
Anion and Cation Dynamics in Polyhydroborate Salts: NMR Studies


Abstract
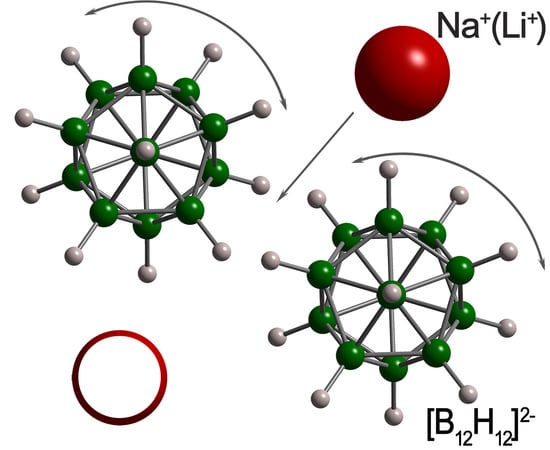
1. Introduction
2. NMR Approach to Dynamical Studies
3. Ultrafast Low-Temperature Reorientational Motion of BH4 Anions
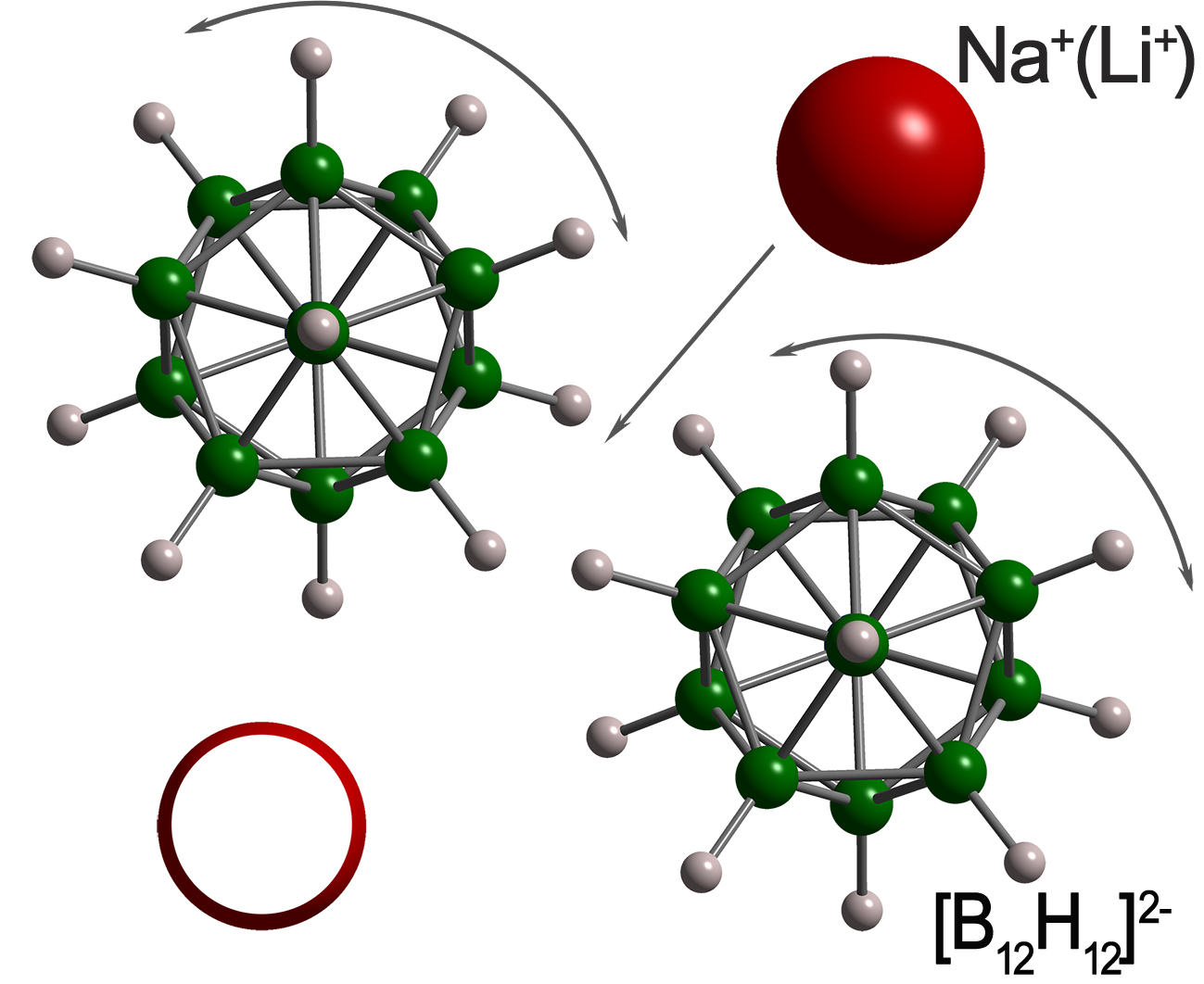
4. Anion Reorientations and Cation Diffusion in B12H12- and B10H10-Based closo-Borates and Related Compounds
5. Effects of Nanoconfinement on Dynamical Properties of Hydroborates
6. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mohtadi, R.; Orimo, S. The Renaissance of Hydrides as Energy Materials. Nat. Rev. Mater. 2016, 2, 16091. [Google Scholar] [CrossRef]
- Hansen, B.R.S.; Paskevicius, M.; Li, H.; Akiba, E.; Jensen, T.R. Metal Boranes: Progress and Applications. Coord. Chem. Rev. 2016, 323, 60–70. [Google Scholar] [CrossRef]
- Orimo, S.; Nakamori, Y.; Elisen, J.R.; Züttel, A.; Jensen, C.M. Complex Hydrides for Hydrogen Storage. Chem. Rev. 2007, 107, 4111–4132. [Google Scholar] [CrossRef] [PubMed]
- Udovic, T.J.; Matsuo, M.; Unemoto, A.; Verdal, N.; Stavila, V.; Skripov, A.V.; Rush, J.J.; Takamura, H.; Orimo, S. Sodium Superionic Conduction in Na2B12H12. Chem. Commun. 2014, 50, 3750–3752. [Google Scholar] [CrossRef]
- Udovic, T.J.; Matsuo, M.; Tang, W.S.; Wu, H.; Stavila, V.; Soloninin, A.V.; Skoryunov, R.V.; Babanova, O.A.; Skripov, A.V.; Rush, J.J.; et al. Exceptional Superionic Conductivity in Disordered Sodium Decahydro-closo-decaborate. Adv. Mater. 2014, 26, 7622–7626. [Google Scholar] [CrossRef]
- Tang, W.S.; Unemoto, A.; Zhou, W.; Stavila, V.; Matsuo, M.; Wu, H.; Orimo, S.; Udovic, T.J. Unparalleled Lithium and Sodium Superionic Conduction in Solid Electrolytes with Large Monovalent Cage-like Anions. Energy Environ. Sci. 2015, 8, 3637–3645. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.S.; Matsuo, M.; Wu, H.; Stavila, V.; Zhou, W.; Talin, A.A.; Soloninin, A.V.; Skoryunov, R.V.; Babanova, O.A.; Skripov, A.V.; et al. Liquid-like Ionic Conduction in Solid Lithium and Sodium Monocarba-Closo-Decaborates near or at Room Temperature. Adv. Energy Mater. 2016, 6, 1502237. [Google Scholar] [CrossRef]
- Yoshida, K.; Sato, T.; Unemoto, A.; Matsuo, M.; Ikeshoji, T.; Udovic, T.J.; Orimo, S. Fast Sodium Ionic Conduction in Na2B10H10-Na2B12H12 Complex Hydride and Application to a Bulk-Type All-Solid-State Battery. Appl. Phys. Lett. 2017, 110, 103901. [Google Scholar] [CrossRef]
- Kim, S.; Oguchi, H.; Toyama, N.; Sato, T.; Takagi, S.; Otomo, T.; Arunkumar, D.; Kuwata, N.; Kawamura, J.; Orimo, S. A Complex Hydride Lithium Superionic Conductor for High-Energy-Density All-Solid-State Lithium Metal Batteries. Nat. Commun. 2019, 10, 1081. [Google Scholar] [CrossRef]
- Duchêne, L.; Remhof, A.; Hagemann, H.; Battaglia, C. Status and Prospects of Hydroborate Electrolytes for All-Solid-State Batteries. Energy Storage Mater. 2020, 25, 782–794. [Google Scholar] [CrossRef]
- Payandeh, S.H.; Asakura, R.; Avramidou, P.; Rentsch, D.; Łodziana, Z.; Černý, R.; Remhof, A.; Battaglia, C. Nido-Borate/Closo-Borate Mixed-Anion Electrolytes for All-Solid-State Batteries. Chem. Mater. 2020, 32, 1101–1110. [Google Scholar] [CrossRef]
- Tsang, T.; Farrar, T.C. Nuclear Magnetic Relaxation Studies of Internal Rotations and Phase Transitions in Borohydrides of Lithium, Sodium, and Potassium. J. Chem. Phys. 1969, 50, 3498–3502. [Google Scholar] [CrossRef]
- Skripov, A.V.; Soloninin, A.V.; Babanova, O.A. Nuclear Magnetic Resonance Studies of Atomic Motion in Borohydrides. J. Alloys Compd. 2011, 509, S535–S539. [Google Scholar] [CrossRef]
