Identification of Human Leukotriene A4 Hydrolase Inhibitors Using Structure-Based Pharmacophore Modeling and Molecular Docking

 ,
,  , and
, and
Abstract
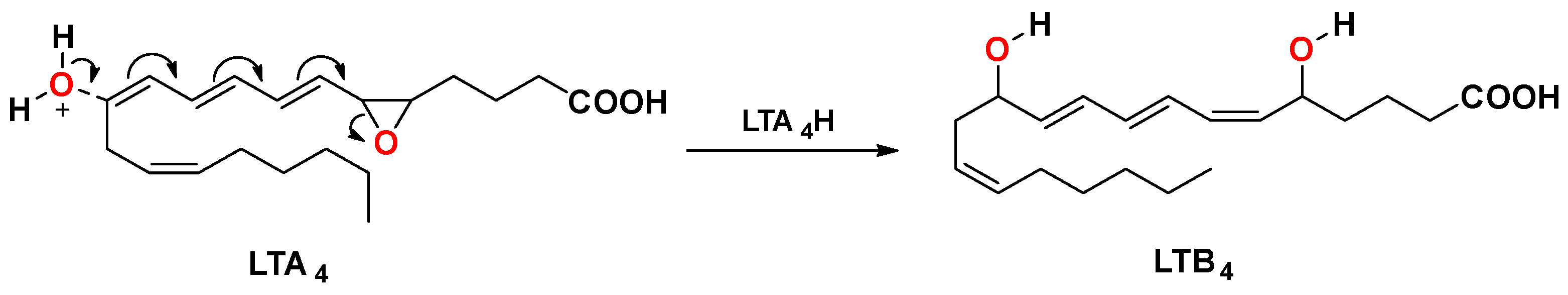
1. Introduction
2. Results and Discussion
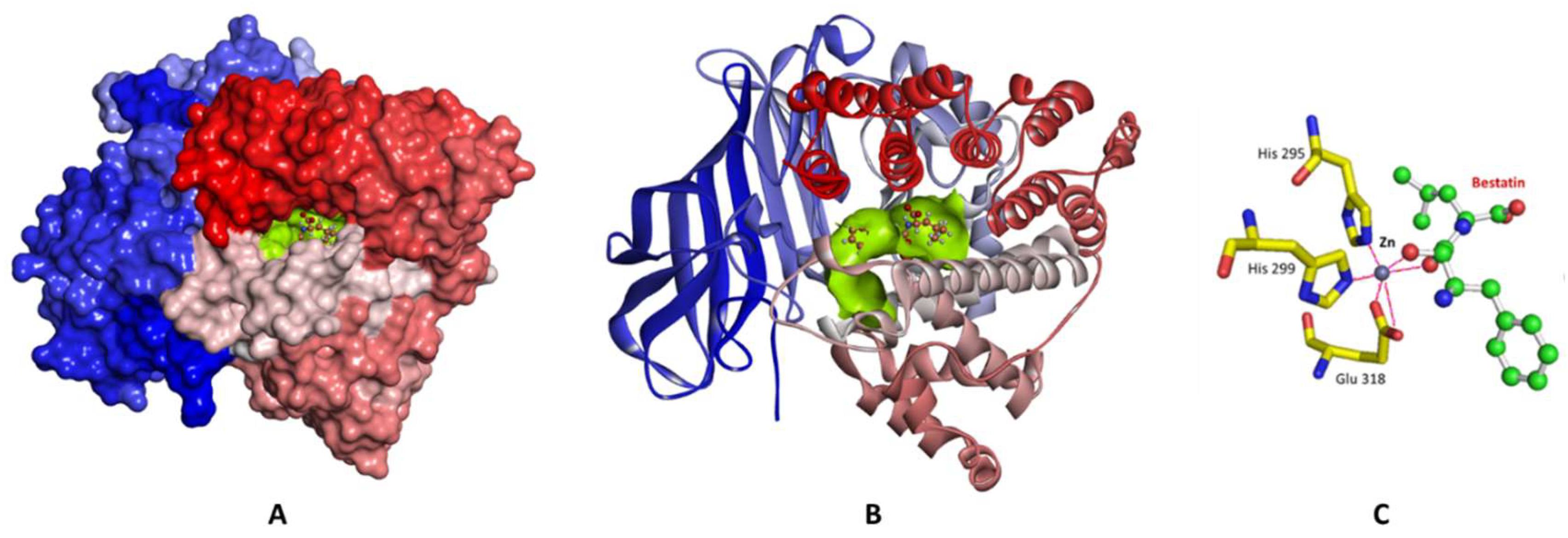
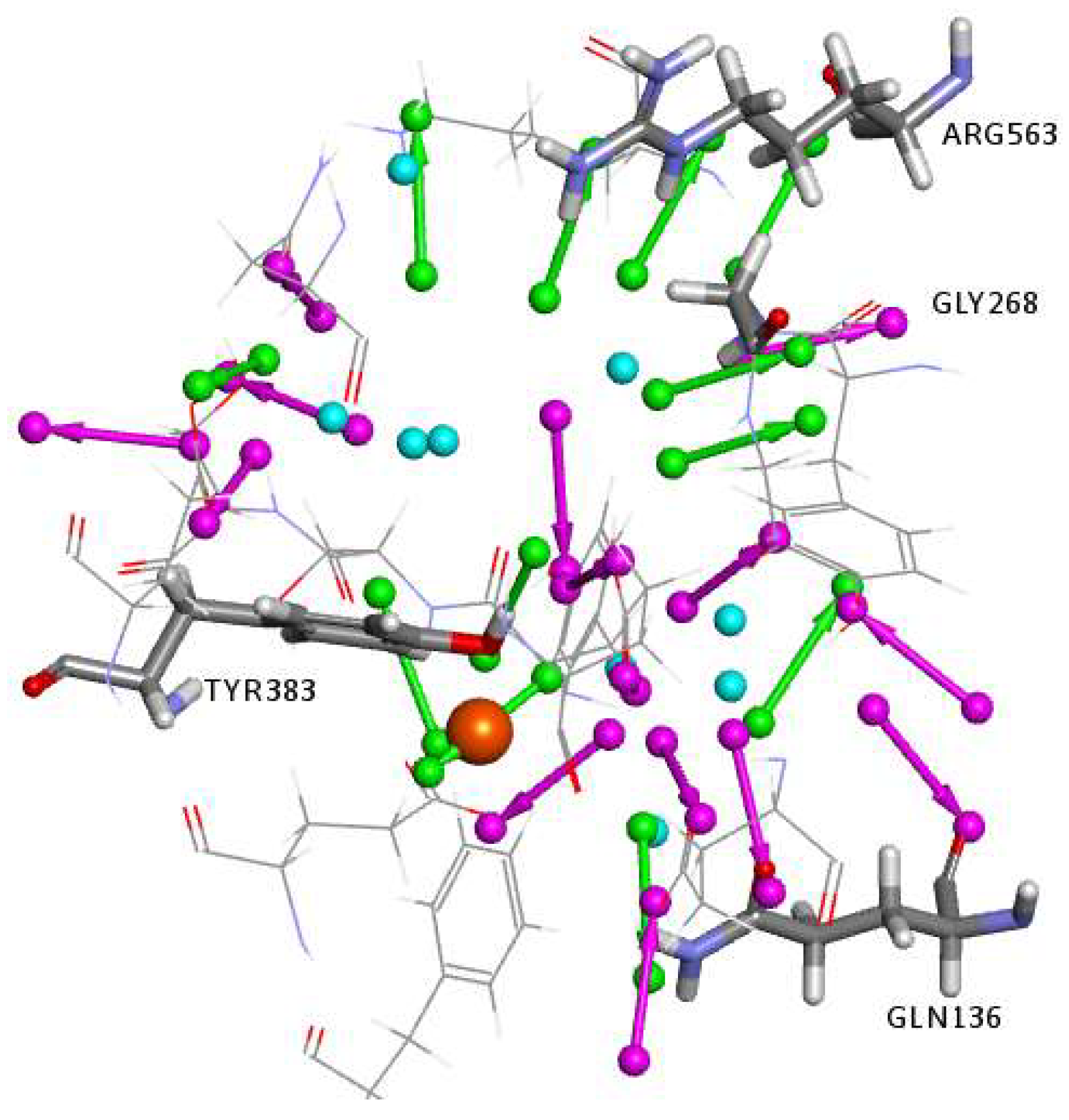
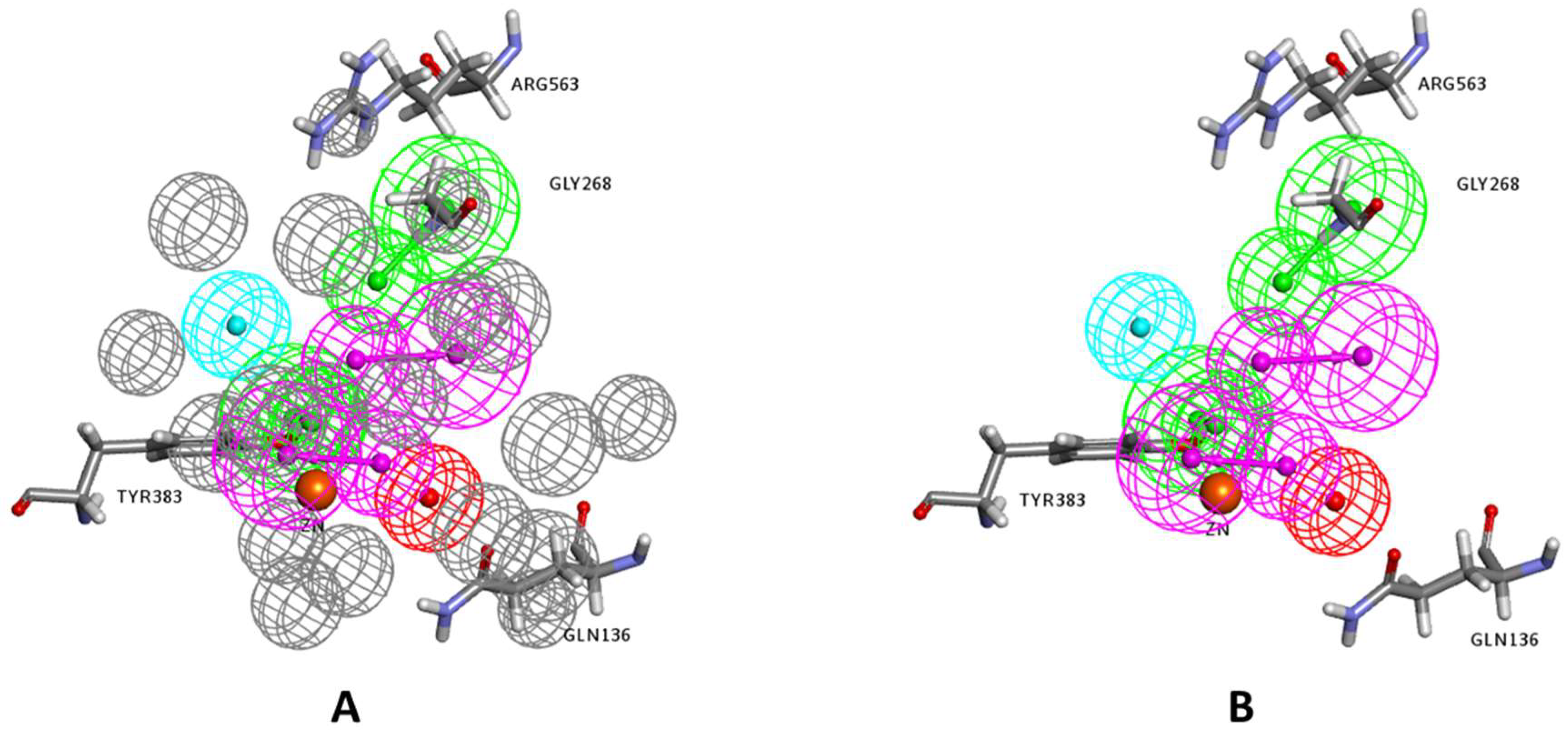
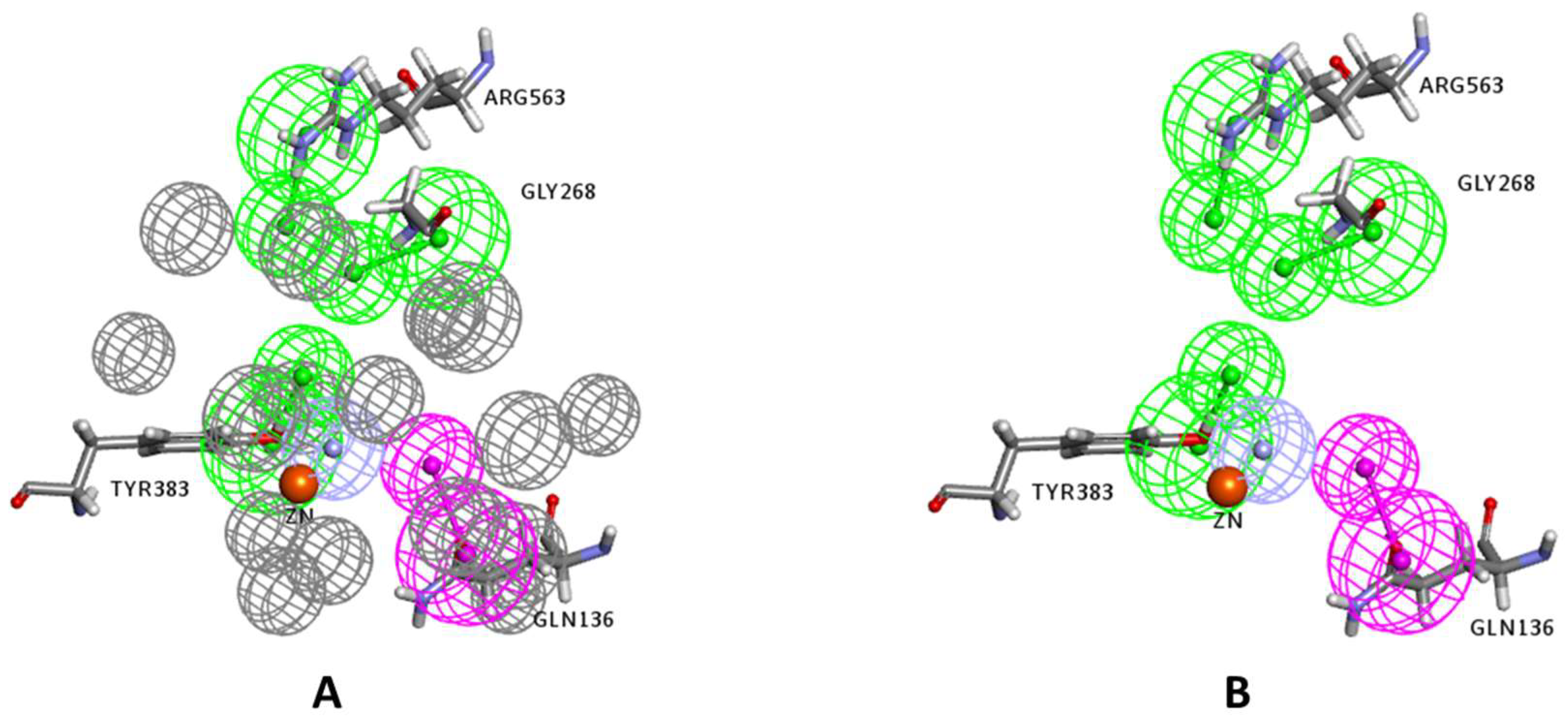
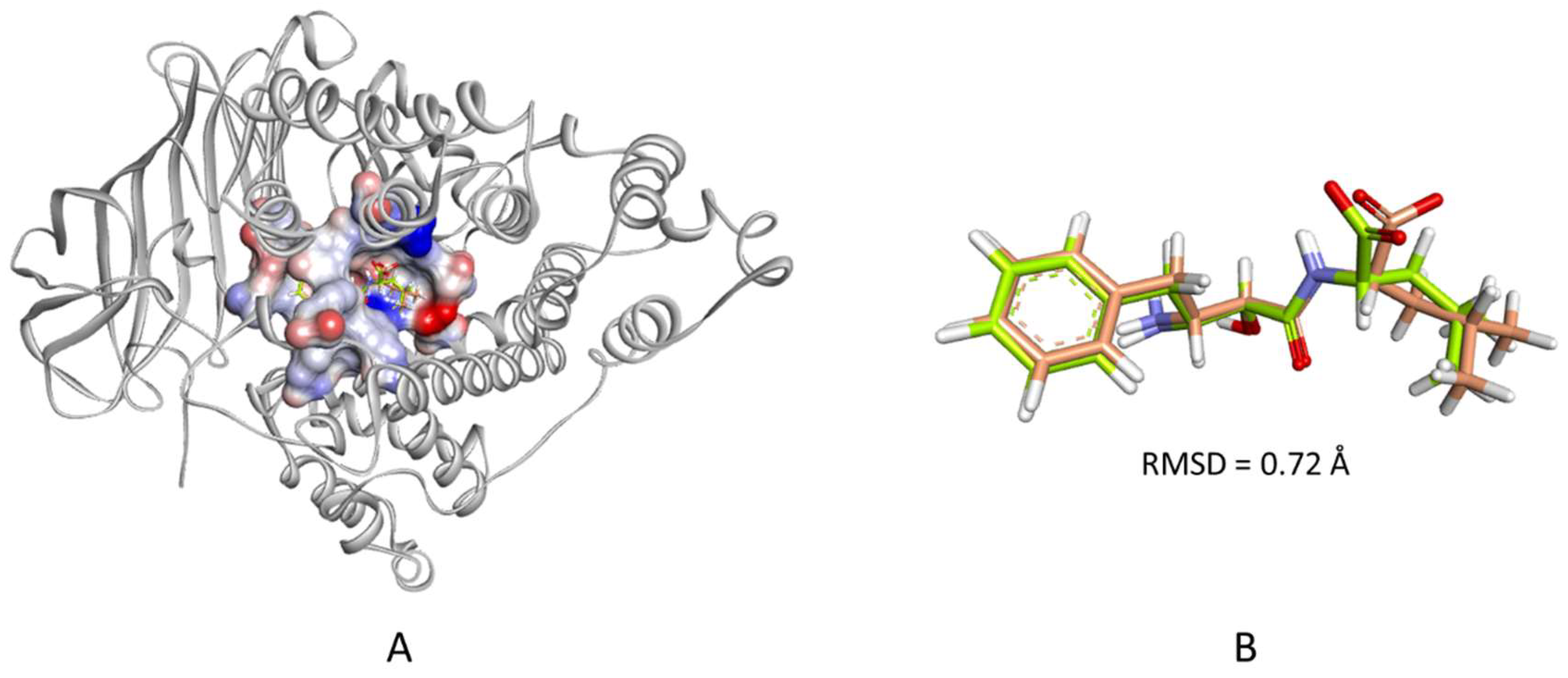
2.1. Structure-Based Pharmacophore Generation
2.2. Virtual Screening of Commercial Databases
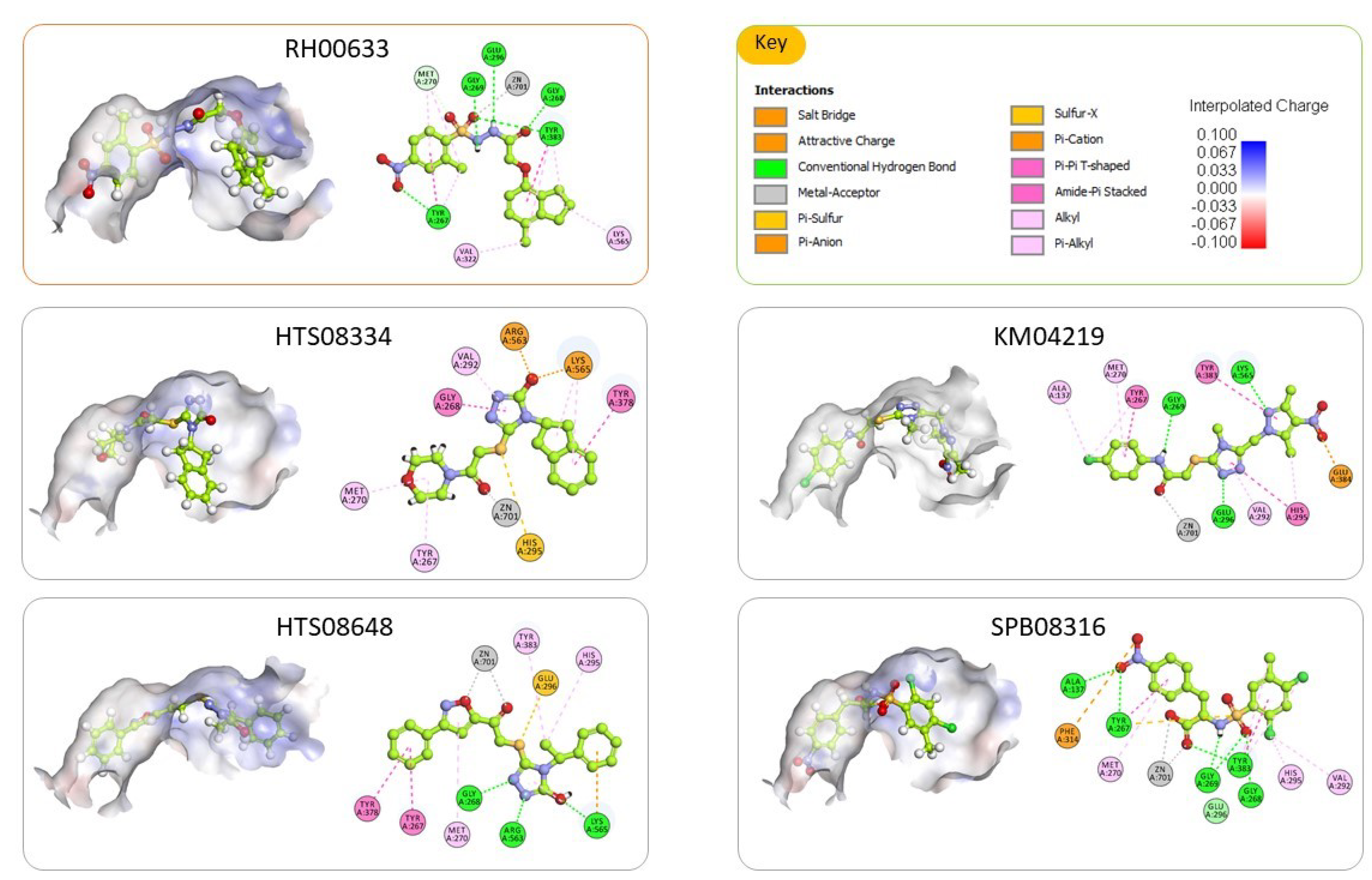
2.3. Molecular Docking and Consensus Scoring
2.4. Inhibition Assay of Hydrolase Activity of LTA4H
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Preparation of the LTA4H Enzyme
3.2.2. Structure-Based Pharmacophore Generation
3.2.3. Virtual Screening of Commercial Databases
3.2.4. Molecular Docking
3.2.5. In Vitro Enzyme Inhibition Assay
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Crooks, S.; Stockley, R. Leukotriene B4. Int. J. Biochem. Cell Boil. 1998, 30, 173–178. [Google Scholar] [CrossRef]
- Samuelsson, B. Leukotrienes: Mediators of immediate hypersensitivity reactions and inflammation. Science 1983, 220, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Ford-Hutchinson, A.W.; Bray, M.A.; Doig, M.V.; Shipley, M.E.; Smith, M.J.H. Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature 1980, 286, 264–265. [Google Scholar] [CrossRef]
- Goodarzi, K.; Goodarzi, M.; Tager, A.M.; Luster, A.D.; Von Andrian, U.H. Leukotriene B4 and BLT1 control cytotoxic effector T cell recruitment to inflamed tissues. Nat. Immunol. 2003, 4, 965–973. [Google Scholar] [CrossRef]
