 1H, H3′a), 4.07–4.05 (m, 1H, H4′a), 3.87 (dd, J = 4.0, 11.3 Hz, 1H, H5′a), 3.71 (dd, J = 3.3, 11.3 Hz, 1H, H5a’), 1.57 (s, 9H, tBuBoc), 0.91 (s, 9H, tBuTBS), 0.89 (s, 9H, tBuTBS), 0.87 (s, 9H, tBuTBS), 0.80 (s, 9H, tBuTBS), 0.77 (s, 9H, tBuTBS), 0.08 (s, 3H, MeTBS), 0.07 (s, 6H, MeTBS), 0.05 (s, 3H, MeTBS), 0.04 (s, 3H, MeTBS), 0.02 (s, 3H, MeTBS), −0.04 (s, 3H, MeTBS), −0.08 (s, 3H, MeTBS), −0.18 (s, 3H, MeTBS), −0.39 (s, 3H, MeTBS). 13C NMR (126 MHz, CDCl3): δ 176.8 (C=O), 152.9 (C2b), 150.3 (2C, Cq), 149.6 (C=O), 146.7 (2C, C2a and Cq), 145.4 (Cq), 142.6 (C8b), 141.9 (CqTriazole), 139.0 (C8a), 135.6 (Cq), 131.9 (CBz), 129.7 (2C, CBz), 128.1 (2C, CBz), 125.6 (CHTriazole), 122.8 (Cq), 122.6 (Cq), 90.4 (C1′b), 88.5 (C1′a), 85.1 (C4′a), 83.6 (C4′b), 82.5 (CqBoc), 76.0 (C2′a), 73.5 (C3′b), 73.3 (C2′b), 71.7 (C3′a), 62.6 (C5′a), 51.9 (C5′b), 43.8 (CH2N), 28.2 (3C, tBuBoc), 26.1 (3C, tBuTBS), 26.0 (3C, tBuTBS), 25.9 (3C, tBuTBS), 25.8 (3C, tBuTBS), 25.7 (3C, tBuTBS), 18.6 (CqTBS), 18.2 (CqTBS), 18.1 (CqTBS), 18.0 (CqTBS), 17.9 (CqTBS), −4.2 (MeTBS), −4.3 (MeTBS), −4.4 (MeTBS), −4.5 (MeTBS), −4.6 (MeTBS), −4.7 (2C, MeTBS), −5.0 (MeTBS), −5.2 (MeTBS), −5.3 (MeTBS). HRMS (ESI) m/z: calcd for C65H108N13O10Si5 [M − H]−: 1370.7188; found: 1370.7138.
Compound 11b: Following the general procedure A for CuAAC, starting from alkyne 3b (50 mg, 0.067 mmol) and azido compound 7 (49 mg, 0.08 mmol) and using Cyclohexane/EtOAc 6:4 as eluent for flash chromatography purification, compound 11b was obtained as a white foam (54 mg, 60%). 1H NMR (500 MHz, CDCl3): δ 8.72 (s, 1H, H2a or H2b), 8.10 (s, 1H, H2a or H2 b), 8.10 (s, 1H, H8a or H8b), 8.01 (s, 1H, NH), 7.93 (s, 1H, H8a or H8b), 7.90 (s, 1H, HTriazole), 5.85–5.83 (m, 2H, H1′a and H1′b), 5.30–5.21 (m, 2H, CH2N), 5.17 (dd, J = 5.8, 4.3 Hz, 1H, H2′b), 4.92 (dd, J = 14.3, 6.5 Hz, 1H, H5′b), 4.62 (dd, J = 14.3, 6.5 Hz, 1H, H5′b), 4.48–4.45 (m, 1H, H3′b), 4.39–4.35 (m, 1H, H4′b), 4.32–4.27 (m, 2H, H2′a, H3′a), 4.09–4.06 (m, 1H, H4′a), 3.98 (dd, J = 11.6, 3.4 Hz, 1H, H5′a), 3.75 (dd, J = 11.6, 2.5 Hz, 1H, H5′a), 1.56 (s, 18H, tBuBoc), 0.92 (s, 9H, tBuTBS), 0.90 (s, 9H, tBuTBS), 0.86 (s, 9H, tBuTBS), 0.85 (s, 9H, tBuTBS), 0.76 (s, 9H, tBuTBS), 0.10 (s, 3H, MeTBS), 0.10 (s, 3H, MeTBS), 0.07 (s, 3H, MeTBS), 0.05 (s, 3H, MeTBS), 0.03 (s, 3H, MeTBS), 0.01 (s, 3H, MeTBS), −0.03 (s, 3H, MeTBS), −0.08 (s, 6H, MeTBS), −0.40 (s, 3H, MeTBS). 13C NMR (126 MHz, CDCl3): δ 153.0 (C2a or C2b), 150.5 (Cq), 150.4 (Cq), 149.6 (2C, C=O), 146.9 (CqTriazole), 146.5 (C2a or C2b), 144.2 (Cq), 142.7 (Cq), 142.3 (C8a or C8b), 138.1 (C8a or C8b), 125.7 (CHTriazole), 123.0 (Cq), 122.3 (Cq), 90.4 (C1′a or C1′b), 88.9 (C1′a or C1′b), 84.1 (C4′a), 83.9 (C4′b), 82.4 (2C, CqBoc), 76.7 (C2′a), 73.5 (C3′b), 73.2 (C2′b), 70.5 (C3′a), 61.7 (C5′a), 51.9 (C5′b), 43.1 (CH2N), 28.3 (3C, tBuBoc), 28.3 (3C, tBuBoc), 26.3 (3C, tBuTBS), 26.0 (3C, tBuTBS), 25.9 (6C, tBuTBS), 25.8 (3C, tBuTBS), 18.7 (CqTBS), 18.2 (CqTBS), 18.1 (CqTBS), 18.0 (CqTBS), 17.9 (CqTBS), −4.1 (MeTBS), −4.4 (2C, 2 MeTBS), −4.5 (MeTBS), −4.6 (MeTBS), −4.7 (MeTBS), −4.9 (MeTBS), −5.1 (MeTBS), −5.2 (MeTBS), −5.3 (MeTBS). HRMS (ESI) m/z: calcd for C63H114N13O11Si5 [M + H]+: 1368.7601; found 1368.7611.