- Skripov, A.V.; Soloninin, A.V.; Filinchuk, Y.; Chernyshov, D. Nuclear Magnetic Resonance Study of the Rotational Motion and the Phase Transition in LiBH4. J. Phys. Chem. C 2008, 112, 18701–18705. [Google Scholar] [CrossRef]
- Corey, R.L.; Shane, D.T.; Bowman, R.C.; Conradi, M.S. Atomic Motions in LiBH4 by NMR. J. Phys. Chem. C 2008, 112, 18706–18710. [Google Scholar] [CrossRef]
- Skripov, A.V.; Soloninin, A.V.; Babanova, O.A.; Skoryunov, R.V. Nuclear Magnetic Resonance Studies of Atomic Motion in Borohydride-Based Materials: Fast Anion Reorientations and Cation Diffusion. J. Alloys Compd. 2015, 645, S428–S433. [Google Scholar] [CrossRef]
- Skripov, A.V.; Babanova, O.A.; Soloninin, A.V.; Stavila, V.; Verdal, N.; Udovic, T.J.; Rush, J.J. Nuclear Magnetic Resonance Study of Atomic Motion in A2B12H12 (A = Na, K, Rb, Cs): Anion Reorientations and Na+ Mobility. J. Phys. Chem. C 2013, 117, 25961–25968. [Google Scholar] [CrossRef]
- Lu, Z.; Ciucci, F. Structural Origin of the Superionic Na Conduction in Na2B10H10 Closo-Borates and Enhanced Conductivity by Na Deficiency for High Performance Solid Electrolyte. J. Mater. Chem. A 2016, 4, 17740–17748. [Google Scholar] [CrossRef]
- Varley, J.B.; Kweon, K.; Mehta, P.; Shea, P.; Heo, T.W.; Udovic, T.J.; Stavila, V.; Wood, B.C. Understanding Ionic Conductivity Trends in Polyborane Solid Electrolytes from Ab Initio Molecular Dynamics. ACS Energy Lett. 2017, 2, 250–255. [Google Scholar] [CrossRef]
- Kweon, K.; Varley, J.B.; Shea, P.; Adelstein, N.; Mehta, P.; Heo, T.W.; Udovic, T.J.; Stavila, V.; Wood, B.C. Structural, Chemical, and Dynamical Frustration: Origins of Superionic Conductivity in Closo-Borate Solid Electrolytes. Chem. Mater. 2017, 29, 9142–9153. [Google Scholar] [CrossRef]
- Dimitrievska, M.; Shea, P.; Kweon, K.E.; Bercx, M.; Varley, J.B.; Tang, W.S.; Skripov, A.V.; Stavila, V.; Udovic, T.J.; Wood, B.C. Carbon Incorporation and Anion Dynamics as Synergistic Drivers for Ultrafast Diffusion in Superionic LiCB11H12 and NaCB11H12. Adv. Energy Mater. 2018, 6, 1703422. [Google Scholar] [CrossRef]
- Soloninin, A.V.; Skripov, A.V.; Buzlukov, A.L.; Stepanov, A.P. Nuclear Magnetic Resonance Study of Li and H diffusion in the High-Temperature Solid Phase of LiBH4. J. Solid State Chem. 2009, 182, 2357–2361. [Google Scholar] [CrossRef]
- Babanova, O.A.; Soloninin, A.V.; Stepanov, A.P.; Skripov, A.V.; Filinchuk, Y. Structural and Dynamical Properties of NaBH4 and KBH4: NMR and Synchrotron X-ray Diffraction Studies. J. Phys. Chem. C 2010, 114, 3712–3718. [Google Scholar] [CrossRef]
- Shane, D.T.; Corey, R.L.; McIntosh, C.; Rayhel, L.H.; Bowman, R.C.; Vajo, J.J.; Gross, A.F.; Conradi, M.S. LiBH4 in Carbon Aerogel Nanoscaffolds: An NMR Study of Atomic Motions. J. Phys. Chem. C 2010, 114, 4008–4014. [Google Scholar] [CrossRef]
- Epp, V.; Wilkening, M. Fast Li Diffusion in Crystalline LiBH4 Due to Reduced Dimensionality: Frequency-Dependent NMR Spectroscopy. Phys. Rev. B 2010, 82, 020301(R). [Google Scholar] [CrossRef]
- Babanova, O.A.; Soloninin, A.V.; Skripov, A.V.; Ravnsbæk, D.B.; Jensen, T.R.; Filinchuk, Y. Reorientational Motion in Alkali-Metal Borohydrides: NMR Data for RbBH4 and CsBH4 and Systematics of the Activation Energy Variations. J. Phys. Chem. C 2011, 115, 10305–10309. [Google Scholar] [CrossRef]
- Jimura, K.; Hayashi, S. Reorientational Motion of BH4 Ions in Alkali Borohydrides MBH4 (M = Li, Na, K) as Studied by Solid-State NMR. J. Phys. Chem. C 2012, 116, 4883–4891. [Google Scholar] [CrossRef]
