
Dried Urine Microsampling Coupled to Liquid Chromatography—Tandem Mass Spectrometry (LC–MS/MS) for the Analysis of Unconjugated Anabolic Androgenic Steroids

 ,
, 
 and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Study of Sampling, Extraction and Separation Parameters
2.1.1. Sampling Assays
2.1.2. Extraction Procedure
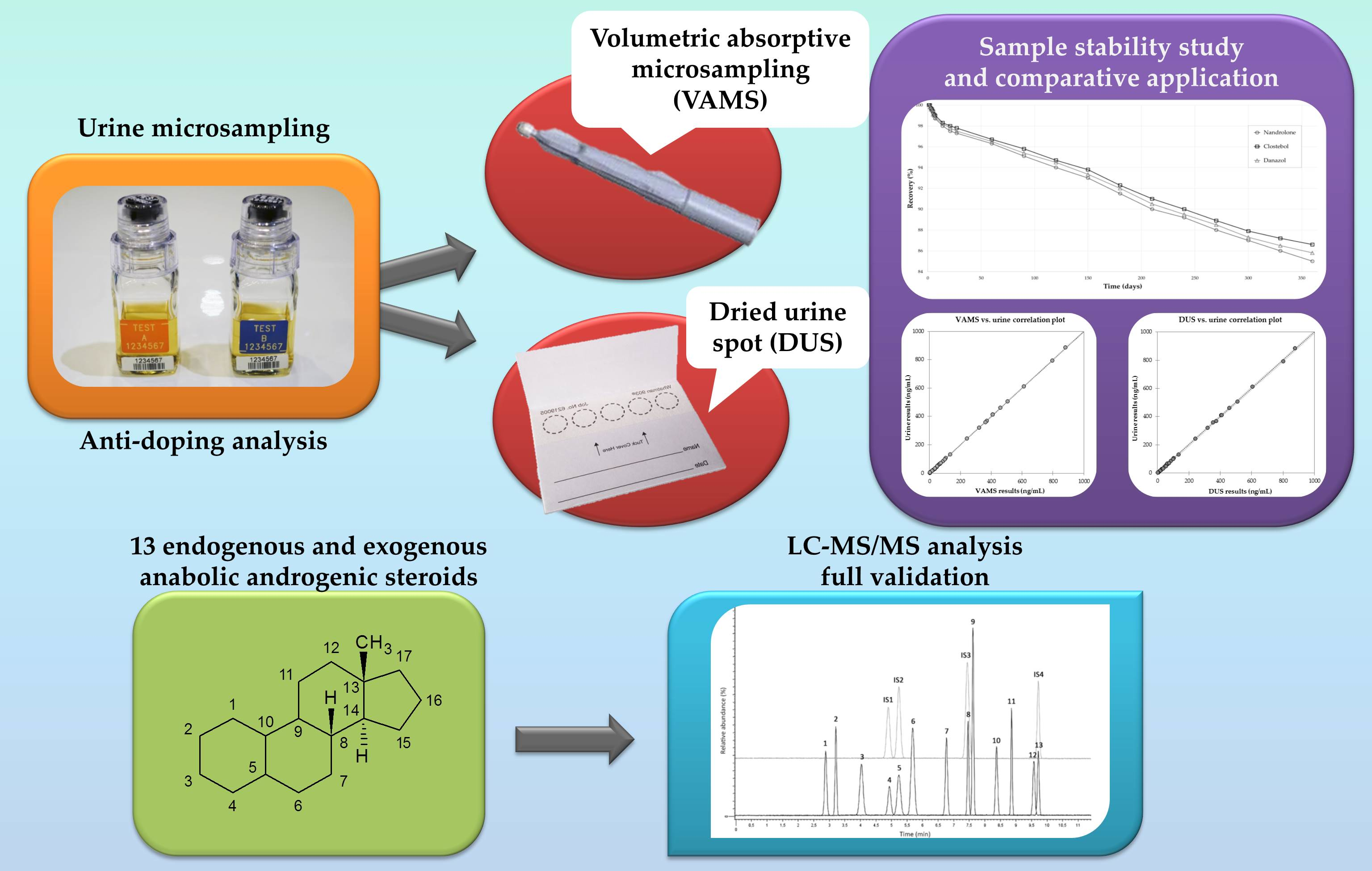
2.2. LC-MS/MS Analysis and Method Validation
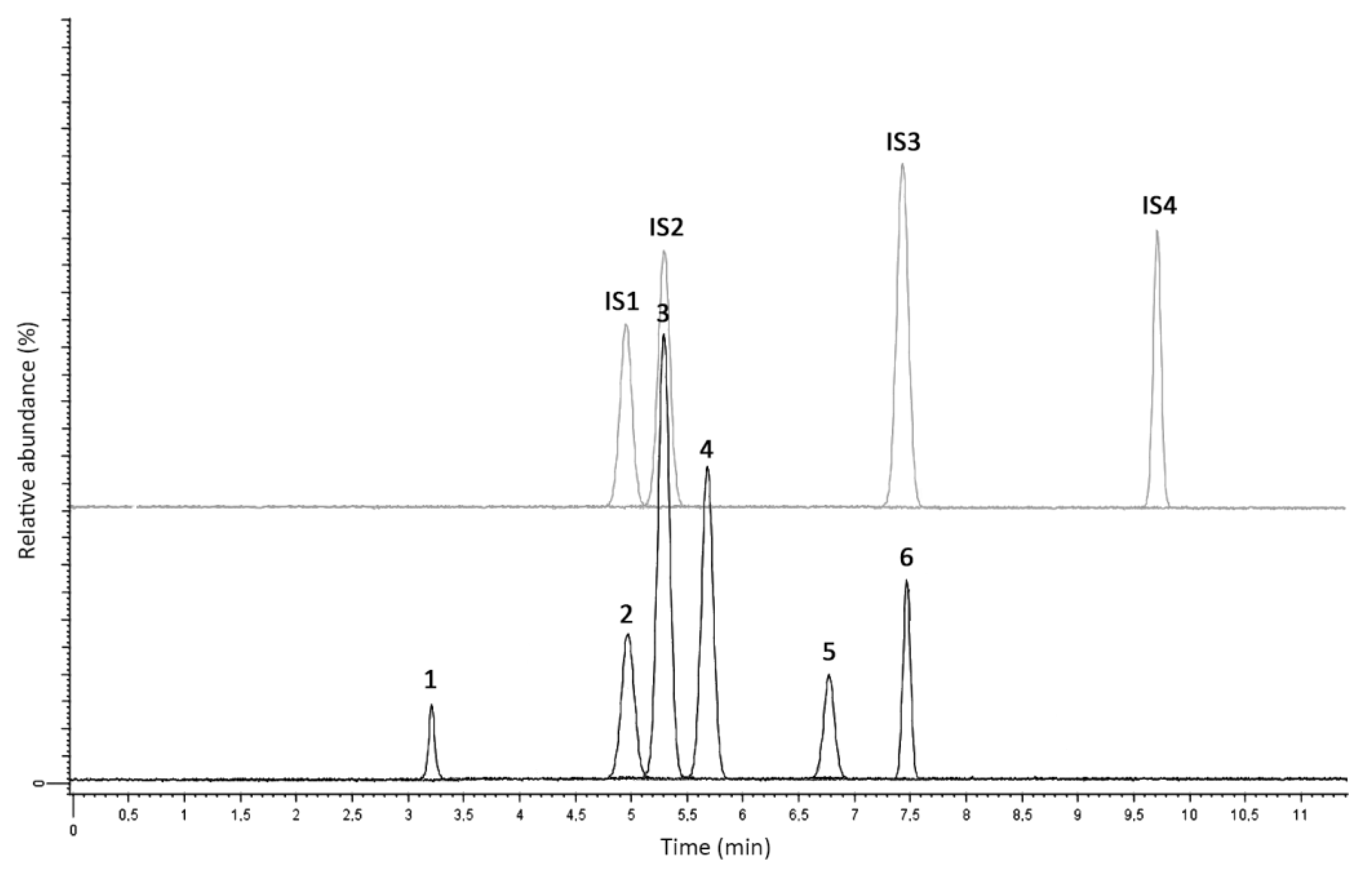
2.2.1. Linearity and Selectivity
2.2.2. Absolute Recovery and Precision
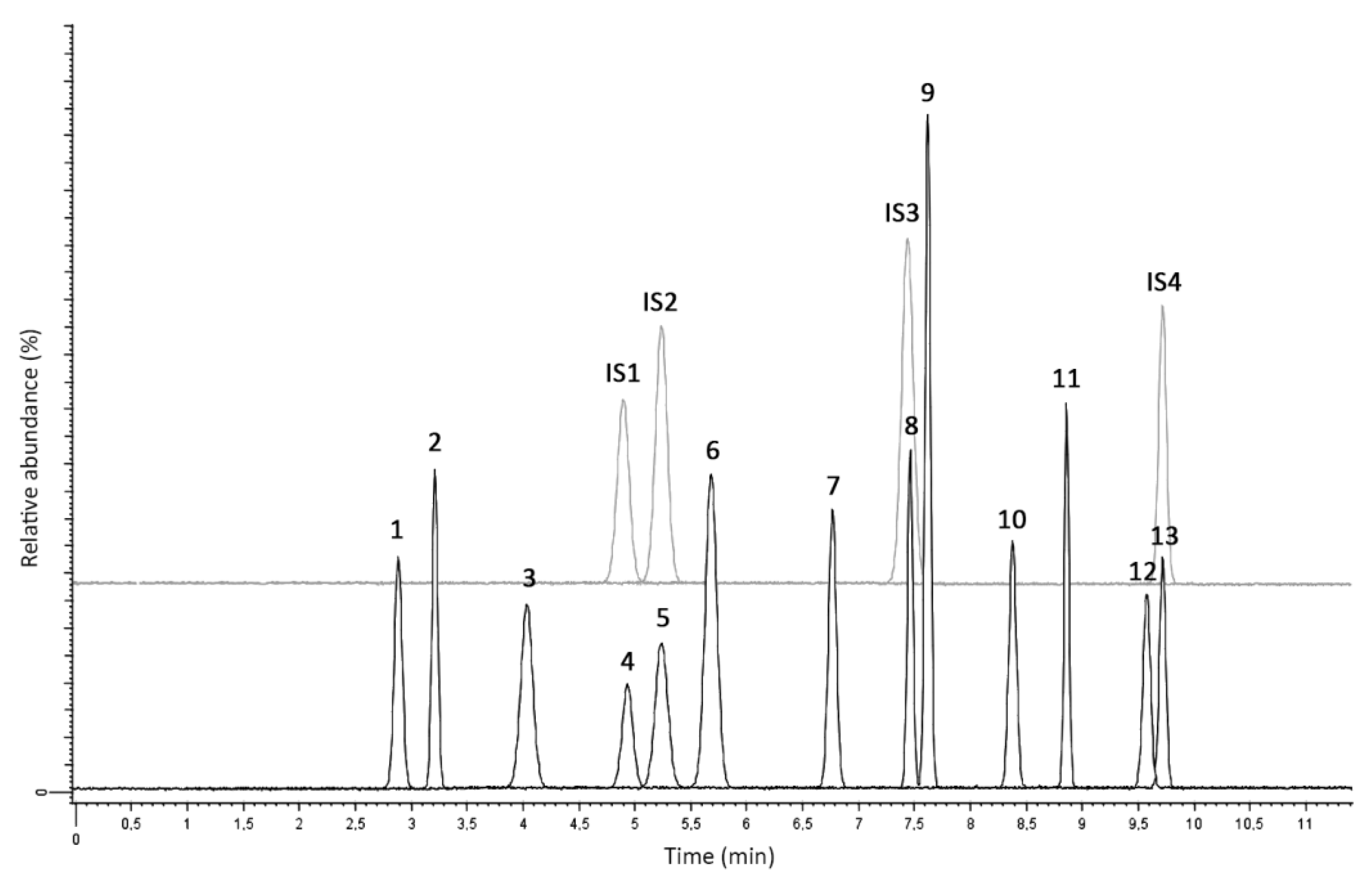
2.2.3. Matrix Effect and Stability
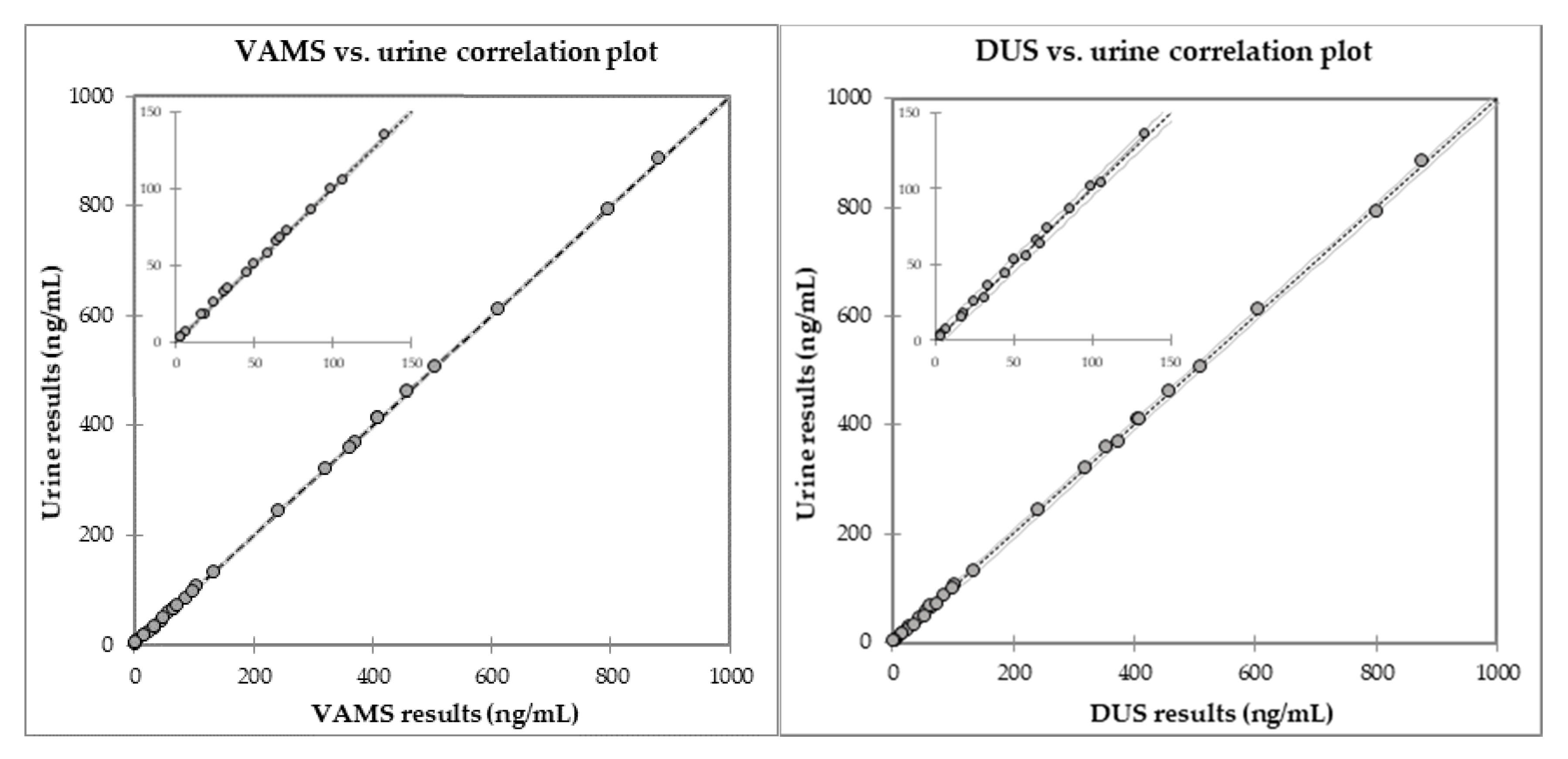
2.3. Comparison with Fluid Samples
3. Materials and Methods
3.1. Chemicals and Solutions
3.2. LC-MS/MS System
3.3. Microsample Collection and Pretreatment
3.3.1. Volumetric Absorptive Microsampling
3.3.2. Dried Urine Spot Microsampling
3.4. Method Validation
3.4.1. Calibration Curves
3.4.2. Absolute Recovery and Precision Assays
3.4.3. Matrix Effect
3.4.4. Selectivity
3.4.5. Stability
3.4.6. Analysis and Comparison with Urine Samples from Real Cases
