
Fermented Ginseng Extract, BST204, Suppresses Tumorigenesis and Migration of Embryonic Carcinoma through Inhibition of Cancer Stem Cell Properties

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
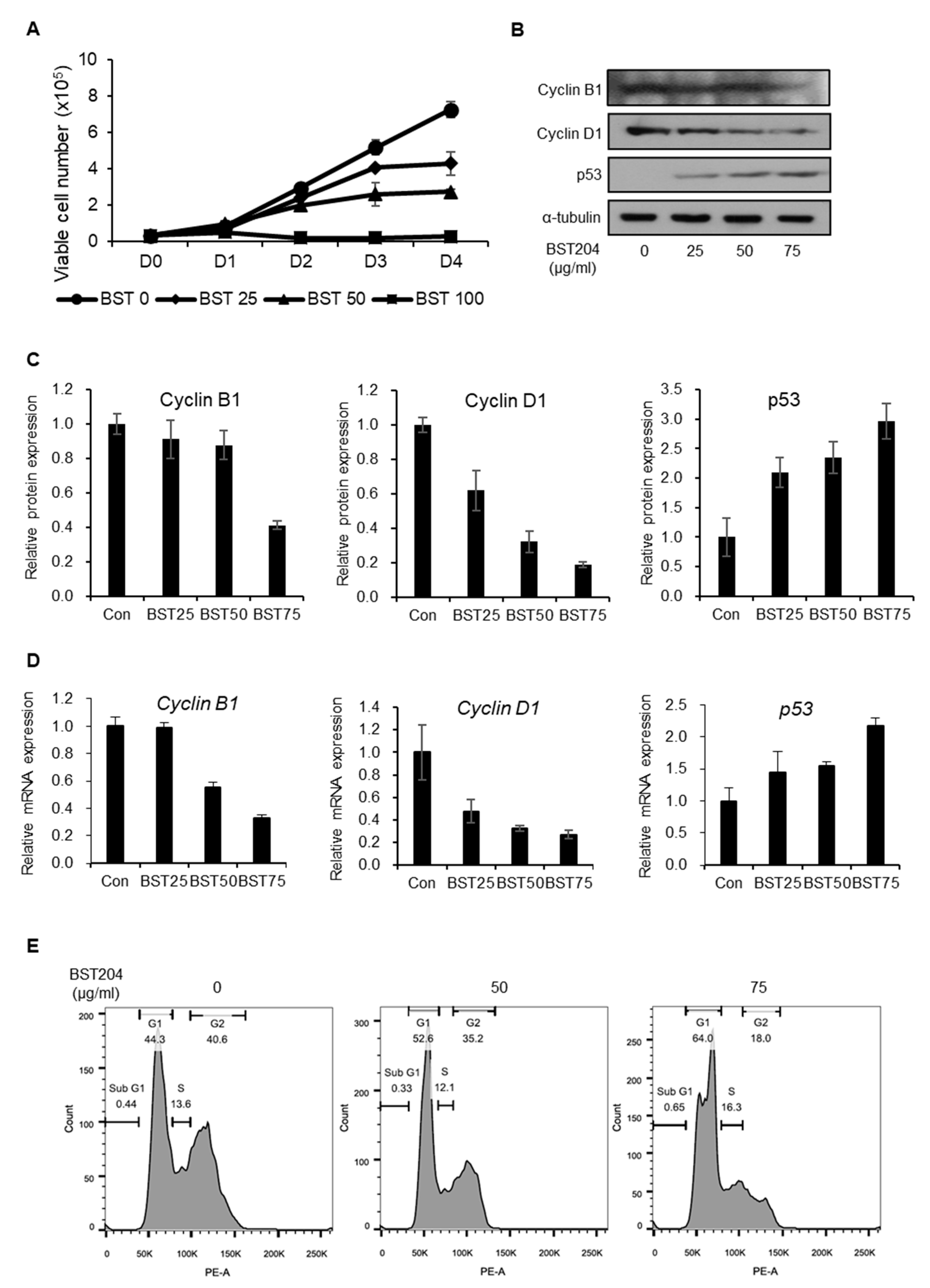
2.1. BST204 Inhibits Proliferation of EC Cells Through G1 Cell Cycle Arrest
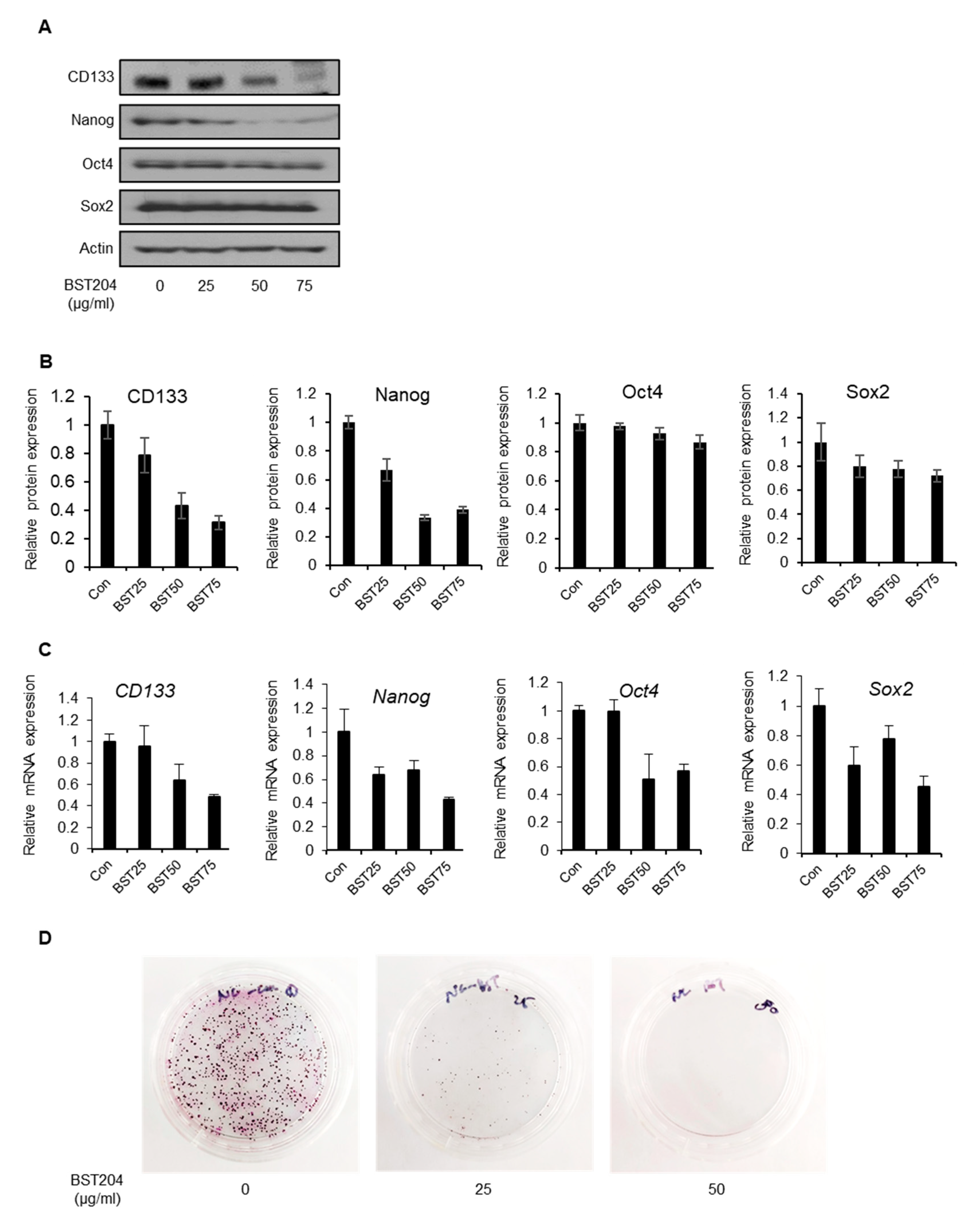
2.2. BST204 Downregulates Cancer Stemness Protein Expression and Inhibits Tumorigenicity of EC Cells
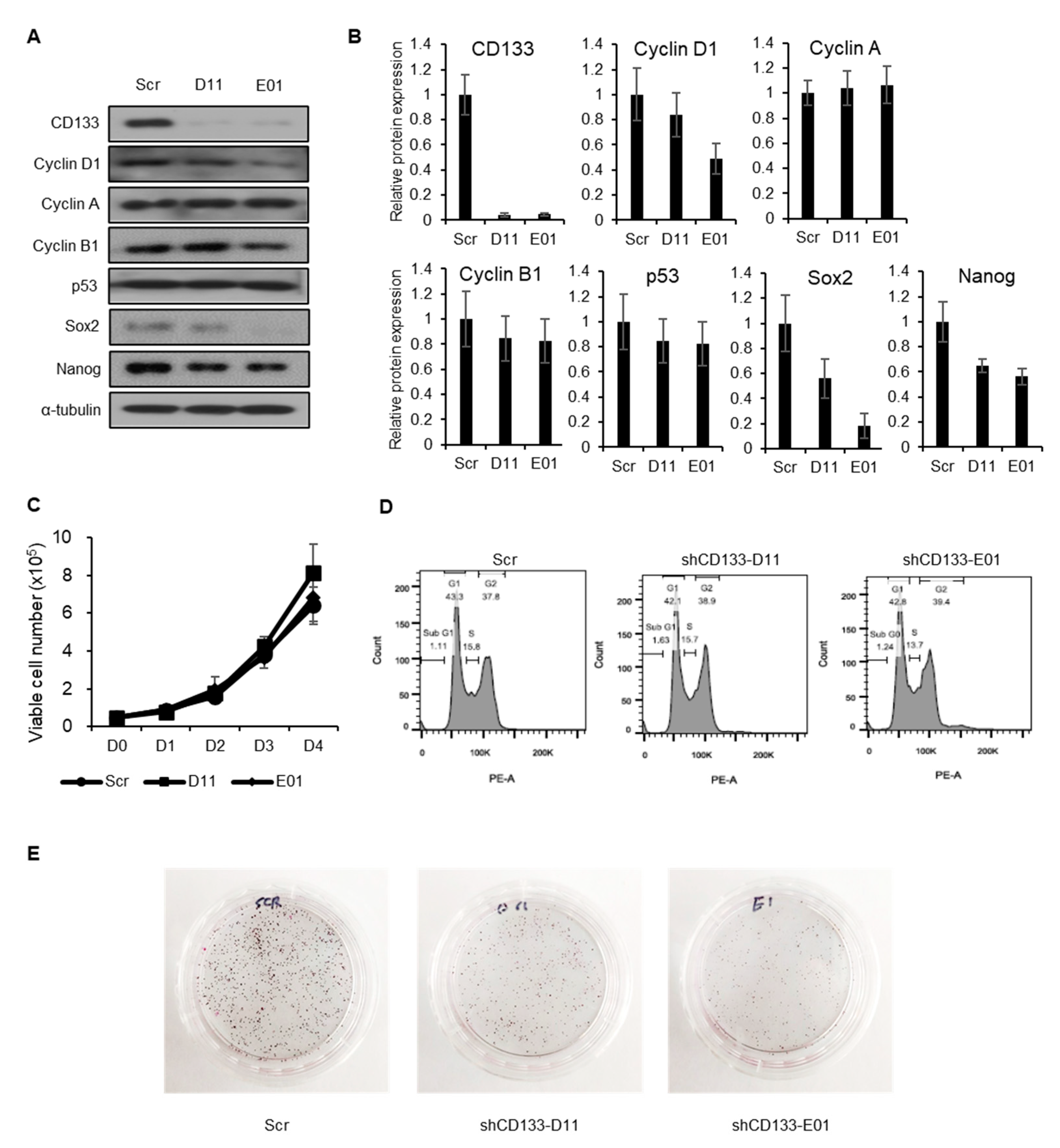
2.3. CD133 Knockdown Leads to Suppression of Tumorigenesis but Not Cancer Cell Proliferation
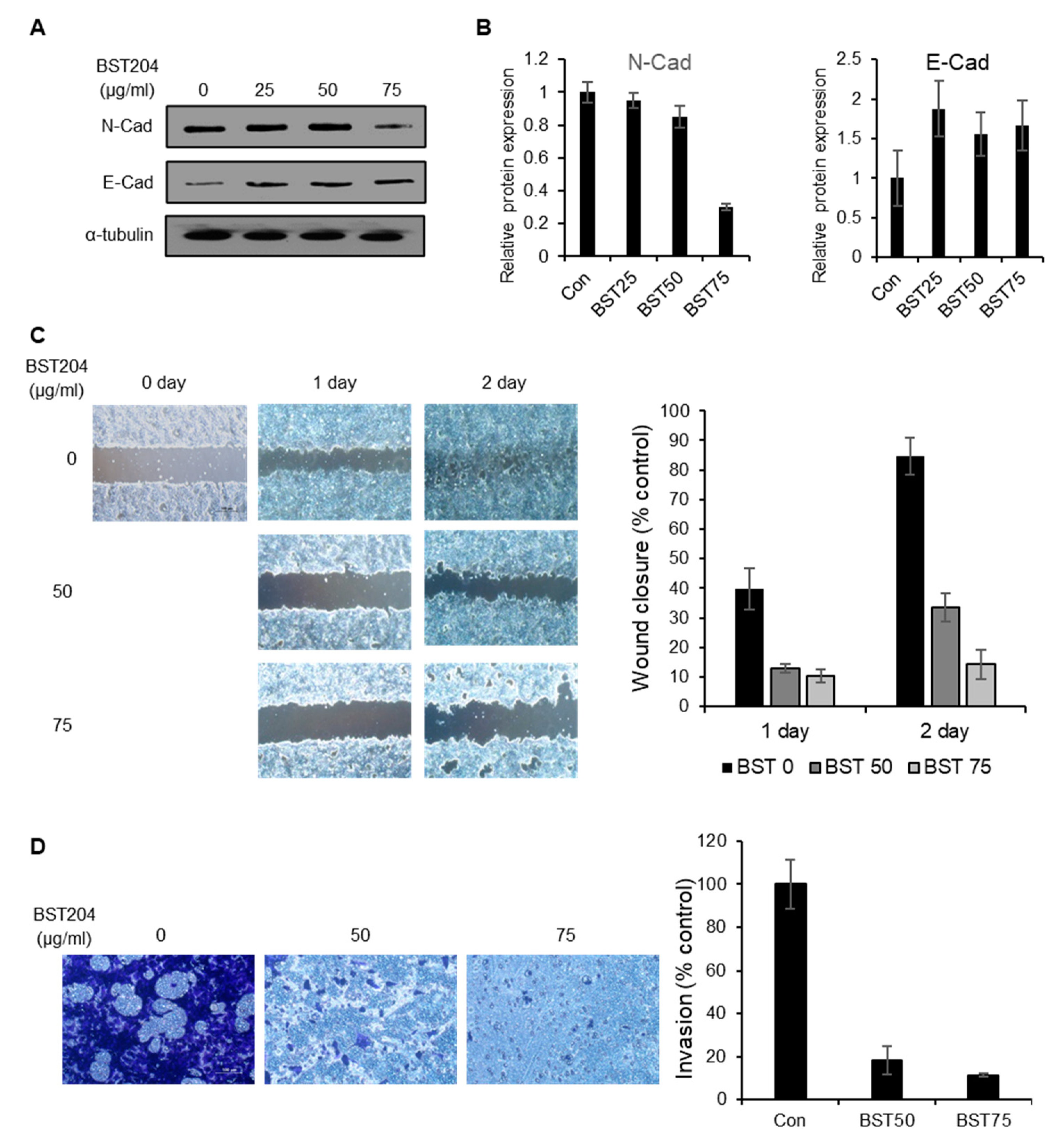
2.4. BST204 Inhibits the Migration and Invasion of Cancer Cells via Suppression of Epithelial–mesenchymal Transition (EMT)
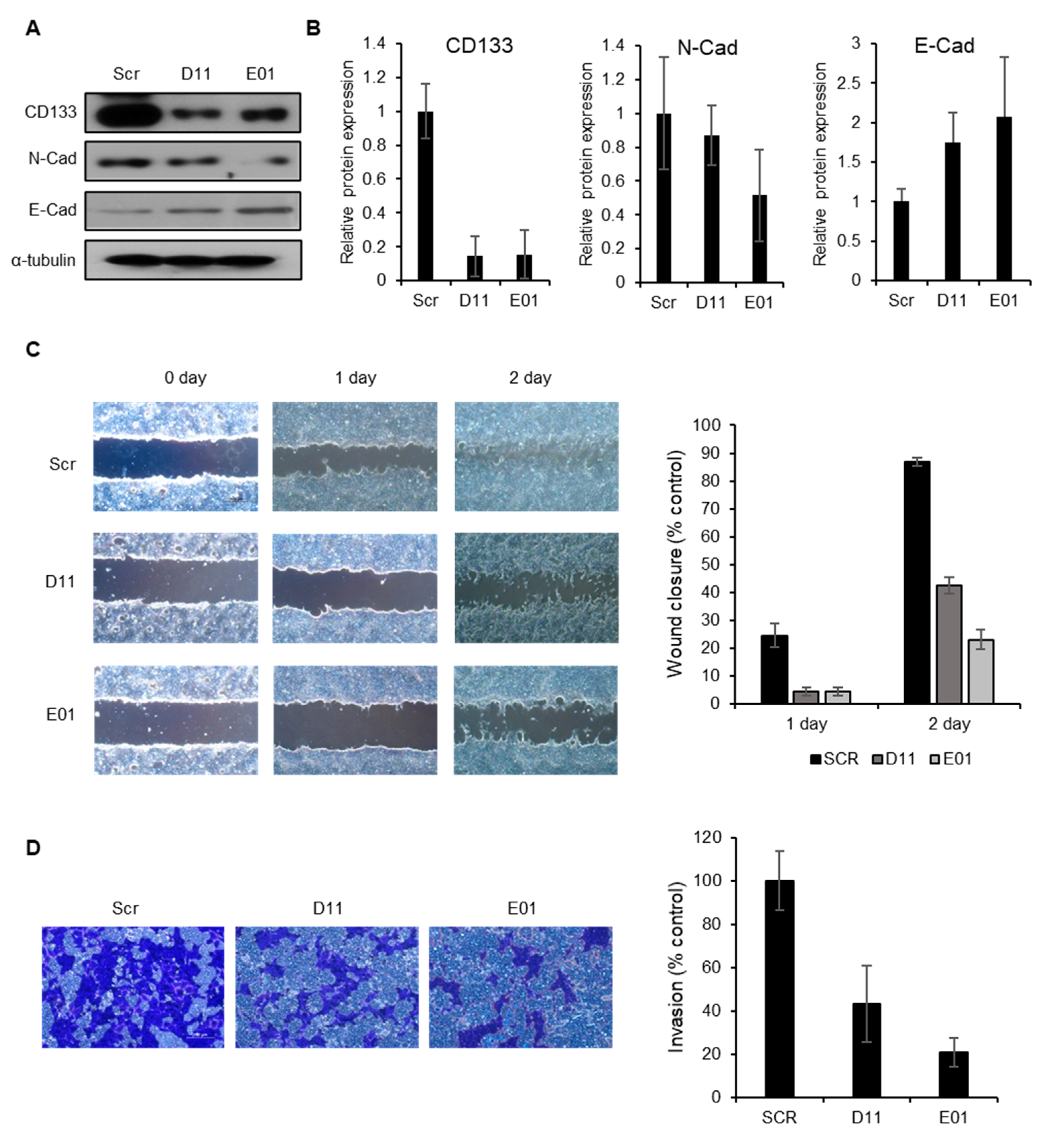
2.5. CD133 Knockdown Leads to Suppression of Migration and Invasion in EC Cells
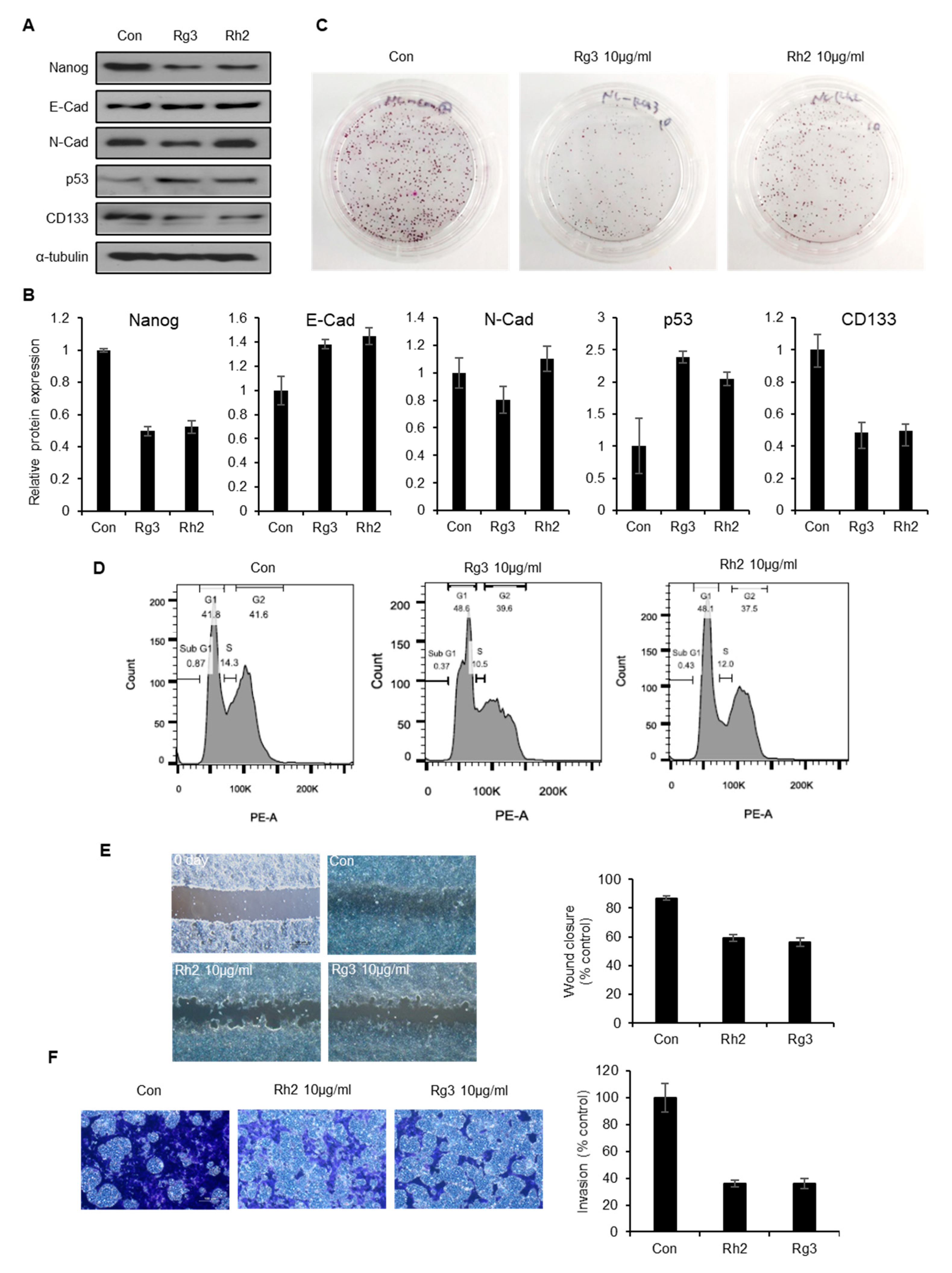
2.6. Single Ginsenoside Components of BST204 have Limited Effects on Suppression of CSC Properties
3. Discussion
4. Materials and Methods
4.1. Preparation of BST204
4.2. Cell Culture and Proliferation Assay
4.3. Immunoblotting
4.4. Reagents and Antibodies
4.5. Wound Healing Assay
4.6. Matrigel Invasion Assay
4.7. Clonogenic Colony Formation Assay
4.8. Flow Cytometry
4.9. Generation of Stable CD133 Knockdown Cell Line
4.10. Quantitative Reverse Transcription Polymerase Chain Reaction (RT-qPCR)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Adorno-cruz, V.; Kibria, G.; Liu, X.; Doherty, M.; Junk, D.J.; Guan, D.; Hubert, C.; Venere, M.; Mulkearns-hubert, E.; Sinyuk, M.; et al. Cancer Stem Cells: Targeting the Roots of Cancer, Seeds of Metastasis, and Sources of Therapy Resistance. Cancer Res. 2015, 75, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Koch, U.; Krause, M.; Baumann, M. Seminars in Cancer Biology Cancer stem cells at the crossroads of current cancer therapy failures—Radiation oncology perspective. Semin. Cancer Biol. 2010, 20, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Vaseem, M.; Kala, M.; Nivsarkar, M. CD90 a potential cancer stem cell marker and a therapeutic target. Cancer Biomark. 