3.1. Chemistry
3.1.1. General Information
Commercially available chemicals were of reagent grade and used as received. The reactions were monitored by thin layer chromatography (TLC), using silica gel plates (Kieselgel 60F
254, E. Merck, Darmstadt, Germany). Column chromatography was performed on silica gel 60 M (0.040–0.063 mm, E. Merck, Darmstadt, Germany). Melting points are uncorrected and were measured on a Büchi (New Castle, DE, USA) Melting Point B-540 apparatus. All
1H and
13C NMR spectra were registered with a Bruker Avance III (Billerica, MA, USA) spectrometer operating at 500.13 and 125.77 MHz for
1H and
13C, respectively, and equipped with a 5 mm probe head with Z-gradient coils. The experiments were performed using pulse programs from the standard Bruker library for samples dissolved in DMSO-
d6, methanol-
d4 or trifluoroacetic acid-
d, depending on the sample. In each case spectra were calibrated at residual solvent resonances. For the temperature
1H NMR spectra a BCU unit was used for temperature control and stabilization. After reaching the required temperature measured samples were equilibrated at this temperature for at least 10 min prior to measurement. Solvent signals in NMR spectra were determined on the basis of literature data [
26]. High resolution mass spectra were performed by the Laboratory of Mass Spectrometry, Institute of Biochemistry and Biophysics PAS, on a LTQ Orbitrap Velos instrument, Thermo Scientific (Waltham, MA, USA). IR spectra were recorded with the Jasco 6200 (Easton, MD, USA) FT/IR spectrometer in the Laboratory of Optical Spectroscopy, Institute of Organic Chemistry PAS (Warsaw, Poland).
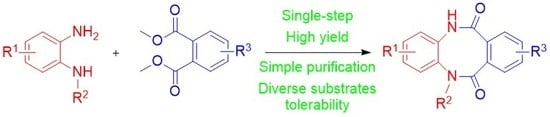
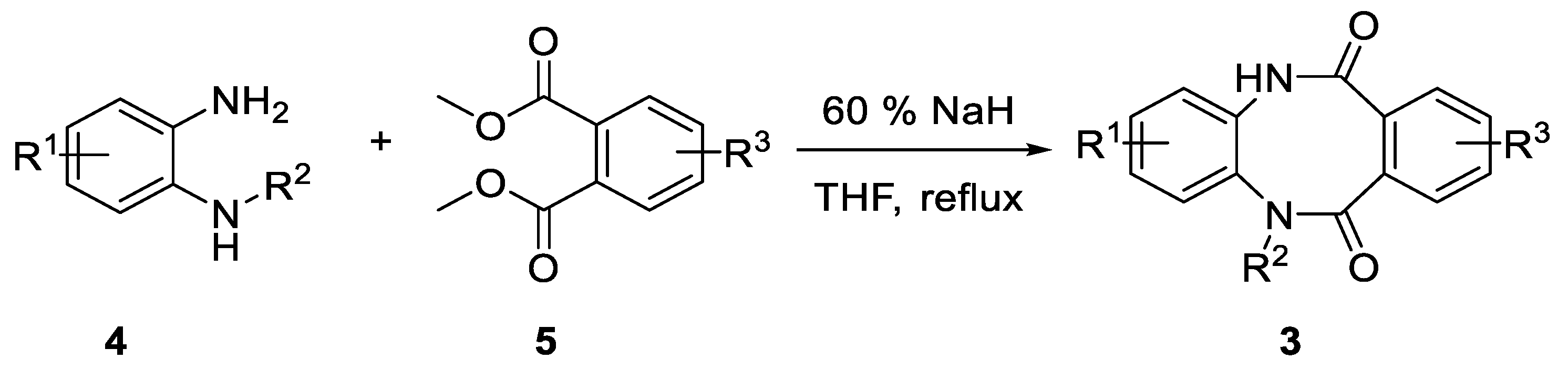
3.1.2. General Procedure for Synthesis 5,12-dihydrodibenzo[b,f][1,4]diazocine-6,11-diones (3a-j)
To a solution of diamine 4a–f (1 equiv.) and diester 5a–e (1 equiv.) in anhydrous THF (5 mL/mmol), 60% sodium hydride in mineral oil (2 equiv.) was added and the resulting mixture was refluxed for 18 h. The reaction mixture was poured into water, acidified with concentrated HCl to pH 2 and extracted with ethyl acetate. The organic phase was washed with brine and dried over anhydrous magnesium sulfate. The drying agent was filtered off and the resulting filtrate was evaporated under reduced pressure. The crude product was purified either by crystallization or by column chromatography.
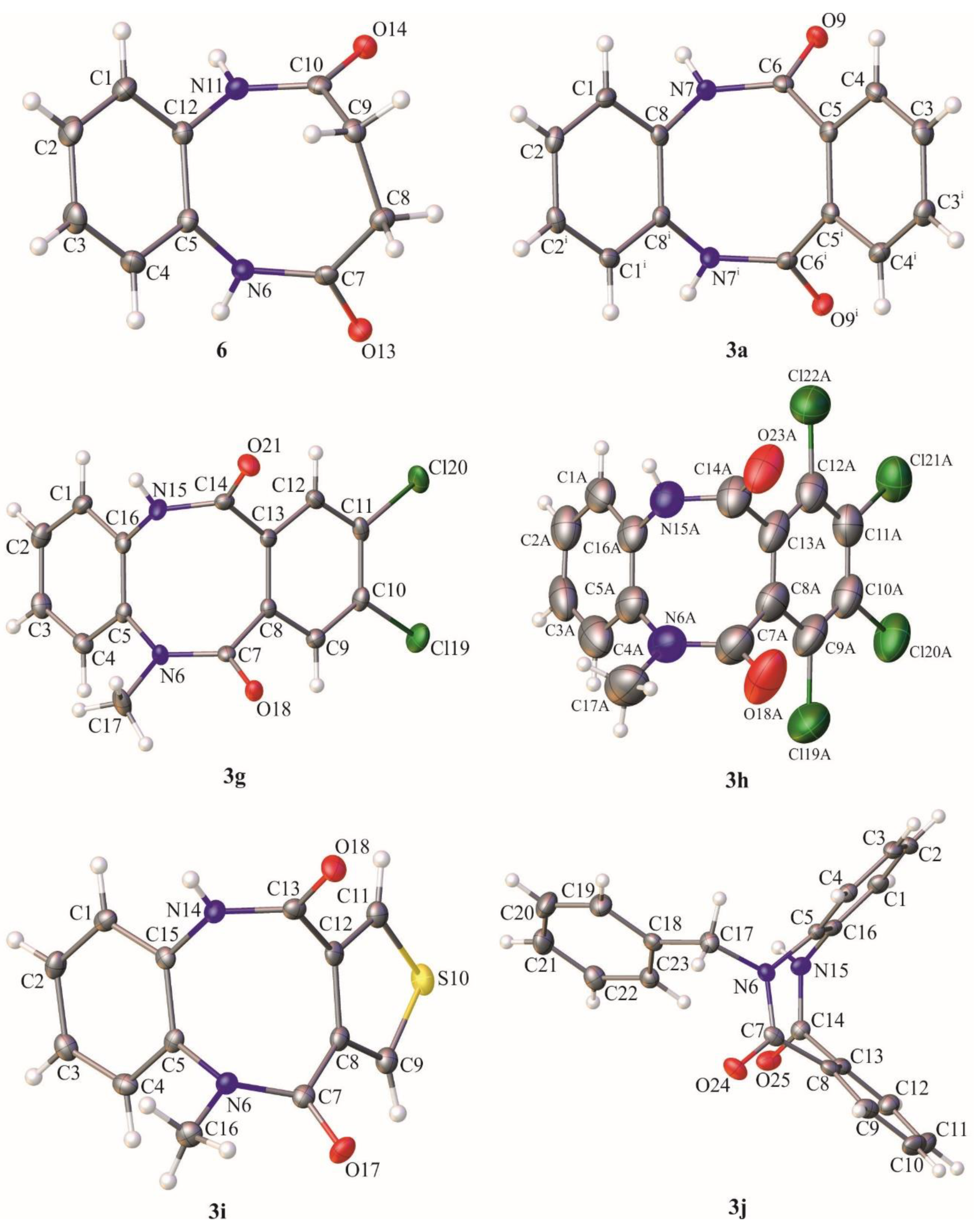
5,12-dihydrodibenzo[b,f][1,4]diazocine-6,11-dione (3a), crystallized from DMF, yield 78%, white crystals, m.p. 286.0–287.0 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.18 (s, 2H, 2 × NH), 7.65–6.85 (m, 8H, HAr); 13C NMR (125 MHz, DMSO-d6) δ 170.4, 134.8, 132.3, 130.2, 127.5, 127.0, 126.6; IR (KBr): cm−1 3178, 3049, 2938, 2901, 2874, 1936, 1825, 1651, 1593, 1573, 1502, 1461, 1403, 1304, 1265, 1239, 1140, 1115, 1043; HRMS (ESI): m/z [M + H]+ calcd for C14H11N2O2: 239.08150, found: 239.08174.