3.4.7. Accuracy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Baker, J.S.; Thomas, N.E.; Davies, B.; Graham, M.R. Anabolic androgenic steroid (AAS) abuse: Not only an elite performance issue? Open Sports Med. J. 2008, 2, 38e9. [Google Scholar] [CrossRef]
- Brennan, B.P.; Kanayama, G.; Pope, H.G., Jr. Performance-enhancing drugs on the web: A growing public-health issue. Am. J. Addict. 2013, 22, 158–161. [Google Scholar] [CrossRef]
- Hartgens, F.; Kuipers, H. Effects of androgenic-anabolic steroids in athletes. Sports Med. 2004, 34, 513–554. [Google Scholar] [CrossRef]
- Pope, H.G., Jr.; Kanayama, G.; Hudson, J.I. Risk factors for illicit anabolicandrogenic steroid use in male weightlifters: A cross-sectional cohort study. Biol. Psychiatry 2012, 71, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Nieschlag, E.; Vorona, E. Doping with anabolic androgenic steroids (AAS): Adverse effects on non-reproductive organs and functions. Rev. Endocr. Metab. Disord. 2015, 16, 199–211. [Google Scholar] [CrossRef]
- Piacentino, D.; Kotzalidis, G.D.; Del Casale, A.; Aromatario, M.R.; Pomara, C.; Girardi, P.; Sani, G. Anabolic-androgenic steroid use and psychopathology in athletes. A systematic review. Curr. Neuropharmacol. 2015, 13, 101–121. [Google Scholar] [CrossRef] [PubMed]
- Pomara, C.; Neri, M.; Bello, S.; Fiore, C.; Riezzo, I.; Turillazzi, E. Neurotoxicity by synthetic androgen steroids: Oxidative stress, apoptosis, and neuropathology: A review. Curr. Neuropharmacol. 2015, 13, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Liu, D. 17β-trenbolone, an anabolic-androgenic steroid as well as an environmental hormone, contributes to neurodegeneration. Toxicol. Appl. Pharmacol. 2015, 282, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Tugyan, K.; Ozbal, S.; Cilaker, S.; Kiray, M.; Pekcetin, C.; Ergur, B.U.; Kumral, A. Neuroprotective effect of erythropoietin on nandrolone decanoate-induced brain injury in rats. Neurosci. Lett. 2013, 533, 28–33. [Google Scholar] [CrossRef] [PubMed]
- World Anti-Doping Agency, The 2020 Prohibited List. Available online: https://www.wada-ama.org/sites/default/files/wada_2020_english_prohibited_list_0.pdf (accessed on 11 May 2020).