2016, 16, 301–307. [Google Scholar]
- Uchihara, T.; Ishimoto, T.; Yonemura, A.; Baba, H. Therapeutic targets against gastric cancer stem cells interacting with tumor microenvironment. J. Cancer Metastasis Treat. 2018, 4, 9. [Google Scholar] [CrossRef]
- Gehling, U.M. In vitro differentiation of endothelial cells from AC133-positive progenitor cells. Blood 2000, 15, 3106–3112. [Google Scholar] [CrossRef]
- Lee, A.; Kessler, J.D.; Read, T.; Kaiser, C.; Corbeil, D.; Huttner, W.B.; Johnson, J.E.; Wechsler-Reya, R.J. Isolation of Neural Stem Cells from the Postnatal Cerebellum. Nat. Neurosci. 2005, 8, 723–729. [Google Scholar] [CrossRef]
- Ledaki, I.; Shepherd, C.J.; Davies, M.; Brewer, D.; Attard, G.; de Bono, J.; Hudson, D.L. Expression Profiling of CD133+ and CD133− Epithelial Cells from Human Prostate. Prostate 2008, 68, 1007–1024. [Google Scholar]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Ricci-vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; Maria, R. De Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef]
- Zhu, Z.; Hao, X.; Yan, M.; Yao, M.; Ge, C.; Gu, J.; Li, J. Cancer stem/progenitor cells are highly enriched in CD133+ CD44+ population in hepatocellular carcinoma. Int. J. Cancer 2010, 126, 2067–2078. [Google Scholar] [CrossRef]
- Immervoll, H.; Hoem, D.; Sakariassen, P.Ø.; Steffensen, O.J.; Molven, A. Expression of the “stem cell marker” CD133 in pancreas and pancreatic ductal adenocarcinomas. BMC Cancer 2008, 14, 1–14. [Google Scholar] [CrossRef]
