
Multiscale Structure of Starches Grafted with Hydrophobic Groups: A New Analytical Strategy

 , , , , and
, , , , and
Abstract
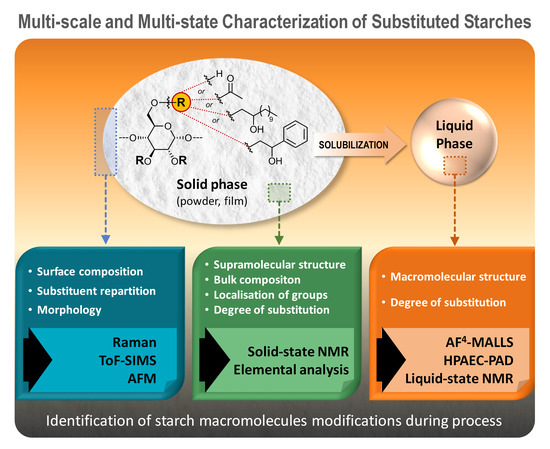
1. Introduction
2. Results and Discussion
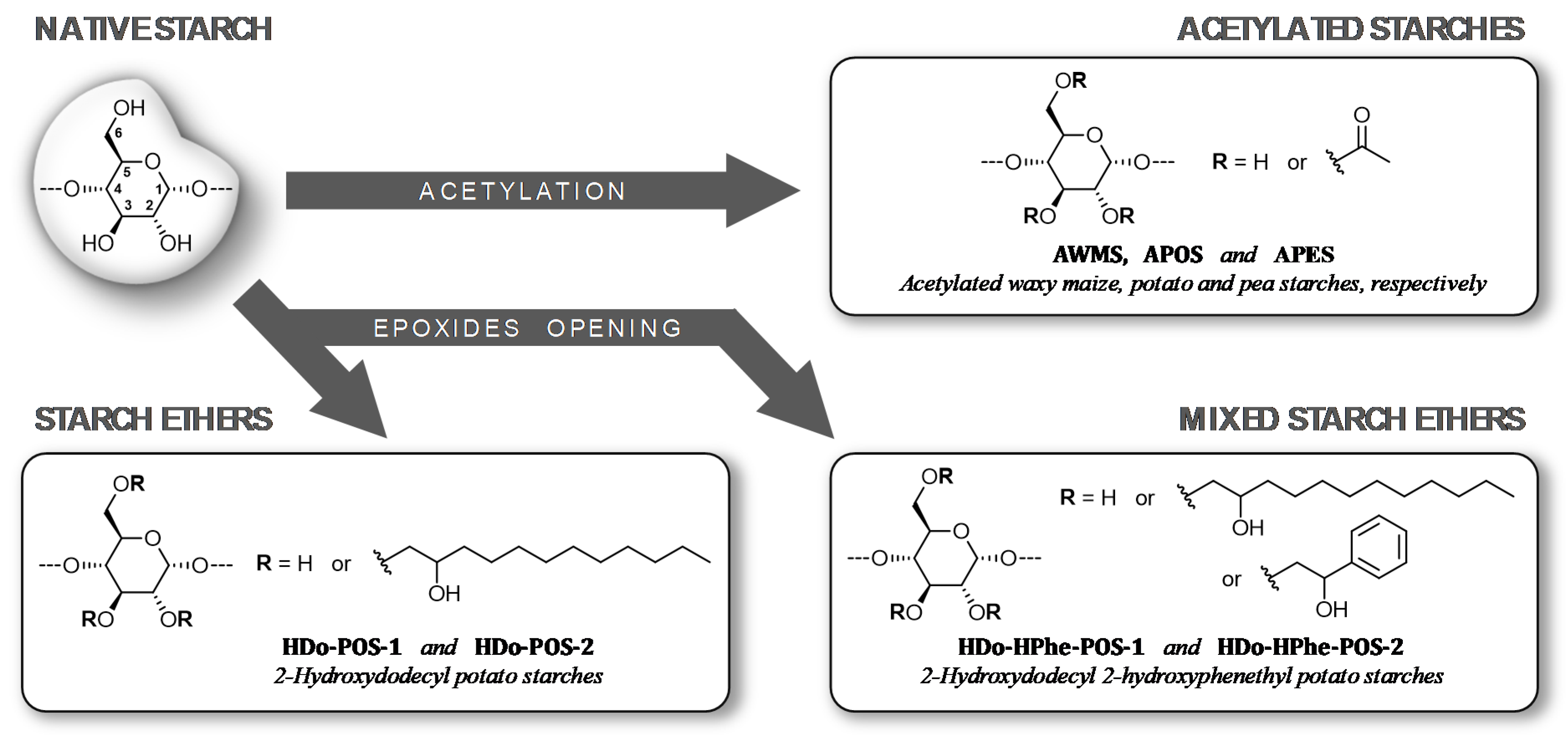
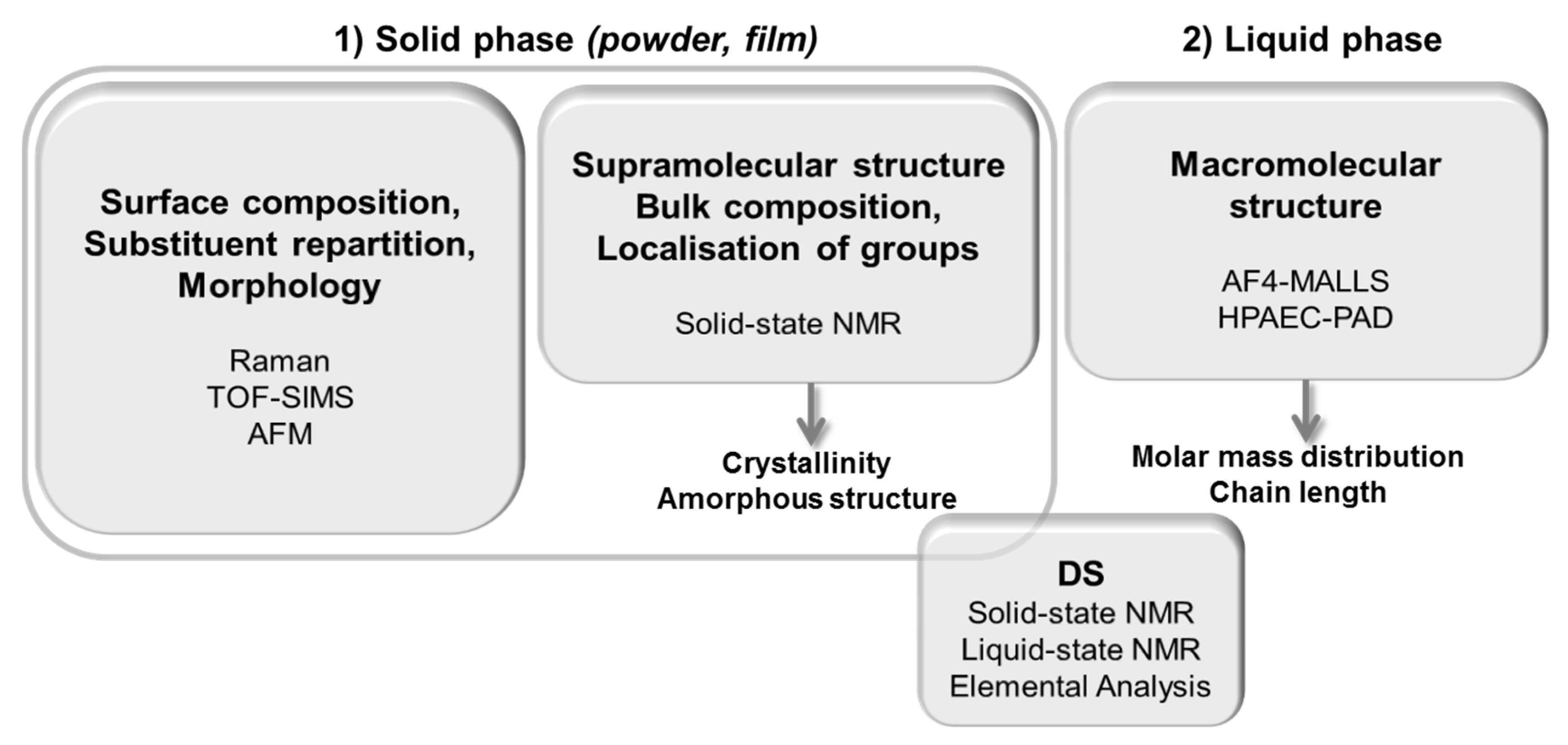