- Wang, J.; Wu, M.; Liu, X.; Xu, Y. Profiling of urinary steroids by gas chromatography-mass spectrometry detection and confirmation of androstenedione administration using isotope ratio mass spectrometry. Steroids 2011, 76, 1560–1565. [Google Scholar] [CrossRef]
- Van Eenoo, P.; Van Gansbeke, W.; De Brabanter, N.; Deventer, K.; Delbeke, F.T. A fast, comprehensive screening method for doping agents in urine by gas chromatography-triple quadrupole mass spectrometry. J. Chromatogr. A 2011, 1218, 3306–3316. [Google Scholar] [CrossRef] [PubMed]
- Raro, M.; Portolés, T.; Pitarch, E.; Sancho, J.V.; Hernández, F.; Garrostas, L.; Marcos, J.; Ventura, R.; Segura, J.; Pozo, O.J. Potential of atmospheric pressure chemical ionization source in gas chromatography tandem mass spectrometry for the screening of urinary exogenous androgenic anabolic steroids. Anal. Chim. Acta 2016, 906, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Cha, E.; Kim, S.; Kim, H.J.; Lee, K.M.; Kim, K.H.; Kwon, O.S.; Lee, J. Sensitivity of GC-EI/MS, GC-EI/MS/MS, LC-ESI/MS/MS, LC-Ag(+) CIS/MS/MS, and GC-ESI/MS/MS for analysis of anabolic steroids in doping control. Drug Test. Anal. 2015, 7, 1040–1049. [Google Scholar] [CrossRef]
- Guo, F.; Shao, J.; Liu, Q.; Shi, J.-B.; Jiang, G.-B. Automated and sensitive determination of four anabolic androgenic steroids in urine by online turbulent flow solid-phase extraction coupled with liquid chromatography-tandem mass spectrometry: A novel approach for clinical monitoring and doping control. Talanta 2014, 125, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Yagi, K.; Ishizaki, A.; Kataoka, H. Determination of anabolic steroids in human urine by automated in-tube solid-phase microextraction coupled with liquid chromatography-mass spectrometry. J. Pharm. Biomed. Anal. 2010, 52, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Jeon, B.W.; Yoo, H.H.; Jeong, E.S.; Kim, H.J.; Jin, C.; Kim, D.H.; Lee, J. LC-ESI/MS/MS method for rapid screening and confirmation of 44 exogenous anabolic steroids in human urine. Anal. Bioanal. Chem. 2011, 401, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, A.; Kuuranne, T.; Kotiaho, T.; Kostiainen, R. Screening of free 17-alkyl-substituted anabolic steroids in human urine by liquid chromatography-electrospray ionization tandem mass spectrometry. Steroids 2004, 69, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Fragkaki, A.G.; Petropoulou, G.; Athanasiadou, I.; Kiousi, P.; Kioukia-Fougia, N.; Archontaki, H.; Bakeas, E.; Angelis, Y.S. Determination of anabolic androgenic steroids as imidazole carbamate derivatives in human urine using liquid chromatography–tandem mass spectrometry. J. Sep. Sci. 2020, 43, 2154–2161. [Google Scholar] [CrossRef]
- Mazzarino, M.; Abate, M.G.; Alocci, R.; Rossi, F.; Stinchelli, R.; Molaioni, F.; de la Torre, X.; Botrè, F. Urine stability and steroid profile: Towards a screening index of urine sample degradation for anti-doping purpose. Anal. Chim. Acta 2011, 683, 221–226. [Google Scholar] [CrossRef]
- Ojanperä, S.; Leinonen, A.; Apajalahti, J.; Lauraeus, M.; Alaja, S.; Moisander, T.; Kettunen, A. Characterization of microbial contaminants in urine. Drug Test. Anal. 2010, 2, 576–581. [Google Scholar] [CrossRef]
- The World Anti-Doping Code. International Standard for Testing and Investigations (ISTI). Available online: https://www.wada-ama.org/sites/default/files/resources/files/isti_march2020_0.pdf (accessed on 10 May 2020).