- Skripov, A.V.; Soloninin, A.V.; Babanova, O.A.; Hagemann, H.; Filinchuk, Y. Nuclear Magnetic Resonance Study of Reorientational Motion in α-Mg(BH4)2. J. Phys. Chem. C 2010, 114, 12370–12374. [Google Scholar] [CrossRef]
- Shane, D.T.; Rayhel, L.H.; Huang, Z.; Zhao, J.-C.; Tang, X.; Stavila, V.; Conradi, M.S. Comprehensive NMR Study of Magnesium Borohydride. J. Phys. Chem. C 2011, 115, 3172–3177. [Google Scholar] [CrossRef]
- Soloninin, A.V.; Babanova, O.A.; Skripov, A.V.; Hagemann, H.; Richter, B.; Jensen, T.R.; Filinchuk, Y. NMR Study of Reorientational Motion in Alkaline-Earth Borohydrides: β and γ Phases of Mg(BH4)2 and α and β Phases of Ca(BH4)2. J. Phys. Chem. C 2012, 116, 4913–4920. [Google Scholar] [CrossRef]
- Eagles, M.; Sun, B.; Richter, B.; Jensen, T.R.; Filinchuk, Y.; Conradi, M.S. NMR Investigation of Nanoporous γ-Mg(BH4)2 and Its Thermally Induced Phase Changes. J. Phys. Chem. C 2012, 116, 13033–13037. [Google Scholar] [CrossRef]
- Jeener, J.; Broekaert, P. Nuclear Magnetic Resonance in Solids: Thermodynamic Effects of a Pair of rf Pulses. Phys. Rev. 1967, 157, 232–240. [Google Scholar] [CrossRef]
- Spiess, H.W. Deuteron Spin Alignment: A Probe for Studying Ultraslow Motions in Solids and Solid Polymers. J. Chem. Phys. 1980, 72, 6755–6762. [Google Scholar] [CrossRef]
- Abragam, A. The Principles of Nuclear Magnetism; Clarendon Press: Oxford, UK, 1961. [Google Scholar]
- Bloembergen, N.; Purcell, E.M.; Pound, R.V. Relaxation Effects in Nuclear Magnetic Resonance Absorption. Phys. Rev. 1948, 73, 679–712. [Google Scholar] [CrossRef]
- Look, D.C.; Lowe, I.J. Effect of Hindered Molecular Rotation between Unequal Potential Wells upon Nuclear Magnetic Resonance Spin-Lattice Relaxation Times and Second Moments. J. Chem. Phys. 1966, 44, 3437–3441. [Google Scholar] [CrossRef]
- Markert, J.T.; Cotts, E.J.; Cotts, R.M. Hydrogen Diffusion in the Metallic Glass a-Zr3RhH3.5. Phys. Rev. B 1988, 37, 6446–6452. [Google Scholar] [CrossRef]
- Stejskal, E.O.; Tanner, J.E. Spin Diffusion Measurements: Spin Echos in the Presence of a Time-Dependent Field Gradient. J. Chem. Phys. 1965, 42, 288–292. [Google Scholar] [CrossRef]
- Price, W.S. NMR Studies of Translational Motion—Principles and Applications; University Press: Cambridge, UK, 2009. [Google Scholar]
- Callaghan, P.T. Translational Dynamics and Magnetic Resonance—Principles of Pulsed Gradient Spin Echo NMR; University Press: Oxford, UK, 2011. [Google Scholar]
- Tanner, J.E. Use of the Stimulated Echo in NMR Diffusion Studies. J. Chem. Phys. 1970, 52, 2523–2526. [Google Scholar] [CrossRef]
- Bée, M. Quasielastic Neutron Scattering, Principles and Applications in Solid State Chemistry, Biology and Materials Science; Adam Hilger: Bristol, UK, 1988. [Google Scholar]
- Hempelmann, R. Quasielastic Neutron Scattering and Solid State Diffusion; Clarendon Press: Oxford, UK, 2000. [Google Scholar]
- Maekawa, H.; Matsuo, M.; Takamura, H.; Ando, M.; Noda, Y.; Karahashi, T.; Orimo, S. Halide-Stabilized LiBH4, a Room-Temperature Lithium Fast-Ion Conductor. J. Am. Chem. Soc. 2009, 131, 894–895. [Google Scholar] [CrossRef]
- Skripov, A.V.; Soloninin, A.V.; Rude, L.H.; Jensen, T.R.; Filinchuk, Y. Nuclear Magnetic Resonance Studies of Reorientational Motion and Li Diffusion in LiBH4-LiI Solid Solutions. J. Phys. Chem. C 2012, 116, 26177–26184. [Google Scholar] [CrossRef]
- Martelli, P.; Remhof, A.; Borgschulte, A.; Ackermann, R.; Strässle, E.J.P.; Ernst, M.; Matsuo, M.; Orimo, S.; Züttel, A. Rotational Motion in LiBH4/LiI Solid Solutions. J. Phys. Chem. A 2011, 115, 5329–5334. [Google Scholar] [CrossRef] [PubMed]