- Lundeen, K.A.; Sun, B.; Karlsson, L.; Fourie, A. Leukotriene B4 receptors BLT1 and BLT2: Expression and function in human and murine mast cells. J. Immunol. 2006, 177, 3439–3447. [Google Scholar] [CrossRef]
- Ott, V.L.; Cambier, J.C.; Kappler, J.; Marrack, P.; Swanson, B.J. Mast cell–dependent migration of effector CD8+ T cells through production of leukotriene B4. Nat. Immunol. 2003, 4, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Jupp, J.; Hillier, K.; Elliott, D.H.; Fine, D.; Bateman, A.C.; Johnson, P.A.; Cazaly, A.M.; Penrose, J.F.; Sampson, A.P. Colonic expression of leukotriene-pathway enzymes in inflammatory bowel diseases. Inflamm. Bowel Dis. 2007, 13, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Rask-Madsen, J. Soluble mediators and the interaction of drugs in IBD. Drugs Today 1998, 34, 45–63. [Google Scholar] [CrossRef]
- Griffiths, R.J.; Pettipher, E.R.; Koch, K.; Farrell, C.A.; Breslow, R.; Conklyn, M.J.; Smith, M.A.; Hackman, B.C.; Wimberly, D.J.; Milici, A.J. Leukotriene B4 plays a critical role in the progression of collagen-induced arthritis. Proc. Natl. Acad. Sci. USA 1995, 92, 517–521. [Google Scholar] [CrossRef]
- Chen, M.; Lam, B.K.; Kanaoka, Y.; Nigrovic, P.A.; Audoly, L.P.; Austen, K.F.; Lee, D.M. Neutrophil-derived leukotriene B4 is required for inflammatory arthritis. J. Exp. Med. 2006, 203, 837–842. [Google Scholar] [CrossRef]
- O’Driscoll, B.R.; Cromwell, O.; Kay, A.B. Sputum leukotrienes in obstructive airways diseases. Clin. Exp. Immunol. 1984, 55, 397–404. [Google Scholar]