- Protti, M.; Mandrioli, R.; Mercolini, L. Perspectives and strategies for anti-doping analysis. Bioanalysis 2019, 11, 149–152. [Google Scholar] [CrossRef]
- Protti, M.; Rudge, J.; Sberna, A.E.; Gerra, G.; Mercolini, L. Dried haematic microsamples and LC–MS/MS for the analysis of natural and synthetic cannabinoids. J. Chromatogr. B 2017, 1044-1045, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Mandrioli, R.; Mercolini, L.; Protti, M. Blood and plasma volumetric absorptive microsampling (VAMS) coupled to LC-MS/MS for the forensic assessment of cocaine consumption. Molecules 2020, 25, 1046. [Google Scholar] [CrossRef] [PubMed]
- Protti, M.; Catapano, M.C.; Samolsky Dekel, B.G.; Rudge, J.; Gerra, G.; Somaini, L.; Mandrioli, R.; Mercolini, L. Determination of oxycodone and its major metabolites in haematic and urinary matrices: Comparison of traditional and miniaturised sampling approaches. J. Pharm. Biomed. Anal. 2018, 152, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Marasca, C.; Protti, M.; Mandrioli, R.; Atti, A.R.; Armirotti, A.; Cavalli, A.; De Ronchi, D.; Mercolini, L. Whole blood and oral fluid microsampling for the monitoring of patients under treatment with antidepressant drugs. J. Pharm. Biomed. Anal. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Mercolini, L.; Protti, M.; Catapano, M.C.; Rudge, J.; Sberna, A.E. LC-MS/MS and volumetric absorptive microsampling for quantitative bioanalysis of cathinone analogues in dried urine, plasma and oral fluid samples. J. Pharm. Biomed. Anal. 2016, 123, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Protti, M.; Vignali, A.; Sanchez Blanco, T.; Rudge, J.; Bugamelli, F.; Ferranti, A.; Mandrioli, R.; Mercolini, L. Enantioseparation and determination of asenapine in biological fluid micromatrices by HPLC with diode array detection. J. Sep. Sci. 2018, 41, 1257–1265. [Google Scholar] [CrossRef] [PubMed]
- Kok, M.G.M.; Fillet, M. Volumetric absorptive microsampling: Current advances and applications. J. Pharm. Biomed. Anal. 2018, 147, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Protti, M.; Mandrioli, R.; Mercolini, L. Tutorial: Volumetric absorptive microsampling (VAMS). Anal. Chim. Acta 2019, 1046, 32–47. [Google Scholar] [CrossRef] [PubMed]
- Protti, M.; Mandrioli, R.; Mercolini, L. Microsampling and LC-MS/MS for anti-doping testing of glucocorticoids in urine. Bioanalysis 2020, 12, 769–782. [Google Scholar] [CrossRef] [PubMed]
- AbuRuz, S.; Millership, J.; Heaney, L.; McElnay, J. Simple liquid chromatography method for the rapid simultaneous determination of prednisolone and cortisol in plasma and urine using hydrophilic lipophilic balanced solid phase extraction cartridges. J. Chromatogr. B 2003, 798, 193–201. [Google Scholar] [CrossRef] [PubMed]
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, Validation of Analytical Procedures: Text and Methodology Q2(R1). Available online: https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf (accessed on 10 May 2020).
Sample Availability: Not available. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Carbon N. | Nandrolone | 1-Androstenedione | DHEA | Testosterone | Epitestosterone | DHT | Methandrostenolone |
|---|---|---|---|---|---|---|---|
| 1 | -H2 | -H | -H2 | -H2 | -H2 | -H2 | -H |
| 2 | -H2 | -H | -H2 | -H2 | -H2 | -H2 | -H |
| 1,2 | -C=C- | -C=C- | |||||
| 3 | =O | =O | -OH β | =O | =O | =O | =O |
| 2,3 | |||||||
| 4 | -H | -H2 | -H2 | -H | -H | -H2 | -H |
| 5 | -H α | -H α | |||||
| 4,5 | -C=C- | -C=C- | -C=C- | -C=C- | |||
| 6 | -H2 | -H2 | -H | -H2 | -H2 | -H2 | -H2 |
| 5,6 | -C=C- | ||||||