- Glumac, P.M.; LeBeau, A.M. The role of CD133 in cancer: A concise review. Clin. Transl. Med. 2018, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.M.; Liu, S.; Lu, H.; Zhang, H.; Zhang, P.J.; Gimotty, P.A.; Guerra, M.; Guo, W.; Xu, X. Acquired cancer stem cell phenotypes through Oct4-mediated dedifferentiation. Oncogene 2012, 31, 4898–4911. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Wu, Y.; Shu, J.; Ling, T.; Wu, J.; Chang, E.E.; Chang, S.; Huang, Y. Aberrant expression and distribution of the OCT-4 transcription factor in seminomas. J. Biomed. Sci. 2007, 14, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Chiou, S. Targeting cancer stem cells: Emerging role of Nanog transcription factor. Onco Targets Ther. 2013, 4, 1207–1220. [Google Scholar]
- Oktem, G.; Bilir, A.; Uslu, R.; Inan, S.V.; Demiray, S.B.; Atmaca, H.; Ayla, S.; Sercan, O.; Uysal, A. Expression profiling of stem cell signaling alters with spheroid formation in CD133 high/CD44 high prostate cancer stem cells. Oncol Lett. 2014, 7, 2103–2109. [Google Scholar] [CrossRef] [PubMed]
- Iida, H.; Suzuki, M.; Goitsuka, R.; Ueno, H. Hypoxia induces CD133 expression in human lung cancer cells by up-regulation of OCT3/4 and SOX2. Int. J. Oncol. 2012, 40, 71–79. [Google Scholar]
- Wang, X.Q.; Ng, R.K.; Ming, X.; Zhang, W.; Chen, L.; Chu, A.C.Y.; Pang, R.; Lo, C.M.; Tsao, S.W.; Liu, X.; et al. Epigenetic Regulation of Pluripotent Genes Mediates Stem Cell Features in Human Hepatocellular Carcinoma and Cancer Cell Lines. PLoS ONE 2013, 8, e72435. [Google Scholar] [CrossRef]
- Park, E.K.; Lee, J.C.; Park, J.W.; Bang, S.Y.; Yi, S.A.; Kim, B.K.; Park, J.H.; Kwon, S.H.; You, J.S.; Nam, S.W.; et al. Transcriptional repression of cancer stem cell marker CD133 by tumor suppressor p53. Cell Death Dis. 2015, 6, e1964. [Google Scholar] [CrossRef]
- You, J.S.; Kang, J.K.; Seo, D.; Park, J.H.; Park, J.W.; Lee, J.C. Depletion of Embryonic Stem Cell Signature by Histone Deacetylase Inhibitor in NCCIT Cells: Involvement of Nanog Suppression. Cancer Res. 2009, 15, 5716–5725. [Google Scholar] [CrossRef] [PubMed]