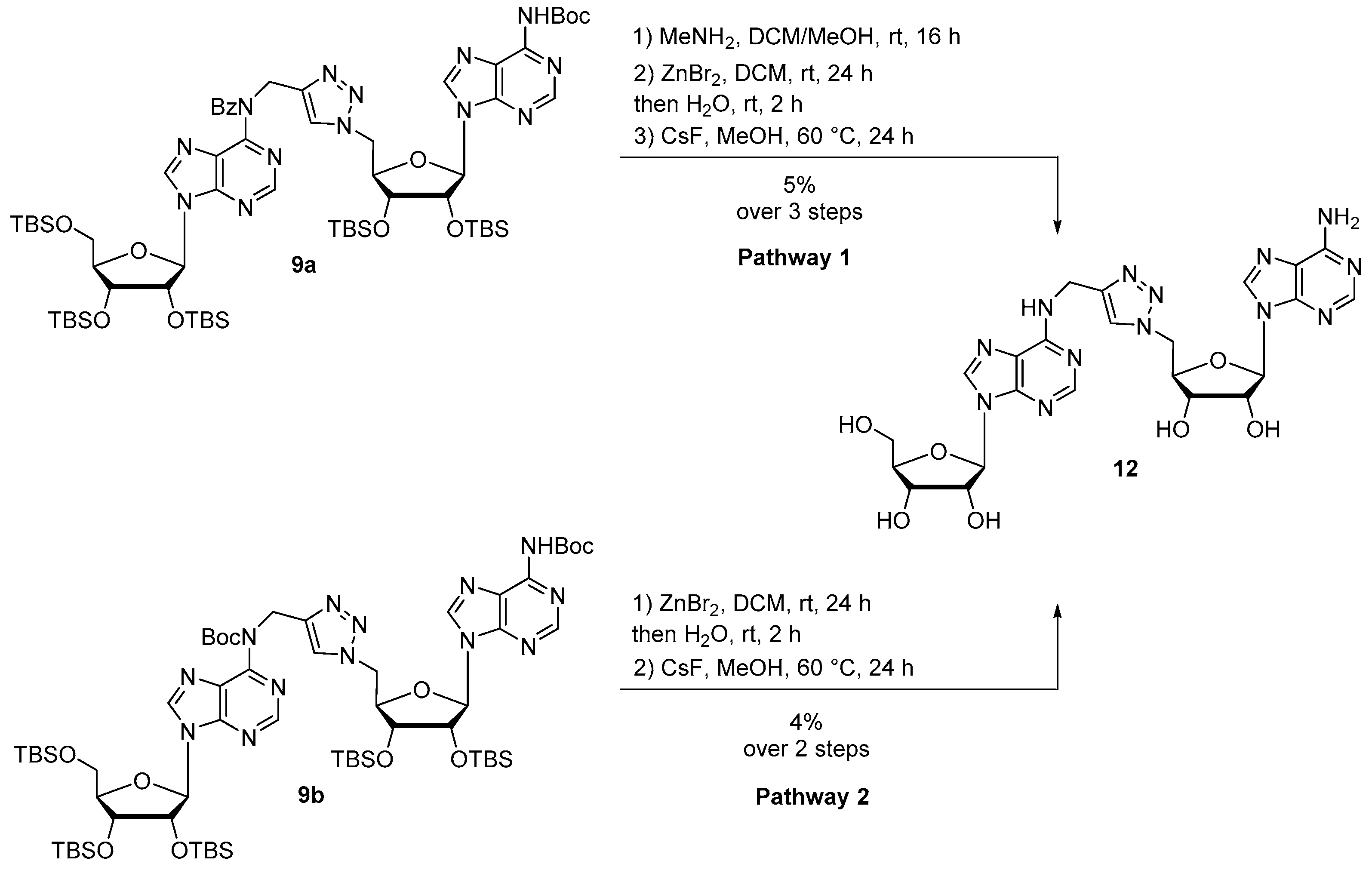
Synthesis of compound 12 following pathway 1: Protected compound 9a (768 mg, 0.56 mmol) was dissolved in DCM/MeOH 4:1 (10 mL) and MeNH2 33% in EtOH (3.6 mL, 28.0 mmol) was added at 0 °C to the solution. The reaction was stirred at room temperature for 16 h, then diluted in DCM and washed with brine. The combined organic layers were dried over MgSO4 and concentrated. The residue was purified by silica gel chromatography (eluent: Cyclohexane/EtOAc 5:5) to provide the debenzoylated compound as a white foam. The residue was then dissolved in DCM (1.4 mL) and ZnBr2 (686 mg, 2.80 mmol) was added. The reaction mixture was vigorously stirred at room temperature for 24 h. Then, water (5.8 mL) was added and the reaction mixture was stirred for 2 additional hours. A work up was performed with DCM and brine. The combined organic layers were dried over MgSO4 and concentrated. The residue was purified by silica gel chromatography (eluent: Cyclohexane/EtOAc 3:7 then DCM/MeOH 9:1) to afford a yellow powder. The resulting compound was engaged in the last deprotection step and was dissolved in MeOH (10 mL) and CsF (8.5 g, 112 mmol) was added. The reaction mixture was stirred at 60 °C for 24 h, concentrated and diluted in water. The residue was purified by HPLC to afford compound 12 as a white foam (17 mg, 5% over 3 steps).