- Montuschi, P.; Peters-Golden, M.L. Leukotriene modifiers for asthma treatment. Clin. Exp. Allergy 2010, 40, 1732–1741. [Google Scholar] [CrossRef]
- Rao, N.L.; Riley, J.P.; Banie, H.; Xue, X.; Sun, B.; Crawford, S.; Lundeen, K.A.; Yu, F.; Karlsson, L.; Fourie, A.M.; et al. Leukotriene A4Hydrolase Inhibition Attenuates Allergic Airway Inflammation and Hyperresponsiveness. Am. J. Respir. Crit. Care Med. 2010, 181, 899–907. [Google Scholar] [CrossRef]
- Bortuzzo, C.; Hanif, R.; Kashfi, K.; Staiano-Coico, L.; Shiff, S.J.; Rigas, B. The effect of leukotrienes B and selected HETEs on the proliferation of colon cancer cells. Biochim. Biophys. Acta (BBA) Lipids Lipid Metab. 1996, 1300, 240–246. [Google Scholar] [CrossRef]
- Yang, P.; Sun, Z.; Chan, D.; Cartwright, C.A.; Vijjeswarapu, M.; Ding, J.; Chen, X.; Newman, R.A. Zyflamend® reduces LTB4 formation and prevents oral carcinogenesis in a 7,12-dimethylbenz[α]anthracene (DMBA)-induced hamster cheek pouch model. Carcinogenesis 2008, 29, 2182–2189. [Google Scholar] [CrossRef]
- Hu, N.; Li, Y.; Zhao, Y.; Wang, Q.; You, J.-C.; Zhang, X.; Ye, L.-H. A novel positive feedback loop involving FASN/p-ERK1/2/5-LOX/LTB4/FASN sustains high growth of breast cancer cells. Acta Pharmacol. Sin. 2011, 32, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, B.; Funk, C.D. Enzymes involved in the biosynthesis of leukotriene B4. J. Boil. Chem. 1989, 264, 19469–19472. [Google Scholar]