2-nitro-5,12-dihydrodibenzo[b,f][1,4]diazocine-6,11-dione (3b), crystallized from ethyl acetate, yield 53%, yellow crystals, m.p. 230.0 °C (decomposition), R.f. = 0.38 (hexane:ethyl acetate 2:8 v/v). 1H NMR (500 MHz, DMSO-d6) δ 10.66 (s, 1H, NH), 10.44 (s, 1H, NH), 8.06 (dd, 1H, J = 2.0 Hz, J = 8.0 Hz, HAr), 7.99 (d, 1H, J = 2.0 Hz, HAr), 7.51–7.30 (m, 5H, HAr); 13C NMR (125 MHz, DMSO-d6) δ 170.2, 170.0, 145.5, 140.9, 135.2, 131.6, 131.4, 130.72, 130.66, 127.6, 126.9, 126.8, 122.6, 122.1; IR (KBr): cm−1 3169, 3077, 3035, 2924, 2862, 1678, 1654, 1593, 1573, 1533, 1501, 1393, 1349, 1238, 1141, 1064; HRMS (ESI): m/z [M + H]+ calcd for C14H10N3O4: 284.06658, found: 284.06699.
2-benzoyl-5,12-dihydrodibenzo[b,f][
1,
4]
diazocine-6,11-dione (
3c), crystallized from ethyl acetate, yield 55%, beige crystals, m.p. 156.0–157.0 °C, R.f. = 0.24 (hexane:ethyl acetate 2:8
v/
v).
1H NMR (500 MHz, DMSO-
d6) δ 10.52 (s, 1H, NH), 10.32 (s, 1H, NH), 7.71–7.63 (m, 3H, H
Ar), 7.59–7.50 (m, 4H, H
Ar), 7.49–7.43 (m, 2H, H
Ar), 7.38–7.30 (m, 3H, H
Ar);
13C NMR (125 MHz, DMSO-
d6) δ 194.0, 170.3, 170.2, 138.6, 136.5, 135.5, 134.7, 132.9, 131.9, 131.8, 130.51,130.49, 129.5 (2 × C), 128.9, 128.6 (2 × C), 128.2, 126.9, 126.80, 120.77; IR (KBr): cm
−1 3171, 3060, 2931, 2873, 1734, 1685, 1659, 1601, 1574, 1492, 1446, 1392, 1314, 1278, 1263, 1239, 1179, 1141, 1125, 1078, 1043; HRMS (ESI):
m/z [M + H]
+ calcd for C
21H
15N
2O
3: 343.10772, found: 343.10731.
8-nitro-5,12-dihydrodibenzo[b,f][1,4]diazocine-6,11-dione (3d), crystallized from ethyl acetate, yield 50%, yellow crystals, m.p. 243.0–244.0 °C, R.f. = 0.40 (hexane:ethyl acetate 2:8 v/v). 1H NMR (500 MHz, DMSO-d6) δ 10.521 (s, 1H, NH), 10.492 (s, 1H, NH), 8.22 (dd, 1H, J = 2.5 Hz, J = 8.5 Hz, HAr), 8.11 (d, 1H, J = 2.5 Hz, HAr), 7.63 (d, 1H, J = 8.5 Hz, HAr), 7.26–7.15 (m, 4H, HAr); 13C NMR (125 MHz, DMSO-d6) δ 168.5, 168.2, 148.3, 137.7, 134.4, 134.3, 133.4, 128.7, 128.01, 127.98, 127.4, 127.2, 125.2, 121.9; IR (KBr): cm−1 3245, 3078, 1674, 1645, 1612, 1582, 1527, 1503, 1487, 1446, 1396, 1346, 1301, 1236, 1149, 1065; HRMS (ESI): m/z [M + H]+ calcd for C14H10N3O4: 284.06658, found: 286.06693.
5-methyl-5,12-dihydrodibenzo[b,f][1,4]diazocine-6,11-dione (3e) crystallized from ethyl acetate, yield 61%, white crystals, m.p. 206.0–207.0 °C, R.f. = 0.36 (hexane:ethyl acetate 2:8 v/v). 1H NMR (500 MHz, DMSO-d6) δ 10.21 (s, 1H, NH), 7.47–7.41 (m, 1H, HAr), 7.40–7.34 (m, 2H, HAr), 7.31–7.19 (m, 4H, HAr), 7.19–7.12 (m, 1H, HAr), 3.30 (s, 3H, Me); 13C NMR (125 MHz, DMSO-d6) δ 170.3, 168.6, 140.4, 135.1, 133.3, 131.7, 130.2, 130.1, 128.3, 128.2, 127.2, 127.1, 126.4, 126.2, 36.6; IR (KBr): cm−1 3213, 3063, 2927, 2883, 1965, 1935, 1828, 1675, 1626, 1594, 1571, 1502, 1488, 1420, 1392, 1346, 1309, 1256, 1236, 1191, 1162, 1140, 1080, 1036, 1007; HRMS (ESI): m/z [M + H]+ calcd for C15H13N2O2: 253.09715, found: 253.09709.