Synthesis of compound 12 following pathway 2: Protected compound 9b (60 mg, 0.046 mmol) was dissolved in DCM (1 mL) and ZnBr2 (58 mg, 0.21 mmol) was added. The reaction mixture was vigorously stirred at room temperature for 24 h. Then, water (2 mL) was added and the reaction mixture was stirred for 2 additional hours. A work up was performed with DCM and brine. The combined organic layers were dried over MgSO4 and concentrated. The residue was purified by silica gel chromatography (eluent Cyclohexane/EtOAc 2:8) to afford a yellow pale powder. The resulting compound was engaged in the last deprotection step and was dissolved in MeOH (10 mL) before adding CsF (480 mg, 3.2 mmol). The reaction mixture was stirred at 60 °C for 24 h, concentrated and purified by HPLC to afford compound 12 as a white foam (1.2 mg, 4% over 2 steps).
Compound 12: 1H NMR (500 MHz, (CD3)2SO): δ 8.35 (s, 1H, H2 or H8), 8.26 (bs, 1 H, NH), 8.23 (s, 1H, H2 or H8), 8.19 (s, 1H, H2 or H8), 8.14 (s, 1H, H2 or H8), 7.81 (s, 1H, HTriazole), 7.28 (bs, 2H, NH2), 5.90–5.89 (m, 2H, H1′a and H1′b), 5.55 (d, J = 5.9 Hz, 1H, OH2′b), 5.43–5.41 (m, 2H, OH2′a and OH3′b), 5.36–5.34 (m, 1H, OH5′a), 5.16 (d, J = 4.6 Hz, 1H, OH3′a), 4.74–4.67 (m, 4H, H5′b and CH2N), 4.67–4.63 (m, 1H, H2′b), 4.63–4.60 (m, 1H, H2′a), 4.25–4.20 (m, 2H, H4′b and H3′b), 4.16–4.14 (m, 1H, H3′a), 3.97–3.95 (m, 1H, H4′a), 3.69–3.65 (m, 1H, H5′a), 3.58–3.53 (m, 1H, H5′a). 13C NMR (126 MHz, (CD3)2SO): δ 156.1 (Cq), 154.2 (Cq), 152.6 (C2 or C8), 152.2 (C2 or C8), 149.3 (Cq), 148.5 (Cq), 145.3 (Cq), 139.9 (C2 or C8), 139.8 (C2 or C8), 123.6 (CHTriazole), 119.8 (Cq), 119.2 (Cq), 87.9 (C1′a or C1′b), 87.7 (C1′a or C1′b), 85.8 (C4′a), 82.5 (C4′b), 73.4 (C2′a), 72.5 (C2′b), 71.0 (C3′b), 70.6 (C3′a), 61.6 (C5′a), 51.3 (C5′b), 35.3 (CH2N). HRMS (ESI) m/z: calcd for C23H26N13O7 [M − H]−: 596.2078; found: 596.2065. HPLC purity: 96.3%; tR = 17.8 min (MeCN/H2O 0:100 to 100:0 over 30 min).
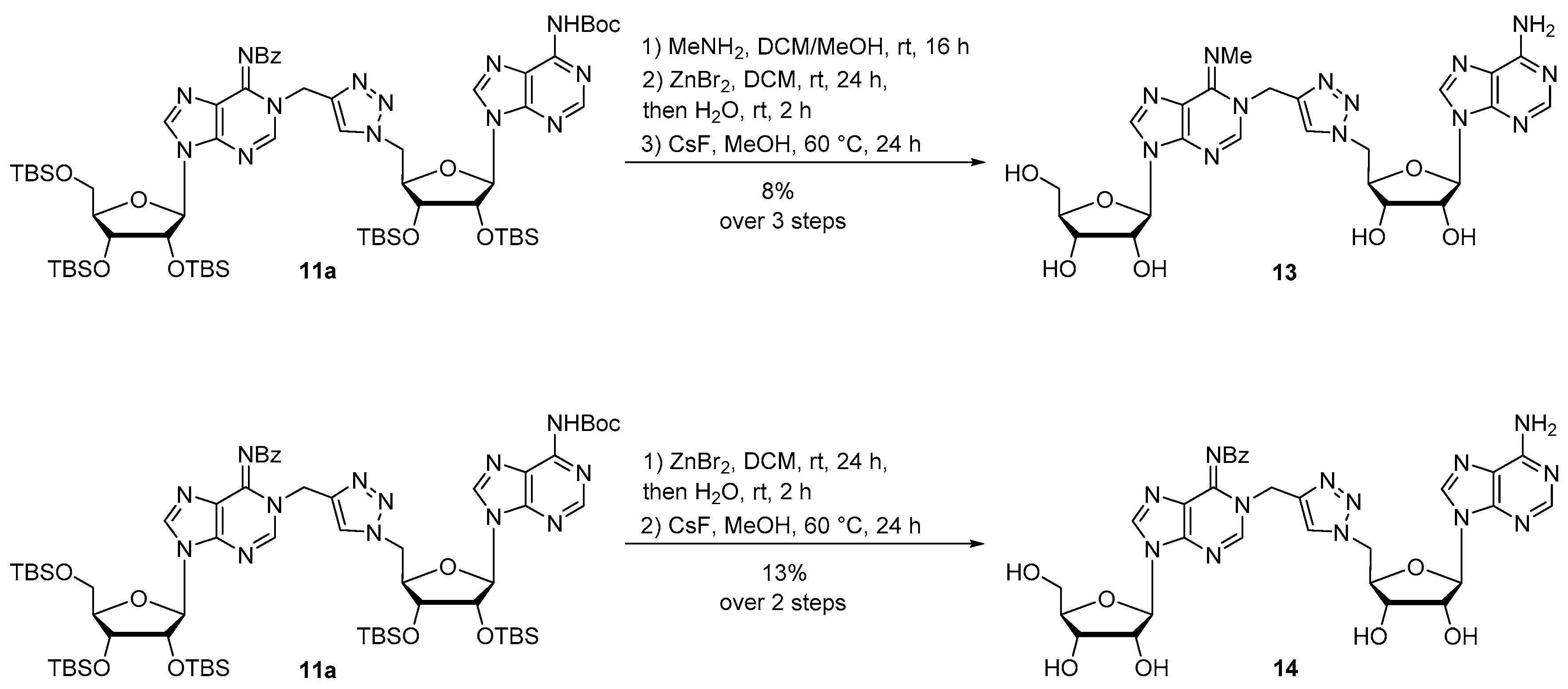