- Kim, J. Pharmacological and medical applications of Panax ginseng and ginsenosides: A review for use in cardiovascular diseases. J. Ginseng Res. 2018, 42, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Baik, M. Change of Ginsenoside Profiles in Processed Ginseng by Drying, Steaming, and Puffing. J. Microbiol. Biotechnol. 2019, 29, 222–229. [Google Scholar]
- Kee, J.; Han, Y.; Mun, J.; Park, S.; Jeon, H.D.; Hong, S. Effect of Korean Red Ginseng extract on colorectal lung metastasis through inhibiting the epithelial e mesenchymal transition via transforming growth factor-b 1/Smad-signaling-mediated Snail/E-cadherin expression. J. Ginseng Res. 2019, 43, 68–76. [Google Scholar] [CrossRef]
- Kim, J.H.; Yi, Y.; Kim, M.; Cho, J.Y. Role of ginsenosides, the main active components of Panax ginseng, in inflammatory responses and diseases. J. Ginseng Res. 2017, 41, 435–443. [Google Scholar] [CrossRef]
- Wu, K.; Huang, J.; Xu, T.; Ye, Z.; Jin, F.; Li, N.; Lv, B. MicroRNA-181b blocks gensenoside Rg3-mediated tumor suppression of gallbladder carcinoma by promoting autophagy flux via CREBRF/CREB3 pathway. Am. J. Transl. Res. 2019, 11, 5776–5787. [Google Scholar]
- Liu, T.; Zuo, L.; Guo, D.; Chai, X.; Xu, J.; Cui, Z.; Wang, Z.; Hou, C. Biomedicine & Pharmacotherapy Ginsenoside Rg3 regulates DNA damage in non-small cell lung cancer cells by activating VRK1/P53BP1 pathway. Biomed. Pharmacother. 2019, 120, 109483. [Google Scholar]
- Liu, Z.; Li, J.; Xia, J.; Jiang, R.; Zuo, G.; Li, X.; Chen, Y.; Xiong, W.; Chen, D. Chemico-Biological Interactions Ginsenoside 20 (s) -Rh2 as potent natural histone deacetylase inhibitors suppressing the growth of human leukemia cells. Chem. Biol. Interact. 2015, 242, 227–234. [Google Scholar] [CrossRef]