2,3-dichloro-5-methyl-5,12-dihydrodibenzo[b,f][1,4]diazocine-6,11-dione (3f) crystallized from THF, yield 55%, white crystals, m.p. 198.0–199.0 °C, R.f. = 0.57 (hexane:ethyl acetate 2:8 v/v). 1H NMR (500 MHz, DMSO-d6) δ 10.35 (s, 1H, NH), 7.91 (s, 1H, HAr); 7.49 (s, 1H, HAr); 7.47–7.40 (m, 2H, HAr), 7.32–7.25 (m, 2H, HAr), 3.30 (s, 3H, Me); 13C NMR (125 MHz, DMSO-d6) δ 170.0, 168.3, 140.3, 135.4, 132.7, 131.1, 130.6, 130.4, 130.3, 130.0, 129.1, 128.3, 126.6, 126.4, 36.6; IR (KBr): cm−1 3177, 3095, 3039, 2894, 1680, 1633, 1592, 1567, 1485, 1427, 1398, 1357, 1305, 1262, 1226, 1138, 1110, 1063, 1019; HRMS (ESI): m/z [M + H]+ calcd for C15H11N2O2Cl2: 321.01921, 323.01626, found: 321.01957, 323.01658.
8,9-dichloro-5-methyl-5,12-dihydrodibenzo[b,f][1,4]diazocine-6,11-dione (3g) crystallized from ethyl acetate, yield 67%, white crystals, m.p. 207.0–208.0 °C, R.f. = 0.53 (hexane:ethyl acetate 2:8 v/v). 1H NMR (500 MHz, DMSO-d6) δ 10.43 (s, 1H, NH), 7.59 (s, 2H, HAr), 7.46 (d, 1H, J = 7.0 Hz, HAr), 7.35–7.24 (m, 2H, HAr), 7.19 (d, 1H, J = 7.0 Hz, HAr), 3.30 (s, 3H, Me); 13C NMR (125 MHz, DMSO-d6) δ 167.8, 166.2, 139.9, 134.6, 133.5, 133.13, 133.05, 132.1, 128.8, 128.63, 128.59, 128.4, 127.43, 127.36, 36.7; IR (KBr): cm−1 3213, 3087, 2931, 1671, 1640, 1593, 1546, 1501, 1424, 1396, 1375, 1316, 1256, 1234, 1130, 1092, 1020; HRMS (ESI): m/z [M + H]+ calcd for C15H11N2O2Cl2: 321.01921, 323.01626, found: 321.01957, 323.01664.
7,8,9,10-tetrachloro-5-methyl-5,12-dihydrodibenzo[b,f][1,4]diazocine-6,11-dione (3h) column chromatography methanol:chloroform 1:9 v/v, yield 15%, white crystals, m.p. 233.0 °C (decomposition), R.f. = 0.67 (hexane:ethyl acetate 2:8 v/v). 1H NMR (500 MHz, DMSO-d6) δ 10.78 (s, 1H, NH), 7.50 (d, 1H, J = 7.0 Hz, HAr), 7.37–7.26 (m, 2H, HAr), 7.23 (d, 1H, J = 7.0 Hz, HAr), 3.31 (s, 3H, Me); 13C NMR (125 MHz, DMSO-d6) δ 164.5, 163.3, 140.1, 134.7, 134.2, 134.1, 133.1, 131.7, 130.0, 129.9, 128.4, 128.2, 127.9, 127.8, 36.7; IR (KBr): cm−1 3347, 3209, 3107, 2936, 2889, 2857, 1678, 1652, 1596, 1537, 1497, 1433, 1401, 1353, 1299, 1264, 1232, 1149, 1098, 1038; HRMS (ESI): m/z [M + H]+ calcd for C15H9N2O2Cl4: 388.94126, 390.93831, 392.93536, found: 388.94181, 390.93890, 392.93563.
5-methyl-5,10-dihydrobenzo[b]thieno[3,4-f][1,4]diazocine-4,11-dione (3i) colum chromatography hexane:ethyl acetate 2:8 v/v, yield 47%, white crystals, m.p. 241.0–242.0 °C, R.f. = 0.32 (hexane:ethyl acetate 2:8 v/v). 1H NMR (500 MHz, DMSO-d6) δ 10.78 (s, 1H, NH), 7.76–7.40 (m, 2H, HAr), 7.47–7.40 (m, 1H, HAr), 7.32–7.23 (m, 2H, HAr), 7.20–7.14 (m, 1H, HAr), 3.27 (s, 3H, Me); 13C NMR (125 MHz, DMSO-d6) δ 166.9, 165.2, 140.0, 135.1, 134.7, 134.2, 128.4, 128.3, 128.2, 127.8, 127.52, 127.48; IR (KBr): cm−1 3176, 3108, 3055, 2904, 1735, 1645, 1587, 1503, 1469, 1366, 1301, 1283, 1243, 1160, 1105, 1044; HRMS (ESI): m/z [M + H]+ calcd for C13H11N2O2S: 259.05357, found: 259.05396.
5-benzyl-5,12-dihydrodibenzo[b,f][1,4]diazocine-6,11-dione (3j) crystallized from ethyl acetate, yield 74%, white crystals, m.p. 205.0–206.0 °C, R.f. = 0.62 (hexane:ethyl acetate 2:8 v/v). 1H NMR (500 MHz, DMSO-d6) δ 10.09 (s, 1H, NH), 7.44–7.37 (m, 2H, HAr), 7.36–7.21 (m, 8H, HAr), 7.19–7.7.11 (m, 2H, HAr), 7.09–7.02 (m, 1H, HAr), 5.30 (d, 1H, J = 15.0 Hz, CH2), 4.81 (d, 1H, J = 15.0 Hz, CH2); 13C NMR (125 MHz, DMSO-d6) δ 170.2, 168.9, 138.9, 136.7, 135.7, 133.2, 131.6, 130.3, 130.1, 128.4 (2 × C), 128.3, 127.8 (3 × C), 127.3, 127.2, 127.0, 126.5, 126.2, 52.4; IR (KBr): cm−1 3160, 3038, 2900, 2869, 1655, 1596, 1572, 1501, 1487, 1430, 1401, 1384, 1360, 1321, 1270, 1224, 1164, 1141, 1121, 1089, 1046, 1025; HRMS (ESI): m/z [M + H]+ calcd for C21H17N2O2: 329.12845, found: 329.12877.