Compound 13: Protected compound 11a (400 mg, 0.3 mmol) was dissolved in DCM/MeOH 4:1 (5 mL) and MeNH2 33% in EtOH (1.8 mL, 15.0 mmol) was added at 0 °C to the solution. The reaction was stirred at room temperature for 16 h, then diluted in DCM and washed with brine. The combined organic layers were dried over MgSO4 and concentrated. The residue was purified by silica gel chromatography (eluent: EtOAc/MeOH 9:1) to provide the debenzoylated compound as a white foam. The residue was then dissolved in DCM (1 mL) and ZnBr2 (117 mg, 0.47 mmol) was added. The reaction mixture was vigorously stirred at room temperature for 24 h. Then, water (2 mL) was added and the reaction mixture was stirred for 2 additional hours. A work up was performed with DCM and brine. The combined organic layers were dried over MgSO4 and concentrated. The resulting compound was engaged in the last deprotection step and was dissolved in MeOH (10 mL) and CsF (2.9 g, 19 mmol) was added. The reaction mixture was stirred at 60 °C for 24 h, concentrated and diluted in water. The residue was purified by HPLC to afford compound 13 as a white foam (15 mg, 8% over 3 steps). 1H NMR (500 MHz, CD3OD): δ 8.35 (2s, 1H, H2a), 8.23 (s, 1H, H8a or H8b), 8.13 (2s, 1H, H8a or H8b), 8.03 (2s, 1H, H2b), 7.84 (s, 0.5H, HTriazole), 7.69 (s, 0.5H, HTriazole), 6.01–5.98 (m, 1H, H1′a), 5.95–5.93 (m, 1H, H1′b), 5.32 (2s, 2H, CH2N), H5′b masked in the residual pick of water, 4.79–4.75 (m, 0.5H, H2′b), 4.71–4.68 (m, 0.5H, H2′b), 4.61 (t, J = 5 Hz, 1H, H2′a), 4.47–4.41 (m, 0.5H, H3′b), 4.37–4.31 (m, 1.5H, H3′b and H3′a), 4.14–4.13 (m, 2H, H4′a and H4′b), 3.88–3.85 (m, 1H, H5′a), 3.78–3.74 (m, 1H, H5′a), 3.56 (2s, 3H, CH3). 13C NMR (126 MHz, CD3OD): δ 178.3 (Cq), 163.0 (Cq), 157.3 (Cq), 153.7 (C8a or C8b), 150.4 (Cq), 150.0 (Cq), 149.8 (Cq), 149.4 (Cq), 147.8 (Cq), 147.5 (Cq), 142.0 (C2a or C2b), 126.6 (CHTriazole), 125.7 (CHTriazole), 126.2 (Cq), 120.8 (Cq), 90.9 (C1′b), 90.4 (C1′a), 87.6 (2C, C4′a and C4′b), 83.9 (C3′a), 76.1 (C2′a), 74.2 (C2′b), 72.2 (C3′b), 62.9 (C5′a), 52.5 (C5′b), 45.1 (CH2N), 43.9 (CH2N), 37.9 (CH3), 35.0 (CH3). LRMS (ESI) m/z: calcd for C24H30N13O7 [M + H]+: 612.23; found: 612.33. HPLC purity: 95.3%; rt = 14.6 min (MeCN/H2O 0:100 to 100:0 over 30 min).