- Verdal, N.; Udovic, T.J.; Rush, J.J.; Wu, H.; Skripov, A.V. Evolution of the Reorientational Motions of the Tetrahydroborate Anions in Hexagonal LiBH4-LiI Solid Solutions by High-Q Quasielastic Neutron Scattering. J. Phys. Chem. C 2013, 117, 12010–12018. [Google Scholar] [CrossRef]
- Frommen, C.; Sørby, M.H.; Ravindran, P.; Vajeeston, P.; Fjellvåg, H.; Hauback, B.C. Synthesis, Crystal Structure, and Thermal Properties of the First Mixed-Metal and Anion-Substituted Rare Earth Borohydride LiCe(BH4)3Cl. J. Phys. Chem. C 2011, 115, 23591–23602. [Google Scholar] [CrossRef]
- Ley, M.B.; Ravnsbæk, D.B.; Filinchuk, Y.; Lee, Y.S.; Janot, R.; Cho, Y.W.; Skibsted, J.; Jensen, T.R. LiCe(BH4)3Cl, a New Lithium-Ion Conductor and Hydrogen Storage Material with Isolated Tetranuclear Anionic Clusters. Chem. Mater. 2012, 24, 1654–1663. [Google Scholar] [CrossRef]
- Ley, M.B.; Boulineau, S.; Janot, R.; Filinchuk, Y.; Jensen, T.R. New Li-Ion Conductors and Solid State Hydrogen Storage Materials: LiM (BH4)3Cl, M = La, Gd. J. Phys. Chem. C 2012, 116, 21267–21276. [Google Scholar] [CrossRef]
- GharibDoust, S.P.; Brighi, M.; Sadykin, Y.; Ravnsbæk, D.B.; Černý, R.; Skibsted, J.; Jensen, T.R. Synthesis, Structure, and Li-Ion Conductivity of LiLa(BH4)3X, X = Cl, Br, I. J. Phys. Chem. C 2017, 121, 19010–19021. [Google Scholar] [CrossRef]
- Skripov, A.V.; Soloninin, A.V.; Ley, M.B.; Jensen, T.R.; Filinchuk, Y. Nuclear Magnetic Resonance Study of BH4 Reorientations and Li Diffusion in LiLa(BH4)3Cl. J. Phys. Chem. C 2013, 117, 14965–14972. [Google Scholar] [CrossRef]
- Soloninin, A.V.; Babanova, O.A.; Skoryunov, R.V.; Skripov, A.V.; Grinderslev, J.; Jensen, T.R. NMR Study of the Dynamical Properties of LiLa(BH4)3Br and LiLa(BH4)3I. Appl. Magn. Reson. 2020, (unpublished, manuscript in preparation). [Google Scholar]
- Soloninin, A.V.; Babanova, O.A.; Medvedev, E.Y.; Skripov, A.V.; Matsuo, M.; Orimo, S. Nuclear Magnetic Resonance Study of Atomic Motion in the Mixed Borohydride-Amide Na2(BH4)(NH2). J. Phys. Chem. C 2014, 118, 14805–14812. [Google Scholar] [CrossRef]
- Chater, P.A.; Anderson, P.A.; Prendergast, J.W.; Walton, A.; Mann, V.S.J.; Book, D.; David, W.I.F.; Johnson, S.R.; Edwards, P.P. Synthesis and Characterization of Amide-Borohydrides: New Complex Light Hydrides for Potential Hydrogen Storage. J. Alloys Compd. 2007, 446, 350–354. [Google Scholar] [CrossRef]
- Somer, M.; Acar, C.; Koz, C.; Kokal, I.; Höhn, P.; Cardoso-Gil, R.; Aydemir, U.; Akselrud, L. α- and β-Na2[BH4][NH2]: Two Modifications of a Complex Hydride in the System NaNH2-NaBH4; Synthesis, Crystal Structures, Thermal Analyses, Mass and Vibrational Spectra. J. Alloys Compd. 2010, 491, 98–105. [Google Scholar] [CrossRef]
- Prager, M.; Heidemann, A. Rotational Tunneling and Neutron Spectroscopy: A Compilation. Chem. Rev. 1997, 97, 2933–2966. [Google Scholar] [CrossRef] [PubMed]
- Verdal, N.; Udovic, T.J.; Rush, J.J.; Stavila, V.; Wu, H.; Zhou, W.; Jenkins, T. Low-Temperature Tunneling and Rotational Dynamics of the Ammonium Cations in (NH4)2B12H12. J. Chem. Phys. 2011, 135, 094501. [Google Scholar] [CrossRef] [PubMed]
- Gradišek, A.; Jepsen, L.H.; Jensen, T.R.; Conradi, M.S. Nuclear Magnetic Resonance Study of Molecular Dynamics in Ammine Metal Borohydride Sr(BH4)2(NH3)2. J. Phys. Chem. C 2016, 120, 24646–24654. [Google Scholar] [CrossRef]
- Skripov, A.V.; Dimitrievska, M.; Babanova, O.A.; Skoryunov, R.V.; Soloninin, A.V.; Morelle, F.; Filinchuk, Y.; Faraone, A.; Wu, H.; Zhou, W.; et al. Low-Temperature Rotational Tunneling of Tetrahydroborate Anions in Lithium Benzimidasolate-Borohydride Li2(bIm)BH4. J. Phys. Chem. C 2019, 123, 20789–20799. [Google Scholar] [CrossRef]