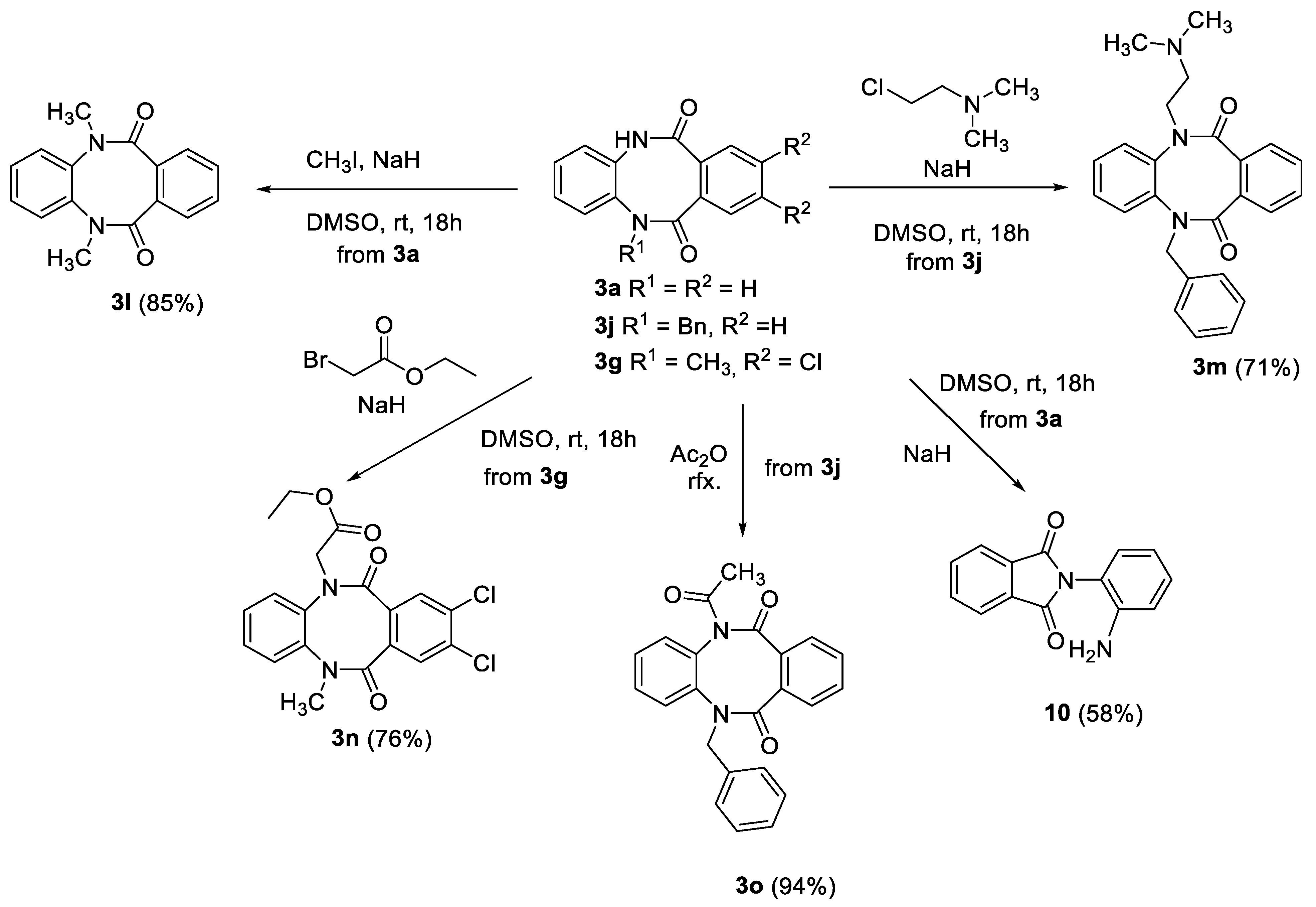
3.1.3. 5,12-dimethyl-5,12-dihydrodibenzo[b,f][1,4]diazocine-6,11-dione (3l)
To a solution of 5,12-dihydrodibenzo[b,f][1,4]diazocine-6,11-dione (3a) (2.0 g, 8.4 mmol, 1.0 equiv.) in anhydrous DMSO (20 mL), 60% sodium hydride in mineral oil (0.74 g, 18.4 mmol, 2.2 equiv.) was added and resulting mixture was stirred for 30 min. After the addition of methyl iodide (1.2 mL, 18.4 mmol 2.2 equiv.), the reaction mixture was stirred for 18 h at room temperature, then was poured into water and the crude product was extracted with ethyl acetate (3 × 50 mL). The organic phase was washed with brine (1 × 50 mL) and dried over anhydrous magnesium sulfate. The crude product was crystallized from ethyl acetate giving 3l as colorless crystals (1.9 g, yield 85%). M.p. 265.0–266.0 °C, R.f. = 0.42 (hexane:ethyl acetate 2:8 v/v). 1H NMR (500 MHz, DMSO-d6) δ 7.48–7.41 (m, 1H, HAr), 7.38–7.32 (m, 1H, HAr), 7.31–7.25 (m, 1H, HAr), 7.24–7.18 (m, 1H, HAr), 3.35 (s, 6H, 2 × Me, overlapped with H2O); 13C NMR (125 MHz, DMSO-d6) δ 168.4, 140.4, 132.8, 130.0, 128.9, 127.2, 126.1, 36.2; IR (KBr): cm−1 3278, 3065, 3045, 2937, 1651, 1593, 1571, 1502, 1486, 1454, 1420, 1388, 1310, 1277, 1162, 1119, 1034, 1003; HRMS (ESI): m/z [M + H]+ calcd for C16H15N2O2: 267.11280, found: 267.11304.
3.1.4. 5-benzyl-12-(2-(dimethylamino)ethyl)-5,12-dihydrodibenzo[b,f][1,4]diazocine-6,11-dione (3m)
To a solution of 5-benzyl-5,12-dihydrodibenzo[b,f][1,4]diazocine-6,11-dione (3j) (1.5 g, 4.6 mmol, 1.0 equiv.) in anhydrous DMSO (30 mL), 60% sodium hydride in mineral oil (0.45 g, 11.4 mmol, 2.5 equiv.) was added, and the resulting mixture was stirred for 30 min. After the addition of 2-chloro-N,N-dimethylethan-1-amine hydrochloride (0.82 g, 5.7 mmol, 1.2 equiv.) the reaction mixture was stirred for 18 h at room temperature, then was poured into water and the crude product was extracted with ethyl acetate (3 × 50 mL). The organic phase was washed with brine (1 × 50 mL) and dried over anhydrous magnesium sulfate. The crude product was purified by column chromatography (triethylamine:ethyl acetate 5:95 v/v) giving 3m as white crystals (1.29 g, yield 71%). M.p. 154.0–155.0 °C, R.f. = 0.38 (triethylamine:ethyl acetate 5:95 v/v). 1H NMR (500 MHz, DMSO-d6) δ 7.40–7.14 (m, 13H, HAr), 5.30 (d, 1H, J = 15.0 Hz, CH2),4.80 (d, 1H, J = 15.0 Hz, CH2), 3.47 (dt, 1H, J = 7.0 Hz, J = 14.0 Hz, CH2), 2.71 (dt, 1H, J = 7.0 Hz, J = 14.0 Hz, CH2), 2.34 (t, 2H, J = 7.0 Hz, CH2), 2.01 (s, 6H, 2 × Me); 13C NMR (125 MHz, DMSO-d6) δ 168.5, 168.3, 140.2, 139.3, 137.2, 133.0, 132.4, 130.1, 130.0, 129.0, 128.8, 128.7 (2 × C), 128.6 (2 × C), 127.7, 127.5 (2 × C), 126.0, 125.9, 56.5, 51.7, 46.4, 45.1; IR (KBr): cm−1 3276, 3061, 2982, 2952, 2858, 2823, 2798, 2775, 1981, 1653, 1583, 1572, 1498, 1486, 1458, 1440, 1401, 1380, 1364, 1330, 1319, 1239, 1218, 1195, 1154, 1120, 1080, 1060, 1042, 1025; HRMS (ESI): m/z [M + H]+ calcd for C25H26N3O2: 400.20195, found: 400.20245.