- Haeggström, J.Z.; Wetterholm, A. Enzymes and receptors in the leukotriene cascade. Cell. Mol. Life Sci. 2002, 59, 742–753. [Google Scholar] [CrossRef] [PubMed]
- Oi, N.; Yamamoto, H.; Langfald, A.; Bai, R.; Lee, M.-H.; Bode, A.M.; Dong, Z. LTA4H regulates cell cycle and skin carcinogenesis. Carcinogenesis 2017, 38, 728–737. [Google Scholar] [CrossRef]
- Chen, X.; Wang, S.; Wu, N.; Yang, C.S. Leukotriene A4 hydrolase as a target for cancer prevention and therapy. Curr. Cancer Drug Targets 2004, 4, 267–283. [Google Scholar] [CrossRef]
- Sun, Z.; Sood, S.; Li, N.; Ramji, D.; Yang, P.; Newman, R.A.; Yang, C.S.; Chen, X. Involvement of the 5-lipoxygenase/leukotriene A4 hydrolase pathway in 7,12-dimethylbenz[a]anthracene (DMBA)-induced oral carcinogenesis in hamster cheek pouch, and inhibition of carcinogenesis by its inhibitors. Carcinogenesis 2006, 27, 1902–1908. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, N.; Wang, S.; Wu, N.; Hong, J.; Jiao, X.; Krasna, M.J.; Beer, D.G.; Yang, C.S. Leukotriene A4 Hydrolase in Rat and Human Esophageal Adenocarcinomas and Inhibitory Effects of Bestatin. J. Natl. Cancer Inst. 2003, 95, 1053–1061. [Google Scholar] [CrossRef]
- Zhao, S.; Yao, K.; Li, D.; Liu, K.; Jin, G.; Yan, M.; Wu, Q.; Chen, H.; Shin, S.H.; Bai, R.; et al. Inhibition of LTA4H by bestatin in human and mouse colorectal cancer. EBioMedicine 2019, 44, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Wang, X.; Zhang, X.; Sun, Z.; Chen, X. Ethanol promotes chemically induced oral cancer in mice through activation of the 5-lipoxygenase pathway of arachidonic acid metabolism. Cancer Prev. Res. 2011, 4, 1863–1872. [Google Scholar] [CrossRef] [PubMed]