| 9 | -H α | -H α | -H α | -H α | -H α | -H α | -H α |
| 10 | -H β | -CH3 β | -CH3 β | -CH3 β | -CH3 β | -CH3 β | -CH3 β |
| 11 | -H2 | -H2 | -H2 | -H2 | -H2 | -H2 | -H2 |
| 17 | -OH β | =O | =O | -OH β | -OH α | -OH β | -OH β, -CH3 α |

| Carbon N. | Norethandrolone | Mesterolone | Clostebol | Stanozolol | Fluoxymesterone | Danazol |
|---|---|---|---|---|---|---|
| 1 | -H2 | -CH3 α | -H2 | -H2 | -H2 | -H2 |
| 2 | -H2 | -H2 | -H2 | -H2 | ||
| 3 | =O | =O | =O | =O | ||
| 2,3 |  |  | ||||
| 4 | -H | -H2 | -Cl | -H2 | -H | -H |
| 5 | -H α | -H α | ||||
| 4,5 | -C=C- | -C=C- | -C=C- | -C=C- | ||
| 6 | -H2 | -H2 | -H2 | -H2 | -H2 | -H2 |
| 9 | -H α | -H α | -H α | -H α | -F α | -H α |
| 10 | -H β | -CH3 β | -CH3 β | -CH3 β | -CH3 β | -CH3 β |
| 11 | -H2 | -H2 | -H2 | -H2 | -OH β | -H2 |
| 17 | -OH β, -CH2CH3 α | -OH β | -OH β | -OH β, -CH3 α | -OH β, -CH3 α | -OH β, -C≡CH α |
| Matrix | Sampling | Drying | Storage | Extraction |
|---|---|---|---|---|
| VAMS | Direct contact | Forced drying (air blowing, room temperature (RT), 20 min) | Dedicated clamshell + resealable bag with desiccant (RT) | 500 µL methanol, 5 min ultrasonication, drying (N2 stream), re-dissolution with 100 µL methanol |
| DUS | Pipetting | Forced drying (MW, 80W, 100 s) | Resealable bag with desiccant (RT) | 500 µL methanol, 5 min ultrasonication, centrifugation (4000 rpm, 5 min, 4 °C), drying (N2 stream), re-dissolution with 100 µL methanol |
| Analyte | Linearity Range, ng/mL | r2 | LOQ, ng/mL | LOD, ng/mL | MDL, ng/mL |
|---|---|---|---|---|---|
| Nandrolone | 1.0–750.0 | 0.9990 | 1.0 | 0.3 | 0.2 |
| 1-Androstenedione | 1.5–500.0 | 0.9993 | 1.5 | 0.5 | 0.5 |
| DHEA | 1.5–750.0 | 0.9992 | 1.5 | 0.5 | 0.5 |
| Testosterone | 1.0–750.0 | 0.9995 | 1.0 | 0.3 | 0.2 |
| Epitestosterone | 1.0–750.0 | 0.9990 | 1.0 | 0.3 | 0.2 |
| DHT | 1.5–750.0 | 0.9992 | 1.5 | 0.5 | 0.5 |
| Methandrostenolone | 1.5–500.0 | 0.9994 | 1.5 | 0.5 | 0.5 |
| Norethandrolone | 1.5–500.0 | 0.9996 | 1.5 | 0.5 | 0.5 |
| Mesterolone | 1.5–500.0 | 0.9990 | 1.5 | 0.5 | 0.5 |
| Clostebol | 1.5–500.0 | 0.9994 | 1.5 | 0.5 | 0.5 |
| Stanozolol | 1.5–750.0 | 0.9997 | 1.5 | 0.5 | 0.5 |
| Fluoxymesterone | 1.5–500.0 | 0.9997 | 1.5 | 0.5 | 0.5 |
| Danazol | 1.5–750.0 | 0.9995 | 1.5 | 0.5 | 0.5 |
| Analyte | Concentration (ng/mL) | Extraction Yield ±SD, % a | Precision, RSD% a | ||||
|---|---|---|---|---|---|---|---|
| Repeatability | Intermediate Precision | ||||||
| DUS | VAMS | DUS | VAMS | DUS | VAMS | ||
| Nandrolone | 1.0 | 73.4 ± 2.0 | 81.0 ± 3.7 | 7.1 | 6.5 | 6.6 | 6.2 |
| 375.0 | 72.1 ± 1.4 | 80.5 ± 2.9 | 6.9 | 6.0 | 6.4 | 5.7 | |
| 750.0 | 72.5 ± 1.5 | 82.3 ± 1.6 | 6.1 | 5.8 | 5.7 | 5.4 | |
| 1-Androstenedione | 1.5 | 71.9 ± 3.3 | 82.6 ± 3.4 | 7.4 | 7.0 | 7.0 | 6.7 |
| 250.0 | 73.4 ± 2.5 | 85.3 ± 2.0 | 7.0 | 6.5 | 6.4 | 6.1 | |
| 500.0 | 75.6 ± 2.7 | 86.2 ± 1.9 | 6.5 | 6.2 | 6.1 | 5.7 | |
| DHEA | 1.5 | 84.3 ± 4.3 | 90.1 ± 3.7 | 7.4 | 7.0 | 7.0 | 6.9 |
| 375.0 | 86.7 ± 2.6 | 90.2 ± 3.4 | 7.2 | 7.0 | 6.6 | 6.4 | |
| 750.0 | 85.4 ± 2.8 | 92.0 ± 2.7 | 7.0 | 6.8 | 6.3 | 6.1 | |
| Testosterone | 1.0 | 71.6 ± 3.4 | 77.3 ± 3.3 | 7.3 | 7.0 | 6.9 | 6.6 |
| 375.0 | 73.1 ± 2.4 | 78.4 ± 3.8 | 6.5 | 6.4 | 6.2 | 5.9 | |
| 750.0 | 75.9 ± 2.6 | 80.3 ± 2.4 | 6.2 | 5.9 | 6.0 | 5.3 | |
| Epitestosterone | 1.0 | 71.3 ± 4.1 | 79.1 ± 4.6 | 7.1 | 6.8 | 6.8 | 6.3 |
| 375.0 | 75.5 ± 2.8 | 79.1 ± 3.1 | 6.3 | 6.2 | 6.4 | 5.8 | |
| 750.0 | 76.4 ± 3.5 | 82.0 ± 2.8 | 6.2 | 5.8 | 5.8 | 5.5 | |
| DHT | 1.5 | 75.0 ± 2.7 | 81.6 ± 2.6 | 7.3 | 7.0 | 7.0 | 6.9 |
| 375.0 | 78.1 ± 2.4 | 80.4 ± 2.7 | 7.2 | 6.9 | 6.8 | 6.6 | |