Compound 14: Compound 11a (107 mg, 0.08 mmol) was dissolved in DCM (1 mL) and ZnBr2 (95 mg, 0.40 mmol) was added. The reaction mixture was vigorously stirred at room temperature for 24 h. Water (2 mL) was added and the reaction mixture was stirred for 2 additional hours. A work up was performed with DCM and brine. The combined organic layers were dried over MgSO4 and concentrated. The residue was purified by silica gel chromatography (eluent: Cyclohexane/EtOAc 2:8) to afford a white foam (52 mg). The purified intermediate was then dissolved in MeOH (5 mL) and CsF (1.2 g, 8 mmol) was added. The reaction mixture was stirred at 60 °C for 24 h, concentrated and diluted in water. The residue was purified by HPLC to afford compound 14 as a white foam (7 mg, 13% over 2 steps). 1H NMR (500 MHz, CD3OD): δ 8.53 (s, 1H, H2 or H8), 8.12 (s, 1H, H2 or H8), 8.06 (s, 1H, H2 or H8), 7.90 (s, 2H, H2 or H8 and HTriazole), 7.83–7.81 (m, 2H, HBz), 7.45–7.42 (m, 1H, HBz), 7.33–7.30 (m, 2H, HBz), 5.96 (d, J = 5.0 Hz, 1H, H1′a), 5.90 (d, J = 5.0 Hz, 1H, H1′b), 5.43 (s, 2H, CH2N), H5′b masked in the residual pick of water, 4.64–4.60 (m, 2H, H2′b and H2′a), 4.47 (t, J = 5.0 Hz, 1H, H3′b), 4.37–4.34 (m, 1H, H4′b), 4.32–4.30 (m, 1H, H3′a), 4.11 (q, J = 3.3 Hz, 1H, H4′a), 3.83 (dd, J = 5.0, 15.0 Hz, 1H, H5′a), 3.74 (dd, J = 5.0, 15.0 Hz, 1H, H5′a). 13C NMR (126 MHz, CD3OD): δ 178.8 (C=O), 157.2 (C2 or C8), 153.7 (C2 or C8), 149.2 (Cq), 148.5 (Cq), 146.9 (Cq), 143.3 (Cq), 141.5 (C2 or C8), 141.2 (Cq), 136.7 (C2 or C8), 133.2 (Cq), 130.6 (CBz), 129.1 (2C, CBz), 129.0 (2C, CBz), 127.0 (CHTriazole), 123.2 (Cq), 120.6 (Cq), 91.0 (C1′a), 90.4 (C1′b), 87.4 (C4′a), 83.4 (C4′b), 76.0 (C2′a), 74.5 (C2′b), 72.2 (C3′b), 72.0 (C3′a), 62.8 (C5′a), 52.4 (C5′b), 45.0 (CH2N). HRMS (ESI) m/z: calcd for C30H32N13O8 [M + H]+: 702.2497; found: 702.2470. HPLC purity: 97.1%; tR = 19.0 min (MeCN/H2O 0:100 to 100:0 over 30 min).
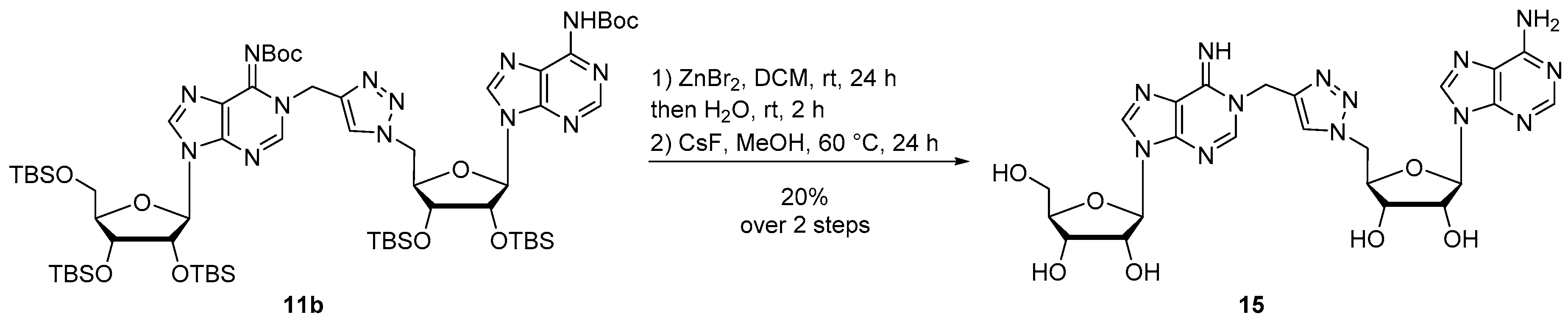
Compound 15: Protected compound 11b (50 mg, 0.036 mmol) was dissolved in DCM (1 mL) and ZnBr2 (44 mg, 0.18 mmol) was added. The reaction mixture was vigorously stirred at room temperature for 24 h then water (2 mL) was added and the reaction mixture was stirred for 2 additional hours. A work up was performed with DCM and brine. The combined organic layers were dried over MgSO4 and concentrated. The residue was purified by silica gel chromatography (eluent Cyclohexane/EtOAc 1:9) to afford a yellow pale powder. The resulting compound was engaged in the last deprotection step and was dissolved in MeOH (10 mL) and CsF (636 mg, 4.2 mmol) was added. The reaction mixture was stirred at 60 °C for 24 h, concentrated and purified by HPLC to afford compound 15 as a white foam (4.3 mg, 20% over 2 steps). 1H NMR (500 MHz, (CD3)2SO): δ 8.27 (s, 1H, H8b), 8.22 (s, 1H, H2a), 8.15 (s, 1H, H2b), 8.14 (s, 1H, H8a), 7.96 (s, 1H, HTriazole), 7.28 (bs, 2H, NH2), 5.89 (d, J = 5.5 Hz, 1H, H1′b), 5.75 (d, J = 5.9 Hz, 1H, H1′a), 5.28–5.17 (m, 2H, CH2N), 4.74–4.70 (m, 2H, H5′b), 4.64–4.61 (m, 1H, H2′b), 4.48–4.44 (m, 1H, H2′a), 4.27–4.20 (m, 2H, H3′b and H4′b), 4.10 (d, J = 4.9 Hz, 1H, H3′a), 3.92 (q, J = 3.8 Hz, 1H, H4′a), 3.64 (dd, J = 12.0, 4.0 Hz, 1H, H5′a), 3.53 (dd, J = 12.0, 3.9 Hz, 1H, H5′a). 13C NMR (126 MHz, (CD3)2SO): δ 156.1 (Cq), 153.2 (Cq), 152.6 (C2b), 149.3 (Cq), 148.2 (C2a), 142.5 (CqTriazole), 141.2 (Cq), 139.8 (C8b), 137.9 (C8a), 124.5 (CHTriazole), 122.8 (Cq), 119.2 (Cq), 87.6 (C1′a or C1′b), 87.6 (C1′a or C1′b), 85.6 (C4′a), 82.3 (C4′b), 73.9 (C2′a), 72.5 (C2′b), 70.9 (C3′b), 70.4 (C3′a), 61.4 (C5′a), 51.4 (C5′a), 41.2 (CH2N). HRMS (ESI) m/z: calcd for C23H28N13O7 [M + H]+: 598.2229; found: 598.2229. HPLC purity: 96.2%; tR = 17.5 min (MeCN/H2O 0:100 to 100:0 over 30 min).