- Park, J.W.; Lee, J.C.; Ann, S.; Seo, D.; Choi, W.S.; Yoo, Y.H.; Park, S.K.; Choi, J.Y.; Um, S.H.; Ahn, S.H.; et al. A Fermented Ginseng Extract, BST204, Inhibits Proliferation and Motility of Human Colon Cancer Cells. Biomol. Ther. 2011, 19, 211–217. [Google Scholar] [CrossRef][Green Version]
- Yi, S.A.; Lee, J.; Park, S.K.; Kim, J.Y.; Park, J.W.; Lee, M.G.; Nam, K.H.; Park, J.H.; Oh, H.; Kim, S.; et al. Fermented ginseng extract, BST204, disturbs adipogenesis of mesenchymal stem cells through inhibition of S6 kinase 1 signaling. J. Ginseng Res. 2018, 44, 58–66. [Google Scholar] [CrossRef]
- Seo, J.Y.; Lee, J.H.; Kim, N.W.; Kim, Y.J.; Ho, S.; Ko, N.Y.; Her, E.; Yoo, Y.H.; Kim, J.W.; Lee, B.Y.; et al. Inhibitory effects of a fermented ginseng extract, BST204, on the expression of inducible nitric oxide synthase and nitric oxide production in lipopolysaccharide-activated murine macrophages. J. Pharm. Pharmacol. 2005, 57, 911–918. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Nie, B.; Pienta, K.J.; Morgan, T.M.; Taichman, R.S. Cancer stem cells and their role in metastasis. Pharmacol. Ther. 2013, 138, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Puram, S.V.; Tirosh, I.; Parikh, A.S.; Patel, A.P.; Yizhak, K.; Gillespie, S.; Rodman, C.; Luo, C.L.; Mroz, E.A.; Emerick, K.S.; et al. Single-Cell Transcriptomic Analysis of Primary and Metastatic Tumor Ecosystems in Head and Neck Cancer. Cell 2017, 171, 1611–1624. [Google Scholar] [CrossRef] [PubMed]
- Polireddy, K.; Dong, R.; McDonald, P.R.; Wang, T.; Luke, B.; Chen, P.; Broward, M.; Roy, A.; Chen, Q. Targeting epithelial-mesenchymal transition for identification of inhibitors for pancreatic cancer cell invasion and tumor spheres formation. PLoS ONE 2016, 11, e0164811. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef]
- Louhichi, T.; Saad, H.; Dhiab, M.B.; Ziadi, S.; Trimeche, M. Stromal CD10 expression in breast cancer correlates with tumor invasion and cancer stem cell phenotype. BMC Cancer 2018, 18, 49. [Google Scholar] [CrossRef]