- Haeggström, J.Z. Leukotriene A4 hydrolase and the committed step in leukotriene B4 biosynthesis. Clin. Rev. Allergy Immunol. 1999, 17, 111–131. [Google Scholar] [CrossRef]
- Haeggström, J.Z.; Wetterholm, A. Leukotriene A4 hydrolase: A key enzyme in chemotactic leukotriene formation. In Novel Inhibitors of Leukotrienes; Springer Science and Business Media LLC: Basel, Switzerland, 1999; pp. 45–61. [Google Scholar]
- Thunnissen, M.M.; Nordlund, P.; Haeggström, J.Z. Crystal structure of human leukotriene A(4) hydrolase, a bifunctional enzyme in inflammation. Nat. Genet. 2001, 8, 131–135. [Google Scholar] [CrossRef]
- Penning, T.D. Inhibitors of Leukotriene A4 (LTA4) Hydrolase as Potential Anti-Inflammatory Agents. Curr. Pharm. Des. 2001, 7, 163–179. [Google Scholar] [CrossRef]
- Caliskan, B.; Banoglu, E. Overview of recent drug discovery approaches for new generation leukotriene A4 hydrolase inhibitors. Expert Opin. Drug Discov. 2012, 8, 49–63. [Google Scholar] [CrossRef]
- Numao, S.; Hasler, F.; Laguerre, C.; Srinivas, H.; Wack, N.; Jäger, P.; Schmid, A.; Osmont, A.; Röthlisberger, P.; Houguenade, J.; et al. Feasibility and physiological relevance of designing highly potent aminopeptidase-sparing leukotriene A4 hydrolase inhibitors. Sci. Rep. 2017, 7, 13591. [Google Scholar] [CrossRef]
- Bhatt, L.; Roinestad, K.; Van, T.; Springman, E. Recent advances in clinical development of leukotriene B4 pathway drugs. Semin. Immunol. 2017, 33, 65–73. [Google Scholar] [CrossRef]