3.1.5. Ethyl 2-(8,9-dichloro-12-methyl-6,11-dioxo-11,12-dihydrodibenzo[b,f] [1,4]diazocin-5(6H)-yl)acetate (3n)
To a solution of 8,9-dichloro-5-methyl-5,12-dihydrodibenzo[b,f][1,4]diazocine-6,11-dione (3g) (2.0 g, 6.2 mmol, 1.0 equiv.) in anhydrous DMSO (20 mL), 60% sodium hydride in mineral oil (0.37 g, 9.4 mmol, 1.5 equiv.) was added and the resulting mixture was stirred for 30 min. After the addition of ethyl bromoacetate (1.0 mL, 9.4 mmol, 1.5 equiv.) the reaction mixture was stirred for 18 h at room temperature, then was poured into water and the crude product was extracted with ethyl acetate (3 × 50 mL). The organic phase was washed with brine (1 × 50 mL) and dried over anhydrous magnesium sulfate. The crude product was purified by column chromatography (hexane:ethyl acetate 7:3 v/v) giving 3n as white crystals (1.93 g, yield 76%). M.p. 114.0–115.0 °C, R.f. = 0.32 (hexane:ethyl acetate 6:4 v/v). 1H NMR (500 MHz, DMSO-d6) δ 7.58 (s, 1H, HAr), 7.52–7.42 (m, 3H, HAr), 7.37–7.29 (m, 2H, HAr), 4.78 (d, 1H, J = 17.0 Hz, CH2), 4.49 (d, 1H, J = 17.0 Hz, CH2), 4.15 (q, 2H, J = 7.0 Hz, CH2), 3.36 (s, 3H, Me, partially overlapped with H2O signal), 1.20 (t, 3H, J = 7.0 Hz, Me); 13C NMR (125 MHz, DMSO-d6) δ 168.1, 166.5, 165.7, 140.2, 138.9, 133.3, 133.0, 132.1, 129.4, 129.3, 128.5, 128.3, 127.6, 127.1, 61.0, 51.0, 36.6, 14.0; IR (KBr): cm−1 3291, 3087, 2986, 2927, 2849, 1757, 1660, 1587, 1548, 1501, 1475, 1449, 1416, 1390, 1353, 1319, 1254, 1204, 1156, 1135, 1096, 1061, 1025, 1011; HRMS (ESI): m/z [M + H]+ calcd for C19H17N2O4Cl2: 407.05599, 409.05304, found: 407.05674, 409.05381.
3.1.6. Acylation of 5-benzyl-5,12-dihydrodibenzo[b,f][1,4]diazocine-6,11-dione (3j)
A suspension of 5-benzyl-5,12-dihydrodibenzo[b,f][1,4]diazocine-6,11-dione (3j) (1.0 g, 3.0 mmol) in acetic anhydride (15 mL) was refluxed for 1 h, then the excess of anhydride was evaporated and crude 5-acetyl-12-benzyl-5,12-dihydrodibenzo[b,f][1,4]diazocine-6,11-dione (3o) was crystallized from mixture of cyclohexane:ethyl acetate 9:1 giving 3o as colorless crystals (1.06 g, 94% yield). M.p. 174.0–175.0 °C, R.f. = 0.54 (hexane:ethyl acetate 6:4 v/v). 1H NMR (500 MHz, DMSO-d6) δ 7.49–7.17 (m, 13H, HAr), 5.21 (d, 1H, J = 14.5 Hz, CH2), 4.78 (d, 1H, J = 14.5 Hz, CH2), 2.31 (s, 3H, Me); 13C NMR (125 MHz, DMSO-d6) δ 171.0, 169.5, 168.3, 139.7, 136.8, 136.3, 132.7, 132.0, 131.7, 130.6, 130.2, 130.1, 128.7 (2 × C), 128.4, 128.3 (2 × C), 127.6, 127.0, 126.52, 126.47, 51.7, 26.4; IR (KBr): cm−1 3064, 3004, 2934, 1949, 1831, 1709, 1649, 1595, 1572, 1496, 1455, 1433, 1393, 1368, 1303, 1278, 1253, 1203, 1156, 1139, 1078, 1045, 1019; HRMS (ESI): m/z [M + H]+ calcd for C23H19N2O3: 371.13902, found: 371.13952.
3.1.7. 1,3,4,6-tetrahydrobenzo[b][1,4]diazocine-2,5-dione (6)
Following the procedure of Elgureo et al. [
20] purified by column chromatography methanol:chloroform 1:9
v/
v, yield 27%, white crystals, m.p. 244.0–245.0 °C, R.f. = 0.22 (methanol:chloroform 1:9
v/
v).
1H NMR (500 MHz, DMSO-
d6) δ 9.55 (2, 2H, 2 × NH); 7.34–7.27 (m, 2H, H
Ar), 7.22–7.15 (m, 2H, H
Ar), 2.33 (bs, 4H, 2 × CH
2);
13C NMR (125 MHz, DMSO-
d6) δ 172.2, 134.8, 127.4, 126.9, 30.5; IR (KBr): cm
−1 3179, 3051, 2920, 1992, 1947, 1681, 1644, 1587, 1504, 1455, 1409, 1332, 1308, 1232, 1176, 1124, 1050, 1010; HRMS (ESI):
m/z [M + H]
+ calcd for C
23H
19N
2O
3: 191.08150, found: 191.08141.