- Leung, K.W.; Wong, A.S. Pharmacology of ginsenosides: A literature review. Chin. Med. 2010, 5, 20. [Google Scholar] [CrossRef]
- Wang, C.Z.; Anderson, S.; Du, W.; He, T.C.; Yuan, C.S. Red ginseng and cancer treatment. Chin. J. Nat. Med. 2016, 14, 7–16. [Google Scholar]
- Park, S.E.; Na, C.S.; Yoo, S.A.; Seo, S.H.; Son, H.S. Biotransformation of major ginsenosides in ginsenoside model culture by lactic acid bacteria. J. Ginseng Res. 2017, 41, 36–42. [Google Scholar] [CrossRef]
- Lee, S.J.; Kim, Y.; Kim, M.G. Changes in the ginsenoside content during the fermentation process using microbial strains. J. Ginseng Res. 2015, 39, 392–397. [Google Scholar] [CrossRef]
- Kim, J.K.; Cui, C.H.; Yoon, M.H.; Kim, S.C.; Im, W.T. Bioconversion of major ginsenosides Rg1 to minor ginsenoside F1 using novel recombinant ginsenoside hydrolyzing glycosidase cloned from Sanguibacter keddieii and enzyme characterization. J. Biotechnol. 2012, 161, 294–301. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Shim, H.S.; Kim, J.Y.; Kim, J.Y.; Park, S.K.; Shim, I. Ginseng Purified Dry Extract, BST204, Improved Cancer Chemotherapy-Related Fatigue and Toxicity in Mice. Evid. Based Complement. Altern. Med. 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.A.; Kim, B.R.; Kim, D.Y.; Jeong, S.; Na, Y.J.; Kim, J.L.; Yun, H.K.; Kim, B.G.; Park, S.H.; Jo, M.J.; et al. Korean Red Ginseng Extract Increases Apoptosis by Activation of the Noxa Pathway in Colorectal Cancer. Nutrients 2019, 11, 2026. [Google Scholar] [CrossRef]
- Koury, J.; Zhong, L.; Hao, J. Targeting Signaling Pathways in Cancer Stem Cells for Cancer Treatment. Stem Cells Int. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Ajani, J.A.; Song, S.; Hochster, H.S.; Steinberg, I.B. Cancer stem cells: The promise and the potential. Semin. Oncol. 