- Low, C.M.; Akthar, S.; Patel, D.F.; Löser, S.; Wong, C.-T.; Jackson, P.L.; Blalock, J.E.; Hare, S.; Lloyd, C.M.; Snelgrove, R.J. The development of novel LTA4H modulators to selectively target LTB4 generation. Sci. Rep. 2017, 7, 44449. [Google Scholar] [CrossRef] [PubMed]
- El-Naggar, M.H.; Mira, A.; Bar, F.A.; Shimizu, K.; Amer, M.M.; Badria, F.A. Synthesis, docking, cytotoxicity, and LTA 4 H inhibitory activity of new gingerol derivatives as potential colorectal cancer therapy. Bioorganic Med. Chem. 2017, 25, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Appiah-Kubi, P.; Soliman, M.E.S. Hybrid Receptor-Bound/MM-GBSA-Per-residue Energy-Based Pharmacophore Modelling: Enhanced Approach for Identification of Selective LTA4H Inhibitors as Potential Anti-inflammatory Drugs. Cell Biophys. 2016, 75, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Hiesinger, K.; Schott, A.; Kramer, J.S.; Blöcher, R.; Witt, F.; Wittmann, S.K.; Steinhilber, D.; Pogoryelov, D.; Gerstmeier, J.; Werz, O.; et al. Design of Dual Inhibitors of Soluble Epoxide Hydrolase and LTA4 Hydrolase. ACS Med. Chem. Lett. 2019, 11, 298–302. [Google Scholar] [CrossRef]
- Rogers, D.; Hopfinger, A.J. Application of Genetic Function Approximation to Quantitative Structure-Activity Relationships and Quantitative Structure-Property Relationships. J. Chem. Inf. Model. 1994, 34, 854–866. [Google Scholar] [CrossRef]
- Al-Shar’I, N.A.; Hassan, M.; Al-Balas, Q.; Almaaytah, A. Identification of Possible Glyoxalase II Inhibitors as Anticancer Agents by a Customized 3D Structure-Based Pharmacophore Model. Jordan J. Pharm. Sci. 2015, 8, 83–103. [Google Scholar]