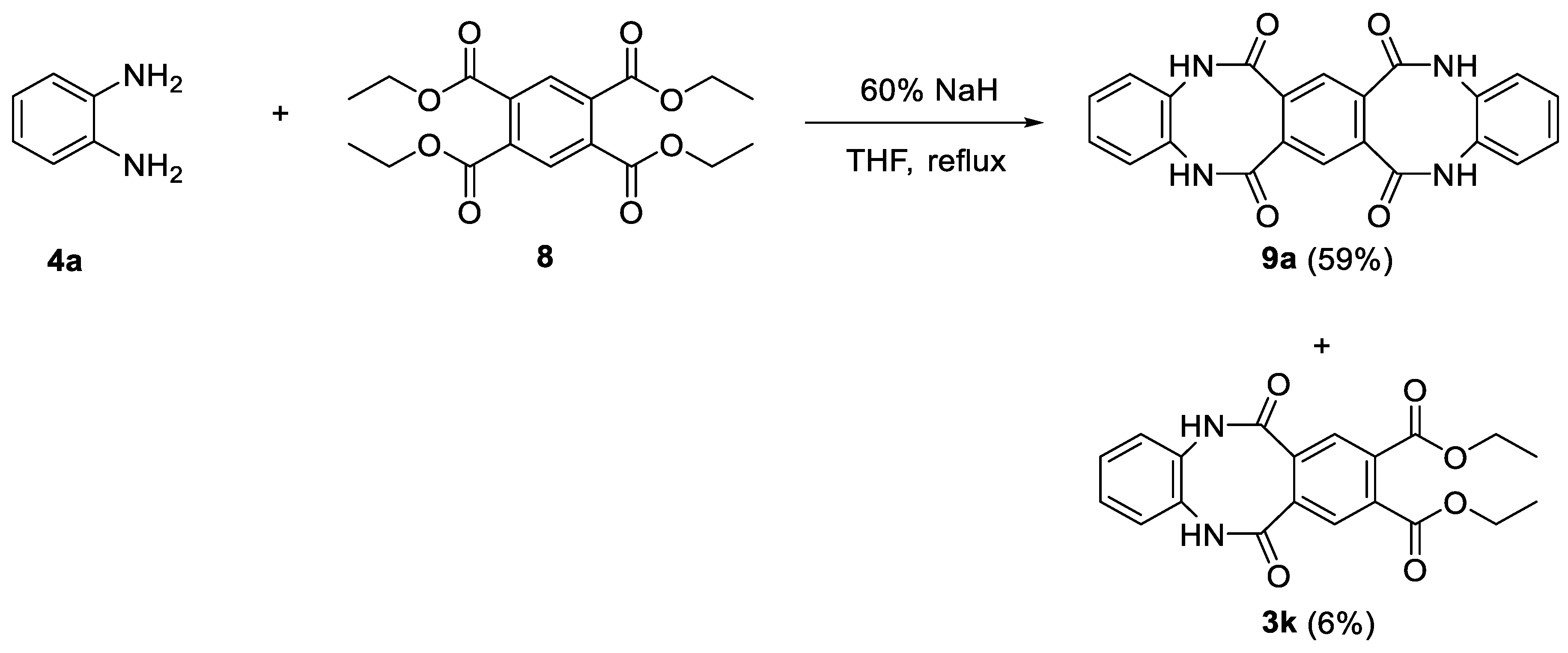
3.1.8. Synthesis of 9a
To a solution of tetraethyl benzene-1,2,4,5-tetracarboxylate (8.0 g, 21.9 mmol, 1.0 equiv.) and phenylenediamine (4.7 g, 43.7 mmol, 2.0 equiv.) in anhydrous THF (100 mL), 60% sodium hydride (3.7 g, 91.8 mmol, 4.2 equiv.) was added and the resulting mixture was refluxed overnight. Then the mixture was poured into water (500 mL), acidified with concentrated HCl to pH 2. The obtained precipitate, containing crude 9a, was filtered through a Schott funnel, washed several times with ethyl acetate and crystallized from the mixture of DMSO:NMP 1:1. The filtrate was extracted with ethyl acetate (3 × 100 mL), the combined organic phases containing tricyclic product 3k were washed with brine (1 × 50 mL) and dried over magnesium sulfate. Evaporation of the solvent gave the crude product which was purified by column chromatography (100% ethyl acetate), giving the final product 3k as a white crystalline solid.
9a: yield 59% (5.13 g), white crystals, m.p. 375.0–376.0 °C. 1H NMR (500 MHz, DMSO-d6) δ 10.16. 10.10 (2 × s, 4H, 4 × NH), 7.30–7.16 (m, 8H, HAr), 7.12–7.00 (m, 2H, HAr); 13C NMR (125 MHz, DMSO-d6) δ, main conformer only: 168.5, 134.3, 133.7, 127.5, 126.9, 124.6; IR (KBr): cm−1 3465, 3189, 3059, 2886, 1669, 1592, 1552, 1503, 1415, 1371, 1302, 1229, 1154, 1113, 1038; HRMS (ESI): m/z [M + H]+ calcd for C22H15N4O4: 399.10878, found: 399.10842.
diethyl 6,11-dioxo-5,6,11,12-tetrahydrodibenzo[b,f][1,4]diazocine-8,9-dicarboxylate (3k): yield 6% (0.5 g), white crystals, m.p. 242.0–243.0 °C, R.f. = 0.28 (hexane:ethyl acetate 2:8 v/v). 1H NMR (500 MHz, DMSO-d6) δ 10.48 (s, 2H, 2 × NH), 7.67 (s, 2H, HAr), 7.26–7.18 (m, 4H, HAr), 4.25 (q, 4H, J = 7.0 Hz, 2 × CH2), 1.24 (t, 6H, J = 7.0 Hz, 2 × Me); 13C NMR (125 MHz, DMSO-d6) δ 168.5, 165.5, 134.6, 134.4, 133.1, 128.0, 127.31, 127.25, 61.9, 13.8; IR (KBr): cm−1 3195, 3062, 2984, 2937, 2901, 1736, 1718, 1665, 1592, 1561, 1501, 1444, 1391, 1363, 1291, 1168, 1144, 1110, 1015; HRMS (ESI): m/z [M + H]+ calcd for C20H19N2O6: 383.12376, found: 383.12371.
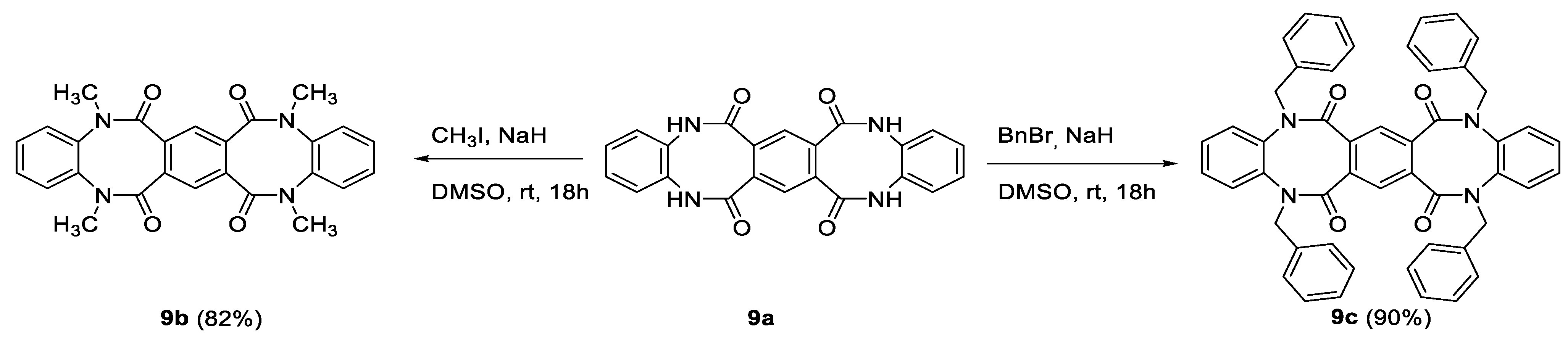
3.1.9. General Procedure for Alkylation of 9a
To a solution of 9a (1 equiv.) in anhydrous DMSO (2 mL/mmol), 60% sodium hydride in mineral oil (5 equiv.) was added. The resulting mixture was stirred for 30 min, then appropriate halide (6 eq.), methyl iodide or benzyl bromide, was added. The reaction mixture was stirred for 18 h at room temperature, then was poured into water and the precipitated product was filtered through a Schott funnel. The obtained solids were washed with ethyl acetate, then with acetone and dried in air to give pure products.