2015, 42, S3–S17. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Rogoff, H.A.; Keates, S.; Gao, Y.; Murikipudi, S.; Mikule, K.; Leggett, D.; Li, W.; Pardee, A.B.; Li, C.J. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc. Natl. Acad. Sci. USA 2015, 112, 1839–1844. [Google Scholar] [CrossRef] [PubMed]
- Beyreis, M.; Gaisberger, M.; Jakab, M.; Neureiter, D.; Helm, K.; Ritter, M.; Kiesslich, T.; Mayr, C. The cancer stem cell inhibitor napabucasin (BBI608) shows general cytotoxicity in biliary tract cancer cells and reduces cancer stem cell characteristics. Cancers 2019, 11, 276. [Google Scholar] [CrossRef]
- Chang, J.H.; Au, H.K.; Lee, W.C.; Chi, C.C.; Ling, T.Y.; Wang, L.M.; Kao, S.H.; Huang, Y.H.; Tzeng, C.R. Expression of the pluripotent transcription factor OCT4 promotes cell migration in endometriosis. Fertil. Steril. 2013, 99, 1332–1339. [Google Scholar] [CrossRef]
- Tamaki, T.; Shimizu, T.; Niki, M.; Shimizu, M.; Nishizawa, T.; Nomura, S. Immunohistochemical analysis of NANOG expression and epithelial-mesenchymal transition in pulmonary sarcomatoid carcinoma. Oncol. Lett. 2017, 13, 3695–3702. [Google Scholar] [CrossRef]
- Nomura, A.; Banerjee, S.; Chugh, R.; Dudeja, V.; Yamamoto, M.; Vickers, S.M.; Saluja, A.K. CD133 initiates tumors, induces epithelial-mesenchymal transition and increases metastasis in pancreatic cancer. Oncotarget 2015, 6, 8313–8322. [Google Scholar] [CrossRef]
- Oh, J.; Yoon, H.J.; Jang, J.H.; Kim, D.H.; Surh, Y.J. The standardized Korean Red Ginseng extract and its ingredient ginsenoside Rg3 inhibit manifestation of breast cancer stem cell–like properties through modulation of self-renewal signaling. J. Ginseng Res. 2019, 43, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Chen, M.; Li, P.; Wu, Y.; Chang, C.; Qiu, Y.; Cao, L.; Liu, Z.; Jia, C. Ginsenoside Rh2 inhibits cancer stem-like cells in skin squamous cell carcinoma. Cell. Physiol. Biochem. 