- Phua, T.T.; Beaudry, B.J.; Peterson, D.T.; Torgeson, D.R.; Barnes, R.G.; Belhoul, M.; Styles, G.A.; Seymour, E.F.W. Paramagnetic Impurity Effects in NMR Determinations of Hydrogen Diffusion and Electronic Structure in Metal Hydrides. Gd3+ in YH2 and LaH2.25. Phys. Rev. B 1983, 28, 6227–6250. [Google Scholar] [CrossRef]
- Horsewill, A.J. Quantum Tunneling Aspects of Methyl Group Rotation Studied by NMR. Prog. Nucl. Magn. Reson. Spectrosc. 1999, 35, 359–389. [Google Scholar] [CrossRef]
- Müller-Warmuth, W.; Schüler, R.; Prager, M.; Kollmar, A. Rotational Tunneling in Methylpyridines as Studied by NMR Relaxation and Inelastic Neutron Scattering. J. Chem. Phys. 1978, 69, 2382–2392. [Google Scholar] [CrossRef]
- Mezei, F. Neutron Spin Echo: Ten Years After. Physica B+C 1983, 120, 51–60. [Google Scholar] [CrossRef]
- Tiritiris, I.; Schleid, T.; Müller, K. Solid-State NMR Studies on Ionic closo-Dodecaborates. Appl. Magn. Reson. 2007, 32, 459–481. [Google Scholar] [CrossRef]
- Verdal, N.; Udovic, T.J.; Rush, J.J.; Cappelletti, R.L.; Zhou, W. Reorientational Dynamics of the Dodecahydro-closo-dodecaborate Anion in Cs2B12H12. J. Phys. Chem. A 2011, 115, 2933–2938. [Google Scholar] [CrossRef] [PubMed]
- Skripov, A.V.; Skoryunov, R.V.; Soloninin, A.V.; Babanova, O.A.; Stavila, V.; Udovic, T.J. Nuclear Magnetic Resonance Study of Anion and Cation Reorientational Dynamics in (NH4)2B12H12. J. Phys. Chem. C 2018, 122, 3256–3262. [Google Scholar] [CrossRef]
- Paskevicius, M.; Pitt, M.P.; Brown, D.H.; Sheppard, D.A.; Chumphongphan, S.; Buckley, C.E. First-Order Phase Transition in the Li2B12H12 System. Phys. Chem. Chem. Phys. 2013, 15, 15825–15828. [Google Scholar] [CrossRef] [PubMed]
- Verdal, N.; Her, J.-H.; Stavila, V.; Soloninin, A.V.; Babanova, O.A.; Skripov, A.V.; Udovic, T.J.; Rush, J.J. Complex High-Temperature Phase Transitions in Li2B12H12 and Na2B12H12. J. Solid State Chem. 2014, 212, 81–91. [Google Scholar] [CrossRef]
- Skripov, A.V.; Belyaev, M.Y.; Rychkova, S.V.; Stepanov, A.P. Nuclear Magnetic Resonance Study of Hydrogen Diffusion in HfV2Hx(Dx) and ZrV2Hx(Dx): Effects of Phase Transitions and Isotope Substitution. J. Phys. Condens. Matter 1991, 3, 6277–6291. [Google Scholar] [CrossRef]
- Skripov, A.V.; Skoryunov, R.V.; Soloninin, A.V.; Babanova, O.A.; Tang, W.S.; Stavila, V.; Udovic, T.J. Anion Reorientations and Cation Diffusion in LiCB11H12 and NaCB11H12: 1H, 7Li, and 23Na NMR Studies. J. Phys. Chem. C 2015, 119, 26912–26918. [Google Scholar] [CrossRef]
- Verdal, N.; Udovic, T.J.; Stavila, V.; Tang, W.S.; Rush, J.J.; Skripov, A.V. Anion Reorientations in the Superionic Conducting Phase of Na2B12H12. J. Phys. Chem. C 2014, 118, 17483–17489. [Google Scholar] [CrossRef]
- Bonnetot, B.; Mongeot, H.; Aboukhassib, A.; Lefebvre, F. Phase Transition Investigations of closo-Hydroborates. Inorg. Chim. Acta 1992, 193, 21–26. [Google Scholar] [CrossRef]
- Soloninin, A.V.; Dimitrievska, M.; Skoryunov, R.V.; Babanova, O.A.; Skripov, A.V.; Tang, W.S.; Stavila, V.; Udovic, T.J. Comparison of Anion Reorientational Dynamics in MCB9H10 and M2B10H10 (M = Li, Na) via Nuclear Magnetic Resonance and Quasielastic Neutron Scattering Studies. J. Phys. Chem. C 2017, 121, 1000–1012. [Google Scholar] [CrossRef]
- Dimitrievska, M.; Stavila, V.; Soloninin, A.V.; Skoryunov, R.V.; Babanova, O.A.; Wu, H.; Zhou, W.; Tang, W.S.; Faraone, A.; Tarver, J.D.; et al. Nature of Decahydro-closo-decaborate Anion Reorientations in an Ordered Alkali-Metal Salt: Rb2B10H10. J. Phys. Chem. C 2018, 122, 15198–15207. [Google Scholar] [CrossRef]