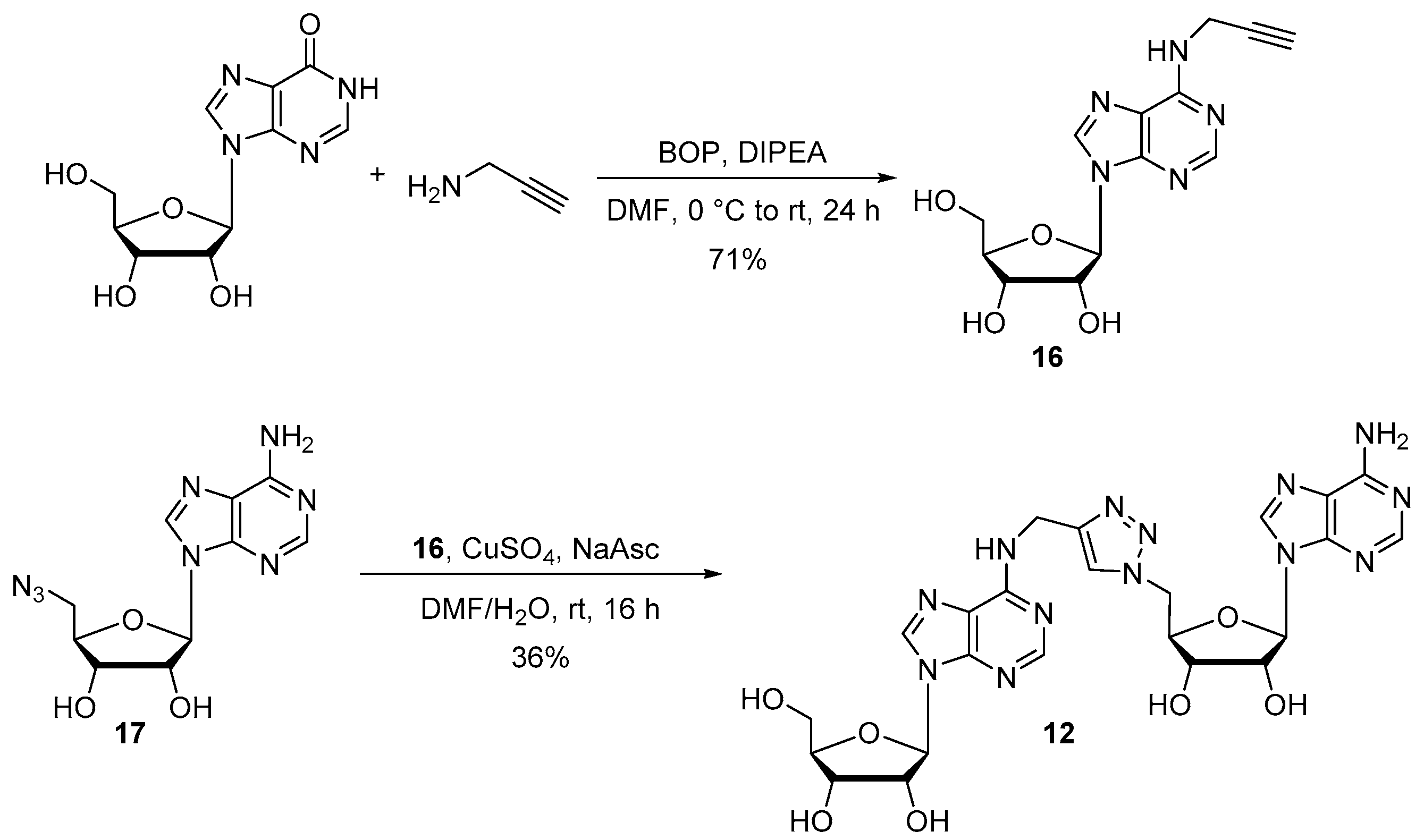
Compound 16: Propargyl amine (1.4 μL, 0.031 mmol), BOP (13 mg, 0.031 mmol) and DIPEA (6.8 μL, 0.039 mmol) were added at 0 °C to a solution of inosine (7 mg, 0.026 mmol) in DMF (1 mL). The reaction mixture was stirred at room temperature for 24 h and then concentrated under reduced pressure. The crude was diluted in water and purified by HPLC to afford compound 16 as a white foam (5.7 mg, 71%). 1H NMR (500 MHz, D2O): δ 8.31 (s, 1H, H8), 8.27 (s, 1H, H2), 6.07 (d, J = 5 Hz, 1H, H1′), H2′ masked in the residual pick of water, 4.47–4.45 (m, 1H, H3′), 4.35 (s, 2H, CH2N), 4.33 (q, J = 3.3 Hz, 1H, H4′), 3.97 (dd, J = 5, 11 Hz, 1H, H5′), 3.88 (dd, J = 5, 10 Hz, 1H, H5′), 2.69 (t, J = 2.5 Hz, 1H, C≡CH). 13C NMR (126 MHz, D2O): δ = 153.9 (C5), 152.3 (C2), 147.8 (C4), 140.4 (C8), 119.6 (C6), 88.4 (C1′), 85.8 (C4′), 80.3 (C≡C-CH2), 73.7 (C2′), 71.9 (C≡CH), 70.6 (C3′), 61.5 (C5′), 30.2 (CH2N). HRMS (ESI) m/z: calcd for C13H14N5O4 [M − H]−: 304.1045; found: 304.1048.