(9b) yield 82%, white crystals, m.p. 400.0 °C, R.f. = 0.56 (methanol:chloroform 1:9 v/v). 1H NMR (500 MHz, CF3COOD) δ 7.42 (s, 2H, HAr), 7.37–7.05 (m, 8H, HAr), 3.46, 3.41 (2 × s, 12H, 4 × Me); 13C NMR (125 MHz, CF3COOD) δ, both conformers: 172.2, 141.0, 140.4, 136.3, 136.2, 133.1, 132.4, 128.8, 128.7, 127.8, 127.2; IR (KBr): cm−1 3464, 3040, 2936, 1649, 1594, 1553, 1503, 1448, 1418, 1383, 1310, 1272, 1177, 1122, 1045; HRMS (ESI): m/z [M + H]+ calcd for C26H23N4O4: 455.17138, found: 455.17135;
(9c) yield 90%, white crystals, m.p. 385.0–386.0 °C, R.f. = 0.67 (hexane:ethyl acetate 2:8 v/v). 1H NMR (500 MHz, DMSO-d6) δ 7.45–7.30 (m, 12H, HAr), 7.28 (s, 2H, HAr), 7.27–7.20 (m, 4H, HAr), 7.17–7.01 (m, 12H, HAr), 4.62 (d, 4H, J = 15.0 Hz, 2 × CH2), 3.96 (d, 4H, J = 15.0 Hz, 2 × CH2); 13C NMR (125 MHz, DMSO-d6) δ 166.7, 138.9, 136.8, 134.0, 129.5, 128.9, 128.4, 127.9, 127.7, 123.9, 51.2; IR (KBr): cm−1 3290, 3084, 3062, 3029, 2935, 1955, 1665, 1651, 1598, 1584, 1495, 1455, 1431, 1399, 1362, 1324, 1298, 1261, 1224, 1201, 1157, 1148, 1119, 1079, 1053, 1030; HRMS (ESI): m/z [M + H]+ calcd for C50H38N4O4: 759.29658, found: 759.29776;
3.1.10. Rearrangement of 5,12-dihydrodibenzo[b,f][1,4]diazocine-6,11-dione (3a) to 2-(2-aminophenyl)isoindoline-1,3-dione (10)
To a suspension of 5,12-dihydrodibenzo[b,f][1,4]diazocine-6,11-dione (3a) (1.0 g, 4.2 mmol, 1 equiv.) in DMSO, 60% (0.18 g, 4.6 mmol, 1.1 equiv.) sodium hydride in mineral oil was added and the resulting mixture was stirred overnight at room temperature. The red clear solution which was formed was poured into water and extracted with ethyl acetate (3 × 50 mL), the organic phase was washed with brine (1 × 50 mL) and dried over anhydrous magnesium sulfate. The crude product was purified by column chromatography (hexane:ethyl acetate 7:3 v/v), giving 10 as a yellow solid (0.58 g, 58% yield). m.p. 195.0–196.0 °C, R.f. = 0.35 (hexane:ethyl acetate 6:4 v/v). 1H NMR (500 MHz, DMSO-d6) δ 7.95–7.83 (m, 4H, HAr), 7.12 (t, 1H, J = 7.5 Hz, HAr), 7.01 (d, 1H, J = 7.5 Hz, HAr), 6.75 (d, 1H, J = 7.5 Hz, HAr), 6.56 (t, 1H, J = 7.5 Hz, HAr), 6.36 (s, 2H, NH2); 13C NMR (125 MHz, DMSO-d6) δ 167.6, 146.5, 134.1, 132.5, 130.0, 129.7, 123.1, 115.9, 115.32, 115.28; IR (KBr): cm−1 3458, 3380, 3030, 1783, 1708, 1623, 1578, 1502, 1461, 1377, 1314, 1271, 1216, 1159, 1143, 1100, 1083, 1049, 1026; HRMS (ESI): m/z [M + H]+ calcd for C14H11N2O2: 239.08150, found: 239.08171;
3.1.11. Synthesis of 4,5-dichloro-N1-methylbenzene-1,2-diamine (4e)
To a solution of 4,5-dichloro-2-nitroaniline (30.0 g, 0.14 mol, 1.0 equiv.) in DMF (150 mL), 60% sodium hydride in mineral oil (6.4 g, 0.16 mol, 1.1 equiv.) was added. After 30 min of stirring in room temperature, methyl iodide (13.5 mL, 0.22 mol, 1.5 equiv.) was added to the reaction mixture and the solution warmed up spontaneously. After 1h, the reaction mixture was poured into water (500 mL) and the product was extracted with ethyl acetate (3 × 100 mL). The organic phase was washed with brine (1 × 50 mL) and dried over anhydrous magnesium sulfate. The drying agent was filtered off, the resulting filtrate was concentrated under reduced pressure and left in the fridge for crystallization. The orange crystals of 4,5-dichloro-N-methyl-2-nitroaniline were formed, which were washed with a small amount of ethyl acetate and dried. Yield: 25.0 g (78%). A slurry of 4,5-dichloro-N-methyl-2-nitroaniline (15.0 g, 0.07 mol, 1.0 equiv.) and iron powder (19.0 g, 0.34 mol, 5.0 equiv.) in a mixture of 95% ethanol (200 mL) and glacial acetic acid (100 mL) was stirred at 60 °C for 2h. The progress of reaction was monitored by TLC, when the reaction was completed; the excess of volatiles was evaporated under reduced pressure. The obtained residue was treated with water and the pH was adjusted to ca. 8 using sodium hydroxide. The crude product was extracted with ethyl acetate (3 × 150 mL), organic phase was washed with brine (1 × 50 mL) and dried over anhydrous magnesium sulfate. Evaporation of the solvent resulted in the 4,5-dichloro-N1-methylbenzene-1,2-diamine (4e), obtained as a brown solid (12.6 g, 97% yield) which was used in the next step without further purification. M.p. 100.0–101.0 °C, R.f. = 0.35 (hexane:ethyl acetate 9:1 v/v). 1H NMR (500 MHz, CDCl3) δ 6.72 (s, 1H, HAr), 6.62 (s, 1H, HAr), 3.30 (bs, 3H, NH2+NH), 2.81 (s, 3H, Me); 13C NMR (125 MHz, CDCl3) δ 138.8, 132.8, 123.2, 120.3, 117.1, 112.0, 31.0; HRMS (ESI): m/z [M + H]+ calcd for C7H9Cl2N2: 191.01373, 193.01078, 175.00785, found: 191.01403, 193.01110, 175.00788;
3.1.12. Synthesis of N1-benzylbenzene-1,2-diamine (4f)