- Dimitrievska, M.; Wu, H.; Stavila, V.; Babanova, O.A.; Skoryunov, R.V.; Soloninin, A.V.; Zhou, W.; Trump, B.A.; Andersson, M.S.; Skripov, A.V.; et al. Structural and Dynamical Properties of Potassium Dodecahydro-monocarba-closo-dodecaborate: KCB11H12. J. Phys. Chem. C 2020. (submitted). [Google Scholar]
- Skripov, A.V.; Majer, G.; Babanova, O.A.; Skoryunov, R.V.; Soloninin, A.V.; Dimitrievska, M.; Udovic, T.J. Na+ Diffusivity in Carbon-Substituted nido- and closo-Hydroborate Salts: Pulsed-Field-Gradient NMR Studies of Na-7-CB10H13 and Na(CB9H10)(CB11H12). J. Alloys Compd. 2020. (submitted). [Google Scholar]
- Duchêne, L.; Lunghammer, S.; Burankova, T.; Liao, W.-C.; Embs, J.P.; Copéret, C.; Wilkening, H.M.R.; Remhof, A.; Hagemann, H.; Battaglia, C. Ionic Conduction Mechanism in the Na2(B12H12)0.5(B10H10)0.5 closo-Borate Solid-State Electrolyte: Interplay of Disorder and Ion-Ion Interactions. Chem. Mater. 2019, 31, 3449–3460. [Google Scholar] [CrossRef]
- Soloninin, A.V.; Skoryunov, R.V.; Babanova, O.A.; Skripov, A.V.; Dimitrievska, M.; Udovic, T.J. Comparison of Anion and Cation Dynamics in a Carbon-Substituted closo-Hydroborate Salt: 1H and 23Na NMR Studies of Solid-Solution Na2(CB9H10)(CB11H12). J. Alloys Compd. 2019, 800, 247–253. [Google Scholar] [CrossRef]
- Tang, W.S.; Dimitrievska, M.; Stavila, V.; Zhou, W.; Wu, H.; Talin, A.A.; Udovic, T.J. Order-Disorder Transitions and Superionic Conductivity in the Sodium nido-Undeca (carba) borates. Chem. Mater. 2017, 29, 10496–10509. [Google Scholar] [CrossRef]
- Tang, W.S.; Matsuo, M.; Wu, H.; Stavila, V.; Unemoto, A.; Orimo, S.; Udovic, T.J. Stabilizing Lithium and Sodium Fast-Ion Conduction in Solid Polyhedral-Borate Salts at Device-Relevant Temperatures. Energy Storage Mater. 2016, 4, 79–83. [Google Scholar] [CrossRef]
- Tang, W.S.; Yoshida, K.; Soloninin, A.V.; Skoryunov, R.V.; Babanova, O.A.; Skripov, A.V.; Dimitrievska, M.; Stavila, V.; Orimo, S.; Udovic, T.J. Stabilizing Superionic-Conducting Structures via Mixed-Anion Solid Solutions of Monocarba-Closo-Borate Salts. ACS Energy Lett. 2016, 1, 659–664. [Google Scholar] [CrossRef]
- Duchêne, L.; Kühnel, R.-S.; Rentsch, D.; Remhof, A.; Hagemann, H.; Battaglia, C. A Highly Stable Sodium Solid-State Electrolyte Based on a Dodeca/Deca-Borate Equimolar Mixture. Chem. Commun. 2017, 53, 4195–4198. [Google Scholar] [CrossRef]
- Brighi, M.; Murgia, F.; Łodziana, Z.; Schouwink, P.; Wołczyk, A.; Černý, R. A Mixed Anion Hydroborate/Carba-Hydroborate as a Room Temperature Na-Ion Solid Electrolyte. J. Power Source 2018, 404, 7–12. [Google Scholar] [CrossRef]
- Skoryunov, R.V.; Babanova, O.A.; Soloninin, A.V.; Skripov, A.V.; Verdal, N.; Udovic, T.J. Effects of Partial Halide Anion Substitution on Reorientational Motion in NaBH4: A Nuclear Magnetic Resonance Study. J. Alloys Compd. 2015, 636, 293–297. [Google Scholar] [CrossRef]
- Heitjans, P.; Indris, S. Diffusion and Ionic Conduction in Nanocrystalline Ceramics. J. Phys. Condens. Matter 2003, 15, R1257–R1289. [Google Scholar] [CrossRef]
- Gross, A.F.; Vajo, J.J.; Van Atta, S.L.; Olso, G.L. Enhanced Hydrogen Storage Kinetics of LiBH4 in Nanoporous Carbon Scaffolds. J. Phys. Chem. C 2008, 112, 5651–5657. [Google Scholar] [CrossRef]
- Ngene, P.; Adelhelm, P.; Beale, A.M.; de Jongh, K.P.; de Jongh, P.E. LiBH4/SBA-15 Nanocomposites Prepared by Melt Infiltration under Hydrogen Pressure: Synthesis and Hydrogen Sorption Properties. J. Phys. Chem. C 2010, 114, 6163–6168. [Google Scholar] [CrossRef]