2015, 36, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Yang, Y.; Yang, Y.; Zhang, Y.; Yue, Z.; Pan, Z.; Ren, X. Ginsenoside Rg3 attenuates cisplatin resistance in lung cancer by downregulating PD-L1 and resuming immune. Biomed. Pharmacother. 2017, 96, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Liu, J.; Wang, Y.; Zhou, Q.; Wang, S.; Wang, X. Ginsenoside Rg3 improves cyclophosphamide-induced immunocompetence in Balb/c mice. Int. Immunopharmacol. 2019, 72, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Guixing, R.; Caie, W.; Cong, T.; Yang, Y. Synergistic effect of combined protopanaxatiol and ginsenoside Rh2 on antiproliferative activity in MDA-MB-231 human breast cancer cells in vitro. Food Agric. Immunol. 2018, 29, 953–963. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.W.; Park, J.H.; Han, J.-W. Fermented Ginseng Extract, BST204, Suppresses Tumorigenesis and Migration of Embryonic Carcinoma through Inhibition of Cancer Stem Cell Properties. Molecules 2020, 25, 3128. https://doi.org/10.3390/molecules25143128
Park JW, Park JH, Han J-W. Fermented Ginseng Extract, BST204, Suppresses Tumorigenesis and Migration of Embryonic Carcinoma through Inhibition of Cancer Stem Cell Properties. Molecules. 2020; 25(14):3128. https://doi.org/10.3390/molecules25143128
Chicago/Turabian StylePark, Jong Woo, Jee Hun Park, and Jeung-Whan Han. 2020. "Fermented Ginseng Extract, BST204, Suppresses Tumorigenesis and Migration of Embryonic Carcinoma through Inhibition of Cancer Stem Cell Properties" Molecules 25, no. 14: 3128. https://doi.org/10.3390/molecules25143128
APA StylePark, J. W., Park, J. H., & Han, J.-W. (2020). Fermented Ginseng Extract, BST204, Suppresses Tumorigenesis and Migration of Embryonic Carcinoma through Inhibition of Cancer Stem Cell Properties. Molecules, 25(14), 3128. https://doi.org/10.3390/molecules25143128