A solution of 1-fluoro-2-nitrobenzene (15.0 mL, 0.14 mol, 1.0 equiv.), benzylamine (17.0 mL, 0.15 mol, 1.1 equiv.) and TEA (25.0 mL, 0.18 mol, 1.3 equiv.) in DMSO (10 mL) was stirred at 100 °C for 18 h. After cooling down, the obtained orange solution was poured into water and the reaction product was extracted with ethyl acetate (3 × 100 mL). The organic phase was washed with brine (1 × 50 mL) and dried over anhydrous magnesium sulfate. Evaporation of the solvent gave N-benzyl-2-nitroaniline as a red solid (29.0 g, 89.5% yield), which was used in the next step without further purification. A slurry of N-benzyl-2-nitroaniline (10.0 g, 44.0 mmol, 1.0 equiv.) and iron powder (12.3 g, 220.0 mmol, 5.0 equiv.) in a mixture of 95% ethanol (100 mL) and glacial acetic acid (50 mL) was stirred at 60 °C for 2 h. The progress of reaction was monitored by TLC, when the reaction was completed, the excess of volatiles was evaporated under reduced pressure. The obtained residue was treated with water, the pH was adjusted to ca. 8 using sodium hydroxide and the reaction product was extracted with ethyl acetate (3 × 150 mL). The organic phase was washed with brine (1 × 50 mL) and dried over anhydrous magnesium sulfate. The drying agent was filtered off and the resulting filtrate was evaporated under reduced pressure, giving N1-benzylbenzene-1,2-diamine (4f) as a brown solid (8.3 g, 96% yield) used in the next step without further purification. M.p. 49.0–50.0 °C, R.f. = 0.33 (hexane:ethyl acetate 9:1 v/v). 1H NMR (500 MHz, CDCl3) δ 7.48–7.43 (m, 2H, HAr), 7.43–7.37 (m, 2H, HAr), 7.36–7.31 (m, 1H, HAr), 6.89-6.83 (m, 1H, HAr), 6.80-6.69 (m, 3H, HAr), 4.35 (s, 2H, CH2), 3.46 (bs, 3H, NH2 + NH); 13C NMR (125 MHz, CDCl3) δ 139.4, 137.7, 134.2, 128.7, 127.9, 127.3, 120.8, 118.9, 116.6, 112.1, 48.7; HRMS (ESI): m/z [M + H]+ calcd for C13H15N2: 199.12298, found: 199.12313;
3.1.13. General Procedure for Synthesis of Phthalic Acids Methyl Esters 5b–d
To a stirred slurry of appropriate phthalic acid (0.1 mol) in methanol (200 mL), concentrated sulphuric acid (20 mL) was added dropwise (caution: exothermic!). The reaction mixture was stirred for ten minutes in room temperature which led to the dissolution of phthalic acid. The obtained solution was refluxed for 18 h, then the excess of methanol was evaporated under reduced pressure and the remaining residue was poured into water. The pH was adjusted to ca. 8 by the addition of solid sodium hydroxide and the reaction product was extracted with ethyl acetate (3 × 100 mL) The organic phase was washed with brine (1 × 50 mL) and dried over anhydrous magnesium sulfate. The drying agent was filtered off and the resulting filtrate was evaporated under reduced pressure. Evaporation of the solvent gave the crude product which was used in next step without further purification.
dimethyl 4-nitrophthalate (5b): yield 76%, beige solid, m.p. 66.0–67.0 °C, R.f. = 0.20 (hexane:ethyl acetate 9:1 v/v). 1H NMR (500 MHz, CDCl3) δ 8.58 (d, 1H, J = 2.5 Hz, HAr), 8.36 (dd, 1H, J = 2.5 Hz, J = 8.5 Hz, HAr), 7.81 (d, 1H, J = 8.5 Hz, HAr), 3.94 (s, 3H, Me); 3.93 (s, 3H, Me); 13C NMR (125 MHz, CDCl3) δ 166.8, 165.5, 148.9, 138.2, 132.7, 130.1, 126.2, 124.5, 53.4; HRMS (ESI): m/z [M + H]+ calcd for C10H10NO6: 240.05026, found: 240.05031;
dimethyl 4,5-dichlorophthalate (5c): yield 91%, pale oil, R.f. = 0.49 (hexane:ethyl acetate 9:1 v/v). 1H NMR (500 MHz, CDCl3) δ 7.77 (s, 2H, HAr), 3.88 (s, 6H, 2 × Me); 13C NMR (125 MHz, CDCl3) δ 166.0, 135.8, 131.4, 131.0, 53.1; HRMS (ESI): m/z [M + H]+ calcd for C10H9O4Cl2: 262.98724, 264.98429, found: 262.98744, 264.98445;
dimethyl 3,4,5,6-tetrachlorophthalate (5d): yield 35%, white solid, m.p. 92.0–93.0 °C, R.f. = 0.49 (hexane:ethyl acetate 9:1 v/v). 1H NMR (500 MHz, CDCl3) δ 3.92 (s, 6H, 2 × Me); 13C NMR (125 MHz, CDCl3) δ 164.3, 136.2, 132.1, 130.7, 53.6; HRMS (ESI): m/z [M + H]+ calcd for C10H7O4Cl4: 330.90930, 332.90635, 334.90340, found: 330.90909, 332.90613, 334.90316.
3.1.14. Synthesis of dimethyl thiophene-3,4-dicarboxylate (5e)
To a slurry of tiophene-3,4-dicarboxylic acid (4.0 g, 23.3 mmol, 1.0 equiv.) and potassium carbonate (8.1 g, 58.3 mmol, 2.5 equiv.) in DMF (20 mL), methyl iodide (3.2 mL, 51.1 mmol, 2.2 equiv.) was added dropwise. The resulting mixture was stirred for 18 h at 40 °C, then poured into water (100 mL) and the pH was adjusted to ca. 8 using sodium hydroxide. The reaction product was extracted with ethyl acetate (3 × 50 mL), the organic phase was washed with brine (1 × 50 mL) and dried over anhydrous magnesium sulfate. The drying agent was filtered off, the resulting filtrate was concentrated under reduced pressure and left in the fridge for crystallization. Big, colorless crystals of 5e were formed (1.83 g, 40% yield). M.p. 59.0–60.0 °C, R.f. = 0.25 (hexane:ethyl acetate 9:1 v/v). 1H NMR (500 MHz, CDCl3) δ 7.84 (s, 2H, HAr), 3.85 (s, 6H, 2 × Me); 13C NMR (125 MHz, CDCl3) δ 163.5, 133.2, 131.9, 52.42; HRMS (ESI): m/z [M + H]+ calcd for C8H9O4S: 201.02161, found: 201.02177;
3.1.15. Synthesis of tetraethyl benzene-1,2,4,5-tetracarboxylate (8)
Following the procedure of Berthold et.al. [
27] the slurry of pyromellitic dianhydride (25.0 g, 0.11 mol) in absolute ethanol (250 mL) was refluxed for 2 h, then sulfuric acid (15 mL) was added dropwise. The reaction mixture was refluxed for an additional 18 h, then the excess ethanol was evaporated. The residue was poured into water and the pH was adjusted to ca. 8 using sodium hydroxide. The reaction product was extracted with ethyl acetate (3 × 100 mL), the organic phase was washed with brine (1 × 50 mL) and dried over anhydrous magnesium sulfate. Evaporation of the solvent gave
8 as an oily residue, which solidified upon storage (27.16 g, 64% yield). M.p. 54.0–55.0 °C, R.f. = 0.28 (hexane:ethyl acetate 9:1
v/
v).
1H NMR (500 MHz, CDCl
3) δ 8.02 (s, 2H, H
Ar), 4.36 (q, 8H, J = 7.0 Hz, 4 × CH
2), 1.35 (t, 12H, J = 7.0 Hz, 4 × Me);
13C NMR (125 MHz, CDCl
3) δ 166.1, 134.4, 129.6, 62.3, 14.1; HRMS (ESI):
m/z [M + H]
+ calcd for C
18H
23O
8: 367.13674, found: 367.13834;












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
