Compound
17: Pyridine (610 μL, 7.48 mmol) and thionyl chloride (1.4 mL, 18.7 mmol) were added at 0 °C, over 5 min to a solution of adenosine (1 g, 3.74 mmol) in MeCN (10 mL). The reaction mixture was stirred at 0 °C for 3 h before being warmed to room temperature and stirred for 16 h. The resulting precipitate was filtered and dissolved in water/MeOH (5:1) and aqueous ammonia (25%, 2 mL) was added. The reaction mixture was stirred at room temperature for 30 min and the solvent was removed under reduced pressure to provide 5′-chloroadenosine [
66]. The resulting 5′-chloroadenosine was then solubilized in DMF (5 mL) and sodium azide (1.2 g, 18.7 mmol) was added. The reaction mixture was heated at 80 °C for 5 h, and cooled to room temperature. The excess of sodium azide was removed by filtration and the filtrate purified by flash chromatography (DCM/MeOH 9:1) to give
17 as a white foam (393 mg, 36% over 2 steps).
1H NMR (500 MHz, CD
3OD): δ 8.29 (s, 1H, H8), 8.21 (s, 1H, H2), 6.03 (s, 1H, H1′), 4.80–4.78 (m, 1H, H2′), 4.40–4.36 (m, 1H, H4′), 4.27 (q,
J = 5 Hz, 0.5H, H3′), 4.18 (q,
J = 5 Hz, 0.5H, H3′), 3.94 (dd,
J = 10, 5 Hz, 0.5H, H5′), 3.84 (dd,
J = 10, 5 Hz, 0.5H, H5′), 3.69–3.62 (m, 1H, H5′). HRMS (ESI)
m/
z: calcd for C
10H
13N
8O
3 [M + H]
+: 293.1110; found: 293.1098. Analytical data were in accordance with the literature [
67].
General procedure B for CuAAC reaction: To a solution of alkyne 16 or 18 (1 eq) in DMF (1 mL), were successively added azido compound 17 (1.5 eq), CuSO4 (0.3 eq, in water 500 µL) and sodium ascorbate (0.6 eq, in water 500 µL). The mixture was stirred at room temperature for 16 h and then concentrated in vacuo. The crude product was purified by HPLC to afford the desired compounds.
Synthesis of compound 12 following general procedure B for CuAAC: Following the general procedure B, starting from alkyne 16 (5.7 mg, 0.019 mmol) and azido compound 17 (8.2 mg, 0.027 mmol), compound 12 was obtained as a white foam (4 mg, 36%).
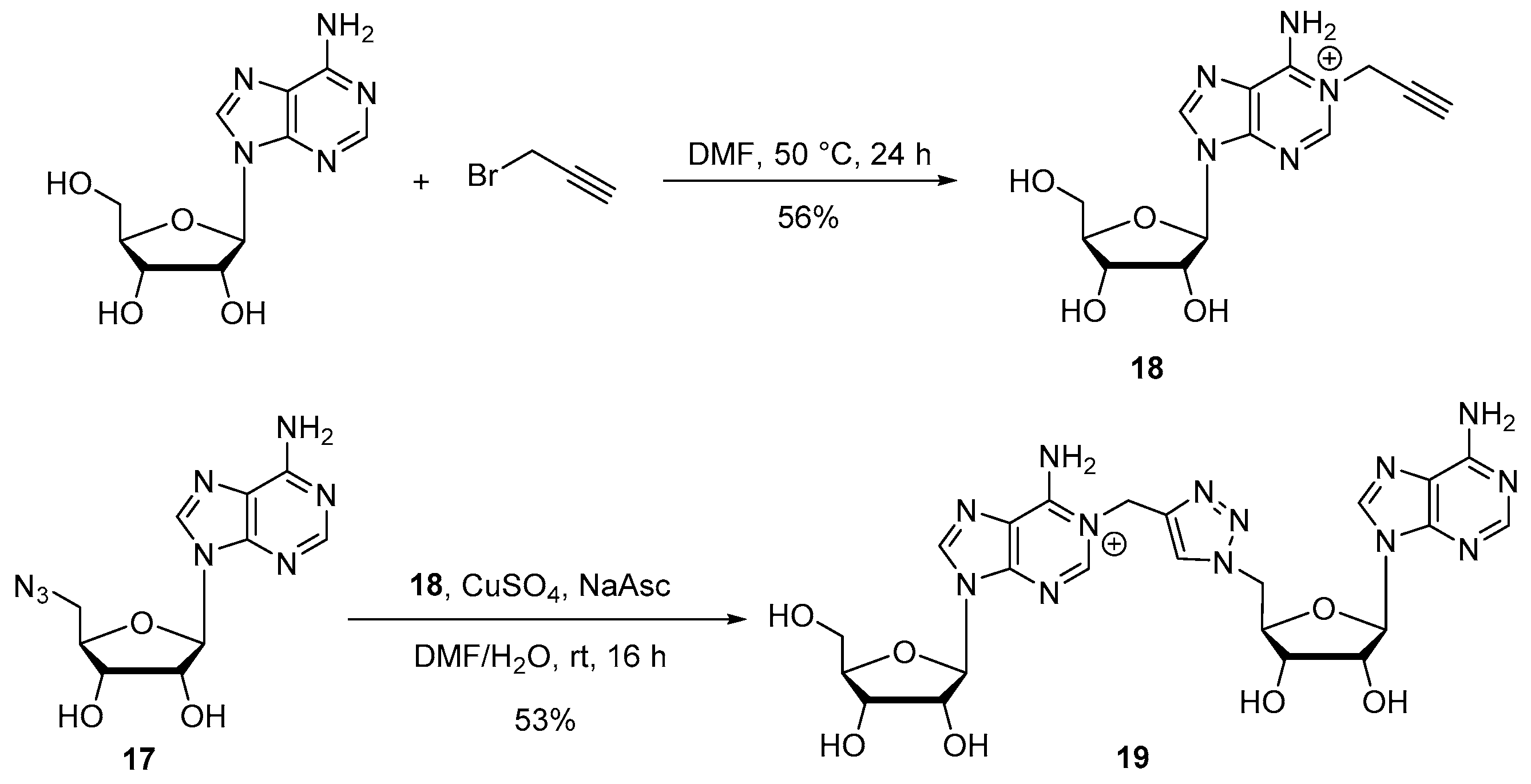