- Al-Shar’I, N.; Al-Balas, Q.A.; Al-Waqfi, R.A.; Hassan, M.A.; Alkhalifa, A.E.; Ayoub, N.M. Discovery of a nanomolar inhibitor of the human glyoxalase-I enzyme using structure-based poly-pharmacophore modelling and molecular docking. J. Comput. Mol. Des. 2019, 33, 799–815. [Google Scholar] [CrossRef]
- Kawai, K.; Nagata, N. Metal–ligand interactions: An analysis of zinc binding groups using the Protein Data Bank. Eur. J. Med. Chem. 2012, 51, 271–276. [Google Scholar] [CrossRef]
- Huang, S.-Y.; Grinter, S.Z.; Zou, X. Scoring functions and their evaluation methods for protein–ligand docking: Recent advances and future directions. Phys. Chem. Chem. Phys. 2010, 12, 12899. [Google Scholar] [CrossRef]
- Wang, R.; Wang, S. How does consensus scoring work for virtual library screening? An idealized computer experiment. J. Chem. Inf. Comput. Sci. 2001, 41, 1422–1426. [Google Scholar] [CrossRef]
- Oda, A.; Tsuchida, K.; Takakura, T.; Yamaotsu, N.; Hirono, S. Comparison of Consensus Scoring Strategies for Evaluating Computational Models of Protein−Ligand Complexes. J. Chem. Inf. Model. 2006, 46, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Batool, M.; Ahmad, B.; Choi, S. A Structure-Based Drug Discovery Paradigm. Int. J. Mol. Sci. 2019, 20, 2783. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, R. Classification of Current Scoring Functions. J. Chem. Inf. Model. 2015, 55, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Discovery Studio; Accelrys Software Inc.: San Diego, CA, USA, 2013.