- Verkuijlen, M.H.W.; Ngene, P.; de Kort, D.W.; Barré, C.; Nale, A.; van Eck, E.R.H.; van Bentum, P.J.M.; de Jongh, P.E.; Kentgens, A.P. Nanoconfined LiBH4 and Enhanced Mobility of Li+ and BH4− Studied by Solid-State NMR. J. Phys. Chem. C 2012, 116, 22169–22178. [Google Scholar] [CrossRef]
- Remhof, A.; Mauron, P.; Züttel, A.; Embs, J.P.; Łodziana, Z.; Ramirez-Cuesta, A.J.; Ngene, P.; de Jongh, P. Hydrogen Dynamics in Nanoconfined Lithiumborohydride. J. Phys. Chem. C 2013, 117, 3789–3798. [Google Scholar] [CrossRef]
- Verdal, N.; Udovic, T.J.; Rush, J.J.; Liu, X.; Majzoub, E.H.; Vajo, J.J.; Gross, A.F. Dynamical Perturbations of Tetrahydroborate Anions in LiBH4 due to Nanoconfinement in Controlled-Pore Carbon Scaffolds. J. Phys. Chem. C 2013, 117, 17983–17995. [Google Scholar] [CrossRef]
- Liu, X.; Majzoub, E.H.; Stavila, V.; Bhakta, R.K.; Allendorf, M.D.; Shane, D.T.; Conradi, M.S.; Verdal, N.; Udovic, T.J.; Hwang, S.-J. Probing the Unusual Anion Mobility of LiBH4 Confined in Highly Ordered Nanoporous Carbon Frameworks via Solid State NMR and Quasielastic Neutron Scattering. J. Mater. Chem. A 2013, 1, 9935–9941. [Google Scholar] [CrossRef]
- Blanchard, D.; Nale, A.; Sveinbjörnsson, D.; Eggenhuisen, T.M.; Verkuijlen, M.H.W.; Vegge, T.; Kentgens, A.P.M.; de Jongh, P.E. Nanoconfined LiBH4 as a Fast Lithium Ion Conductor. Adv. Funct. Mater. 2015, 25, 184–192. [Google Scholar] [CrossRef]
- Zou, H.; Gradišek, A.; Emery, S.B.; Vajo, J.J.; Conradi, M.S. LiBH4 in Aerogel: Ionic Motions by NMR. J. Phys. Chem. C 2017, 121, 15114–15119. [Google Scholar] [CrossRef]
- Lefevr, J.; Cervini, L.; Griffin, J.M.; Blanchard, D. Lithium Conductivity and Ion Dynamics in LiBH4/SiO2 Solid Electrolytes Studied by Solid-State NMR and Quasi-Elastic Neutron Scattering and Applied in Lithium-Sulfur Batteries. J. Phys. Chem. C 2018, 122, 15264–15275. [Google Scholar] [CrossRef]
- Lambregts, S.F.H.; van Eck, E.R.H.; Suwarno; Ngene, P.; de Jongh, P.E.; Kentgens, A.P.M. Phase Behavior and Ion Dynamics of Nanoconfined LiBH4 in Silica. J. Phys. Chem. C 2019, 123, 25559–25569. [Google Scholar] [CrossRef]
- Zettl, R.; de Kort, L.; Gombotz, M.; Wilkening, H.M.R.; de Jongh, P.E.; Ngene, P. Combined Effects of Anion Substitution and Nanoconfinement on the Ionic Conductivity of Li-Based Complex Hydrides. J. Phys. Chem. C 2020, 124, 2806–2816. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Rentsch, D.; Battaglia, C.; Remhof, A. Synthesis, Stability and Li-Ion Mobility of Nanoconfined Li2B12H12. Dalton Trans. 2017, 46, 12434–12437. [Google Scholar] [CrossRef] [PubMed]
- Skripov, A.V.; Soloninin, A.V.; Babanova, O.A.; Skoryunov, R.V.; Stavila, V.; Dimitrievska, M.; Psurek, M.; Udovic, T.J. Dynamical Properties of a Carbon-Substituted closo-Hydroborate in Nanoporous Silica: NMR and QENS Studies of NaCB11H12 in SBA-15. J. Phys. Chem. C 2020, (unpublished, manuscript in preparation). [Google Scholar]
- Matsuo, M.; Nakamori, Y.; Orimo, S.; Maekawa, H.; Takamura, H. Lithium Superionic Conduction in Lithium Borohydride Accompanied by Structural Transition. Appl. Phys. Lett. 2007, 91, 224103. [Google Scholar] [CrossRef]
- Yan, Y.; Grinderslev, J.B.; Lee, Y.-S.; Jørgensen, M.; Cho, Y.W.; Černý, R.; Jensen, T.R. Ammonia-Assisted Fast Li-Ion Conductivity in a New Hemiammine Lithium Borohydride, LiBH4·1/2NH3. Chem. Commun. 2020, 56, 3971–3974. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Activation Energy (meV) | T Range (K) | Method | Reference |
|---|---|---|---|---|
| Li2B12H12 (LT phase) | 1400 (50) | 490–590 | NMR | [69] |
| Na2B12H12 (LT phase) | 770 (20) | 400–520 | NMR | [17] |
| Na2B12H12 (HT phase) | 270 (40) | 523–570 | NMR | [17] |