| 750.0 | 78.5 ± 1.9 | 82.7 ± 1.9 | 6.3 | 6.7 | 6.0 | 6.3 | |
| Methandrostenolone | 1.5 | 74.8 ± 3.2 | 83.2 ± 4.0 | 7.0 | 6.7 | 6.5 | 6.2 |
| 250.0 | 78.6 ± 2.4 | 85.9 ± 3.5 | 6.6 | 6.3 | 6.3 | 6.0 | |
| 500.0 | 76.3 ± 1.8 | 84.6 ± 2.8 | 6.3 | 6.1 | 5.9 | 5.7 | |
| Norethandrolone | 1.5 | 74.9 ± 3.6 | 77.3 ± 2.6 | 7.0 | 6.6 | 6.6 | 6.2 |
| 250.0 | 73.5 ± 2.9 | 77.5 ± 2.9 | 6.6 | 6.3 | 6.2 | 5.9 | |
| 500.0 | 74.0 ± 2.7 | 79.3 ± 1.9 | 6.2 | 5.9 | 5.8 | 5.5 | |
| Mesterolone | 1.5 | 73.2 ± 2.6 | 80.8 ± 3.3 | 7.5 | 7.0 | 7.1 | 6.8 |
| 250.0 | 77.2 ± 2.5 | 81.3 ± 3.6 | 7.4 | 6.7 | 7.0 | 6.4 | |
| 500.0 | 78.3 ± 1.7 | 79.9 ± 2.8 | 6.9 | 6.5 | 6.4 | 6.2 | |
| Clostebol | 1.5 | 73.6 ± 4.0 | 81.5 ± 3.1 | 7.4 | 6.9 | 7.0 | 7.0 |
| 250.0 | 75.7 ± 3.6 | 82.5 ± 2.7 | 7.0 | 6.8 | 6.8 | 6.6 | |
| 500.0 | 76.6 ± 2.9 | 83.8 ± 3.0 | 6.9 | 6.6 | 6.3 | 6.3 | |
| Stanozolol | 1.5 | 70.4 ± 3.2 | 82.7 ± 3.3 | 5.7 | 5.4 | 6.3 | 6.0 |
| 375.0 | 71.8 ± 3.5 | 85.2 ± 2.5 | 5.6 | 5.2 | 6.1 | 5.8 | |
| 750.0 | 74.4 ± 2.9 | 86.7 ± 2.7 | 5.1 | 5.0 | 5.7 | 5.5 | |
| Fluoxymesterone | 1.5 | 71.1 ± 3.2 | 80.6 ± 3.4 | 7.2 | 7.0 | 6.9 | 6.7 |
| 250.0 | 74.4 ± 2.7 | 83.4 ± 2.8 | 7.1 | 6.9 | 6.9 | 6.6 | |
| 500.0 | 75.1 ± 2.6 | 83.7 ± 1.9 | 7.0 | 6.8 | 6.5 | 6.3 | |
| Danazol | 1.5 | 72.8 ± 3.4 | 80.8 ± 4.6 | 6.6 | 6.5 | 6.3 | 6.2 |
| 375.0 | 70.6 ± 2.6 | 81.3 ± 4.3 | 6.9 | 6.5 | 6.4 | 6.1 | |
| 750.0 | 71.6 ± 2.8 | 84.5 ± 2.6 | 6.4 | 6.2 | 6.0 | 5.8 | |
| IS1 | 20.0 | 82.7 ± 2.5 | 89.7 ± 1.8 | 4.2 | 4.6 | 3.8 | 3.4 |
| IS2 | 20.0 | 80.9 ± 1.9 | 88.4 ± 1.8 | 4.5 | 4.8 | 3.6 | 3.2 |
| IS3 | 20.0 | 83.5 ± 2.0 | 88.0 ± 2.0 | 4.3 | 4.7 | 3.8 | 3.5 |
| IS4 | 20.0 | 85.7 ± 2.2 | 90.1 ± 1.9 | 4.3 | 4.8 | 3.8 | 3.1 |
| Analyte | Concentration (ng/mL) | Matrix Effect, %RE a | Stability, % Recovery a,b | ||||
|---|---|---|---|---|---|---|---|
| 3 Months | 1 Year | ||||||
| DUS | VAMS | DUS | VAMS | DUS | VAMS | ||
| Nandrolone | 1.0 | 11.3 | 9.2 | 91 | 92 | 73 | 84 |
| 375.0 | 11.2 | 8.3 | 90 | 90 | 75 | 85 | |
| 750.0 | 9.0 | 8.3 | 92 | 93 | 77 | 87 | |
| 1-Androstenedione | 1.5 | 10.3 | 8.2 | 88 | 90 | 72 | 88 |
| 250.0 | 9.3 | 7.5 | 89 | 90 | 73 | 87 | |
| 500.0 | 10.1 | 6.6 | 88 | 89 | 75 | 88 | |
| DHEA | 1.5 | 9.8 | 8.8 | 89 | 89 | 75 | 83 |
| 375.0 | 9.5 | 6.9 | 90 | 91 | 77 | 84 | |
| 750.0 | 8.2 | 6.8 | 90 | 90 | 77 | 86 | |
| Testosterone | 1.0 | 6.2 | 8.7 | 87 | 90 | 73 | 84 |
| 375.0 | 7.2 | 6.1 | 88 | 91 | 74 | 85 | |
| 750.0 | 6.2 | 7.1 | 89 | 91 | 76 | 85 | |
| Epitestosterone | 1.0 | 8.0 | 7.0 | 87 | 90 | 73 | 87 |
| 375.0 | 7.0 | 5.0 | 89 | 92 | 73 | 88 | |
| 750.0 | 63 | 4.2 | 90 | 93 | 75 | 88 | |
| DHT | 1.5 | 7.9 | 5.1 | 89 | 91 | 74 | 84 |
| 375.0 | 5.9 | 3.2 | 90 | 91 | 74 | 86 | |
| 750.0 | 5.4 | 3.2 | 90 | 94 | 76 | 87 | |
| Methandrostenolone | 1.5 | 9.8 | 9.2 | 88 | 90 | 73 | 85 |
| 250.0 | 10.1 | 9.2 | 89 | 90 | 75 | 86 | |
| 500.0 | 9.1 | 8.2 | 90 | 90 | 74 | 86 | |
| Norethandrolone | 1.5 | 9.1 | 8.8 | 87 | 91 | 73 | 87 |
| 250.0 | 9.2 | 7.9 | 88 | 93 | 74 | 88 | |
| 500.0 | 8.3 | 6.6 | 90 | 92 | 75 | 88 | |
| Mesterolone | 1.5 | 10.4 | 7.3 | 87 | 90 | 74 | 86 |
| 250.0 | 8.5 | 7.2 | 88 | 89 | 73 | 87 | |
| 500.0 | 6.5 | 7.3 | 88 | 90 | 74 | 85 | |
| Clostebol | 1.5 | 11.8 | 8.1 | 88 | 90 | 74 | 84 |
| 250.0 | 11.4 | 8.5 | 90 | 90 | 75 | 87 | |
| 500.0 | 9.0 | 6.7 | 90 | 93 | 75 | 87 | |
| Stanozolol | 1.5 | 10.3 | 6.8 | 86 | 90 | 73 | 84 |
| 375.0 | 8.2 | 4.7 | 87 | 91 | 74 | 85 | |
| 750.0 | 7.3 | 4.6 | 89 | 91 | 75 | 84 | |
| Fluoxymesterone | 1.5 | 9.4 | 7.6 | 87 | 90 | 72 | 84 |
| 250.0 | 9.4 | 7.6 | 89 | 92 | 72 | 84 | |
| 500.0 | 7.3 | 6.3 | 91 | 93 | 73 | 86 | |