Compound 18: Propargyl bromide (80% in toluene) (143 µL, 1.8 mmol) was added to a solution of adenosine (100 mg, 0.37 mmol) in DMF (1 mL) and the reaction mixture was stirred at 50 °C for 24 h. After removal of the solvent under reduced pressure, the crude was diluted in water and purified by HPLC to afford compound 18 as a white foam (64 mg, 56%). 1H NMR (500 MHz, D2O): δ 8.73 (s, 1H, H2), 8.61 (s, 1H, H8), 6.20 (d, J = 5 Hz, 1H, H1′), 5.27 (s, 2H, CH2N), 4.84–4.82 (t, J = 5 Hz, 1H, H2′), 4.50 (t, J = 5 Hz, 1H, H3′), 4.33–4.31 (m, 1H, H4′), 3.97 (dd, J = 5, 10 Hz, 1H, H5′), 3.90 (dd, J = 5, 10 Hz, 1H, H5′), 3.19 (bs, 1H, C≡CH). 13C NMR (126 MHz, D2O): δ 150,2 (C6), 146.9 (C2), 146.8 (C4), 143.4 (C8), 119.6 (C5), 88.6 (C1′), 85.5 (C4′), 78.7 (C≡CH), 74.2 (C2′), 73.1 (C≡CH), 70.1 (C3′), 61.1 (C5′), 40.8 (CH2N). HRMS (ESI) m/z: calcd for C13H16N5O4 [M]+: 306.1196; found: 306.1193.
Compound 19: Following the general procedure B for CuAAC, starting from alkyne 18 (6.3 mg, 0.020 mmol) and azido compound 17 (9.1 mg, 0.03 mmol), compound 19 was obtained as a white foam (6.5 mg, 53%). 1H NMR (500 MHz, CD3OD): δ 8.62 (s, 2H, H2a and H8b), 8.14 (s, 1H, H2b), 8.05 (s, 1H, H8a), 7.96 (s, 1H, HTriazole), 6.09 (d, J = 5 Hz, 1H, H1′b), 5.95 (d, J = 5 Hz, 1H, H1′a), 5.52 (s, 2H, CH2N), 4.92–4.96 (m, 1H, H5′b), H5′b masked in the residual pick of water, 4.76–4.74 (m, 1H, H2′a), 4.62 (t, J = 5 Hz, 1H, H2′b), 4.48 (t, J = 5 Hz, 1H, H3′a), 4.38–4.33 (m, 2H, H3′b and H4′a), 4.15 (q, J = 3.1 Hz, 1H, H4′b), 3.89–3.85 (m, 1H, H5′a), 3.76–3.70 (m, 1H, H5′a). 13C NMR (126 MHz, CD3OD): δ 157.2 (Cq), 154.0 (C2b), 152.2 (Cq), 150.3 (Cq), 148.4 (C2a), 148.0 (Cq), 144.1 (C8b), 141.7 (C8a), 140.8 (CqTriazole), 126.7 (CHTriazole), 121.3 (Cq), 120.7 (Cq), 91.2 (C1′a), 90.4 (C1′b), 87.6 (C4′b), 83.6 (C4′a), 76.5 (C2′b), 74.5 (C2′a), 72.4 (C3′a), 71.8 (C3′b), 62.5 (C5′a), 52.7 (C5′b), 45.9 (CH2N). HRMS (ESI) m/z: calcd for C23H28N13O7 [M + H]+: 598.2234; found: 598.2222. HPLC purity: 97.6%; tR = 14.6 min (MeCN/H2O 0:100 to 100:0 over 30 min).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