| 259 (22) | 480–620 | QENS | [72] | |
| K2B12H12 | 1070 (54) | 270–370 | NMR a | [65] |
| 800 (8) | 366–564 | NMR | [17] | |
| Rb2B12H12 | 910 (46) | 220–340 | NMR a | [65] |
| 549 (5) | 315–560 | NMR | [17] | |
| (NH4)2B12H12 | 930 (40) | 250–350 | NMR a | [65] |
| 486 (8) | 297–474 | NMR | [67] | |
| Cs2B12H12 | 600 (30) | 180–300 | NMR a | [65] |
| 333 (15) | 430–530 | QENS | [66] | |
| 427 (4) | 260–570 | NMR | [17] | |
| Na2B10H10 (HT phase) | 180 (30) | 375–435 | NMR | [74] |
| 124 (2) | 365–500 | QENS | [74] | |
| Rb2B10H10 | 522 (7) b | 219–573 | NMR | [75] |
| 288 (3) and 197 (2) c | 400–680 | QENS | [75] | |
| LiCB11H12 (LT phase) | 409 (11) | 278–384 | NMR | [71] |
| LiCB11H12 (HT phase) | 177 (7) | 390–435 | NMR | [71] |
| NaCB11H12 (LT phase) | 409 (7) | 278–376 | NMR | [71] |
| NaCB11H12 (HT phase) | 177 (8) | 380–435 | NMR | [71] |
| KCB11H12 (LT phase) | 330 (20) | 258–332 | NMR | [76] |
| KCB11H12 (HT phase) | 191 (4) | 341–435 | NMR | [76] |
| 149 (2) | 345–470 | QENS | [76] | |
| LiCB9H10 (LT phase) | 302 (7) b | 258–332 | NMR | [74] |
| LiCB9H10 (HT phase) | 299 (5) | 359–418 | NMR | [74] |
| 170 (2) | 330–410 | QENS | [74] | |
| NaCB9H10 (LT phase) | 234 (6) b | 179–278 | NMR | [74] |
| NaCB9H10 (HT phase) | 205 (5) | 287–376 | NMR | [74] |
| 130 (2) | 290–445 | QENS | [74] | |
| Na-7-CB10H13 (LT phase) | 330 (20) | 209–315 | NMR | [77] |
| Na-7-CB10H13 (HT phase) | 219 (5) | 332–384 | NMR | [77] |
| Na2(B12H12)0.5(B10H10)0.5 | 240 (10) | 300–390 | QENS | [78] |
| Na2(CB9H10)(CB11H12) | 430 (40) and 147 (9) c | 278–376 | NMR | [79] |
| Compound | Activation Energy (meV) | T Range (K) | Method | Reference |
|---|---|---|---|---|
| Na2B12H12 (LT phase) | 450 (10) | 458–517 | NMR | [17] |
| Na2B12H12 (HT phase) | 410 (25) | 523–580 | NMR | [17] |
| Na2B10H10 (LT phase) | 750 (20) | 298–367 | NMR | [5] |
| Na2B10H10 (HT phase) | 190 (10) | 375–435 | NMR | [5] |
| LiCB11H12 (LT phase) | 422 (6) | 332–376 | NMR | [71] |
| LiCB11H12 (HT phase) | 92 (7) | 392–426 | NMR | [71] |
| NaCB11H12 (LT phase) | 327 (11) | 340–367 | NMR | [71] |
| NaCB11H12 (HT phase) | 152 (8) | 376–418 | NMR | [43] |
| LiCB9H10 (HT phase) | 55 (9) | 358–418 | NMR | [7] |
| NaCB9H10 (HT phase) | 153 (7) | 293–401 | NMR | [7] |
| Na-7-CB10H13 (LT phase) | 320 (9) | 198–315 | NMR | [77] |
| Na-7-CB10H13 (HT phase) | 116 (7) | 333–385 | NMR | [77] |
| 134 (3) | 320–403 | PFG-NMR | [77] | |
| Li(CB9H10)0.7(CB11H12)0.3 | ~295 | 298–333 | PFG-NMR | [9] |
| Na2(B12H12)0.5(B10H10)0.5 | 240 (10) | 180–320 | NMR | [78] |
| Na2(CB9H10)(CB11H12) | 353 (11) | 138–349 | NMR | [79] |
| 135 (8) | 350–435 | NMR | [79] | |
| 118 (1) | 298–403 | PFG-NMR | [77] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skripov, A.V.; Soloninin, A.V.; Babanova, O.A.; Skoryunov, R.V. Anion and Cation Dynamics in Polyhydroborate Salts: NMR Studies. Molecules 2020, 25, 2940. https://doi.org/10.3390/molecules25122940
Skripov AV, Soloninin AV, Babanova OA, Skoryunov RV. Anion and Cation Dynamics in Polyhydroborate Salts: NMR Studies. Molecules. 2020; 25(12):2940. https://doi.org/10.3390/molecules25122940
Chicago/Turabian StyleSkripov, Alexander V., Alexei V. Soloninin, Olga A. Babanova, and Roman V. Skoryunov. 2020. "Anion and Cation Dynamics in Polyhydroborate Salts: NMR Studies" Molecules 25, no. 12: 2940. https://doi.org/10.3390/molecules25122940
APA StyleSkripov, A. V., Soloninin, A. V., Babanova, O. A., & Skoryunov, R. V. (2020). Anion and Cation Dynamics in Polyhydroborate Salts: NMR Studies. Molecules, 25(12), 2940. https://doi.org/10.3390/molecules25122940