| Danazol | 1.5 | 10.4 | 8.9 | 88 | 91 | 73 | 84 |
| 375.0 | 9.3 | 6.3 | 89 | 91 | 71 | 86 | |
| 750.0 | 8.2 | 5.0 | 90 | 92 | 72 | 86 | |
| Subject n. | Matrix | Concentration Found, ng/mL a | Other Identified AAS (Concentration, ng/mL) | |||
|---|---|---|---|---|---|---|
| Testosterone | Epitestosterone | DHT | DHEA | |||
| Urine | 106.1 | 31.0 | 64.6 | 321.5 | Clostebol (6.8) | |
| 1 | DUS | 104.3 | 27.6 | 65.9 | 317.9 | Clostebol (7.4) |
| VAMS | 105.4 | 33.1 | 65.3 | 320.4 | Clostebol (6.5) | |
| Urine | 58.2 | 24.6 | 18.2 | 412.6 | Nandrolone (3.6) | |
| 2 | DUS | 55.9 | 25.9 | 17.5 | 409.6 | Nandrolone (3.0) |
| VAMS | 57.6 | 25.5 | 17.9 | 410.5 | Nandrolone (3.5) | |
| Urine | 368.4 | 66.9 | 85.9 | 359.6 | / | |
| 3 | DUS | 374.9 | 63.2 | 86.9 | 353.2 | / |
| VAMS | 370.6 | 67.3 | 86.5 | 361.6 | / | |
| Urine | 612.3 | 44.8 | 49.9 | 245.1 | Stanozolol (98.6) | |
| 4 | DUS | 605.1 | 44.0 | 53.6 | 242.3 | Stanozolol (101.2) |
| VAMS | 610.3 | 45.2 | 50.5 | 243.1 | Stanozolol (99.3) | |
| Urine | 71.1 | 33.4 | 16.8 | 133.4 | Clostebol (3.2) | |
| 5 | DUS | 74.3 | 36.1 | 15.3 | 135.6 | Clostebol (3.8) |
| VAMS | 72.0 | 35.0 | 17.3 | 135.0 | Clostebol (3.0) | |
| Analyte | Q1 (m/z) | Q3 (m/z) Quantifier | Q3 (m/z) Qualifier | Cone Voltage (V) | Collision Energy (eV) | tR (min) |
|---|---|---|---|---|---|---|
| Nandrolone | 275.0 | 109.0 | 239.1 | 25 | 25 | 4.07 |
| 1-Androstenedione | 287.5 | 97.1 | 109.1 | 29 | 21 | 3.22 |
| DHEA | 289.1 | 271.2 | 213.2 | 29 | 21 | 4.91 |
| Testosterone | 289.5 | 109.2 | 97.1 | 25 | 25 | 5.25 |
| Epitestosterone | 289.5 | 109.2 | 97.1 | 25 | 25 | 5.19 |
| DHT | 291.4 | 121.0 | 255.2 | 25 | 25 | 7.48 |
| Methandrostenolone | 301.5 | 121.2 | 283.2 | 25 | 25 | 7.63 |
| Norethandrolone | 303.2 | 285.0 | 267.0 | 25 | 25 | 8.38 |
| Mesterolone | 305.0 | 269.0 | 229.2 | 34 | 16 | 8.87 |
| Clostebol | 323.0 | 143.0 | 131.0 | 25 | 25 | 6.76 |
| Stanozolol | 329.5 | 81.1 | 107.1 | 55 | 31 | 9.72 |
| Fluoxymesterone | 337.0 | 241.0 | 131.1 | 25 | 25 | 2.88 |
| Danazol | 338.2 | 148.0 | 109.1 | 25 | 25 | 9.49 |
| IS1 | 292.0 | 112.0 | - | 25 | 25 | 4.91 |
| IS2 | 277.2 | 219.2 | - | 25 | 25 | 5.25 |
| IS3 | 294.0 | 258.0 | - | 25 | 25 | 7.48 |
| IS4 | 294.1 | 276.2 | - | 29 | 21 | 9.72 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Protti, M.; Marasca, C.; Cirrincione, M.; Sberna, A.E.; Mandrioli, R.; Mercolini, L. Dried Urine Microsampling Coupled to Liquid Chromatography—Tandem Mass Spectrometry (LC–MS/MS) for the Analysis of Unconjugated Anabolic Androgenic Steroids. Molecules 2020, 25, 3210. https://doi.org/10.3390/molecules25143210
Protti M, Marasca C, Cirrincione M, Sberna AE, Mandrioli R, Mercolini L. Dried Urine Microsampling Coupled to Liquid Chromatography—Tandem Mass Spectrometry (LC–MS/MS) for the Analysis of Unconjugated Anabolic Androgenic Steroids. Molecules. 2020; 25(14):3210. https://doi.org/10.3390/molecules25143210
Chicago/Turabian StyleProtti, Michele, Camilla Marasca, Marco Cirrincione, Angelo E. Sberna, Roberto Mandrioli, and Laura Mercolini. 2020. "Dried Urine Microsampling Coupled to Liquid Chromatography—Tandem Mass Spectrometry (LC–MS/MS) for the Analysis of Unconjugated Anabolic Androgenic Steroids" Molecules 25, no. 14: 3210. https://doi.org/10.3390/molecules25143210
APA StyleProtti, M., Marasca, C., Cirrincione, M., Sberna, A. E., Mandrioli, R., & Mercolini, L. (2020). Dried Urine Microsampling Coupled to Liquid Chromatography—Tandem Mass Spectrometry (LC–MS/MS) for the Analysis of Unconjugated Anabolic Androgenic Steroids. Molecules, 25(14), 3210. https://doi.org/10.3390/molecules25143210