- Wu, G.; Robertson, D.H.; Brooks, C.L.; Vieth, M. Detailed analysis of grid-based molecular docking: A case study of CDOCKER—A CHARMm-based MD docking algorithm. J. Comput. Chem. 2003, 24, 1549–1562. [Google Scholar] [CrossRef] [PubMed]
- Böhm, H.-J. LUDI: Rule-based automatic design of new substituents for enzyme inhibitor leads. J. Comput. Mol. Des. 1992, 6, 593–606. [Google Scholar] [CrossRef]
- Böhm, H.-J. The computer program LUDI: A new method for the de novo design of enzyme inhibitors. J. Comput. Mol. Des. 1992, 6, 61–78. [Google Scholar] [CrossRef]
- Wei, D.; Jiang, X.; Zhou, L.; Chen, J.; Chen, Z.; He, C.; Yang, K.; Liu, Y.; Pei, J.; Lai, L. Discovery of Multitarget Inhibitors by Combining Molecular Docking with Common Pharmacophore Matching. J. Med. Chem. 2008, 51, 7882–7888. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available at Maybridge Chemical Holdings Ltd., UK. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pharmacophore Summary | |||
|---|---|---|---|
| Pharmacophore | Number of Features | Feature Set | Selectivity Score |
| Pharmacophore_01 | 6 | AADDHP | 12.549 |
| Pharmacophore_02 | 5 | AADDP | 11.034 |
| Pharmacophore_03 | 5 | ADDHP | 11.034 |
| Pharmacophore_04 | 5 | ADDHP | 11.034 |
| Pharmacophore_05 | 5 | AADHP | 10.120 |
| Pharmacophore_06 | 5 | AADHP | 10.120 |
| Pharmacophore_07 | 5 | AADDH | 9.640 |
| Pharmacophore_08 | 4 | ADDP | 9.519 |
| Pharmacophore_09 | 4 | ADDP | 9.519 |
| Pharmacophore_10 | 4 | DDHP | 9.519 |
| Code | Structure | Fit Value * | Consensus Score |
|---|---|---|---|
| KM04219 |  | 3.542 | 3 |
| SPB08316 |  | 3.084 | 3 |
| RH00633 |  | 2.432 | 3 |
| HTS08334 |  | 2.144 | 3 |
| HTS08648 |  | 2.052 | 3 |
| Compound Code | % of LTA4H Inhibition (at 25 μM) a |
|---|---|
| KM04219 | 44.5 |
| SPB08316 | 51.9 ± 0.84 |
| RH00633 | 73.6 ± 0.25 |
| HTS08334 | 45 ± 17.1 |
| HTS08648 | 8 ± 14.6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Audat, S.A.; Al-Shar’i, N.A.; Al-Oudat, B.A.; Bryant-Friedrich, A.; Bedi, M.F.; Zayed, A.L.; Al-Balas, Q.A. Identification of Human Leukotriene A4 Hydrolase Inhibitors Using Structure-Based Pharmacophore Modeling and Molecular Docking. Molecules 2020, 25, 2871. https://doi.org/10.3390/molecules25122871
Audat SA, Al-Shar’i NA, Al-Oudat BA, Bryant-Friedrich A, Bedi MF, Zayed AL, Al-Balas QA. Identification of Human Leukotriene A4 Hydrolase Inhibitors Using Structure-Based Pharmacophore Modeling and Molecular Docking. Molecules. 2020; 25(12):2871. https://doi.org/10.3390/molecules25122871
Chicago/Turabian StyleAudat, Suaad A., Nizar A. Al-Shar’i, Buthina A. Al-Oudat, Amanda Bryant-Friedrich, Mel F. Bedi, Aref L. Zayed, and Qosay A. Al-Balas. 2020. "Identification of Human Leukotriene A4 Hydrolase Inhibitors Using Structure-Based Pharmacophore Modeling and Molecular Docking" Molecules 25, no. 12: 2871. https://doi.org/10.3390/molecules25122871
APA StyleAudat, S. A., Al-Shar’i, N. A., Al-Oudat, B. A., Bryant-Friedrich, A., Bedi, M. F., Zayed, A. L., & Al-Balas, Q. A. (2020). Identification of Human Leukotriene A4 Hydrolase Inhibitors Using Structure-Based Pharmacophore Modeling and Molecular Docking. Molecules, 25(12), 2871. https://doi.org/10.3390/molecules25122871