2.1. Development of the Experimental Strategy
2.1.1. Determination of the Degree of Substitution
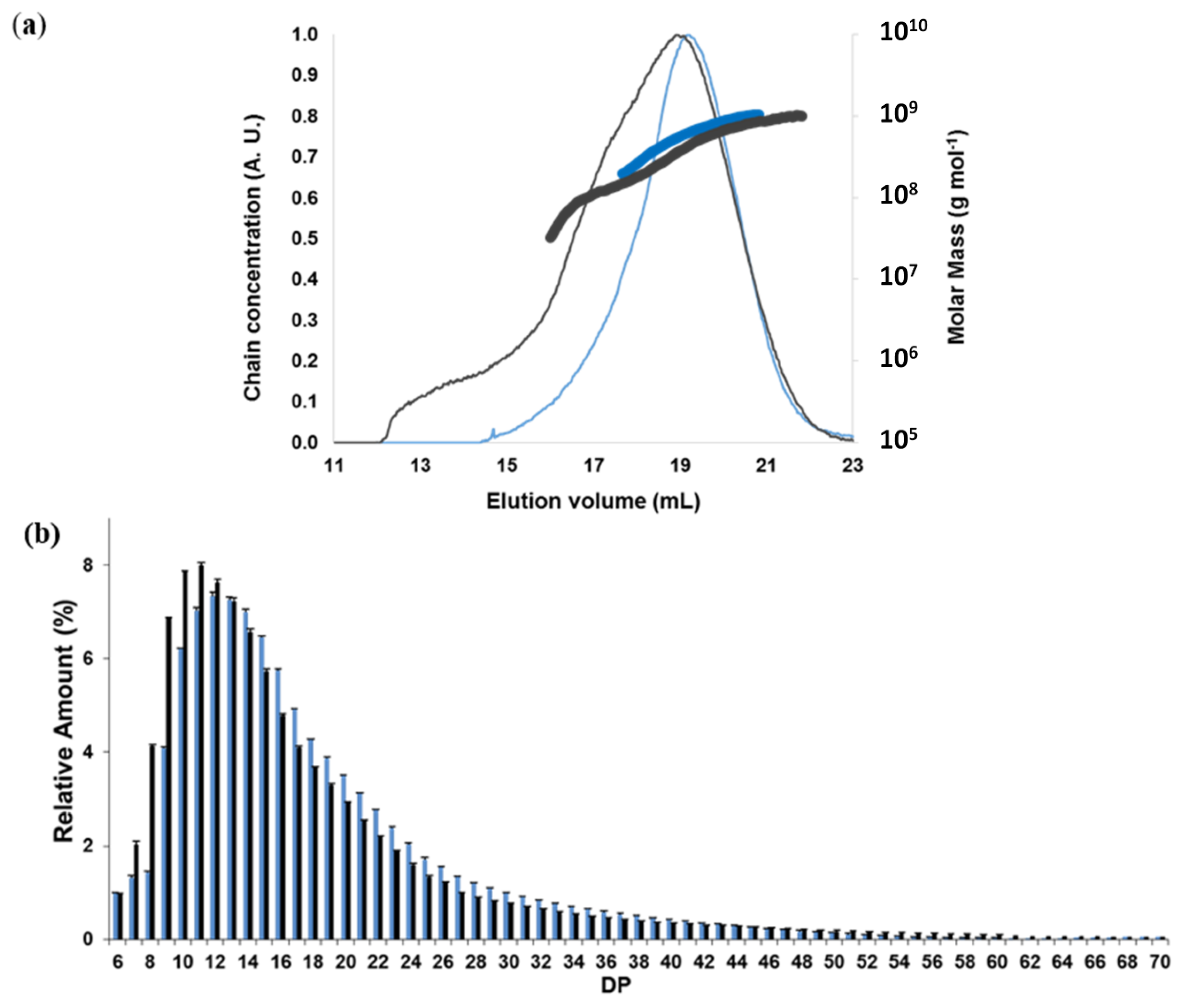
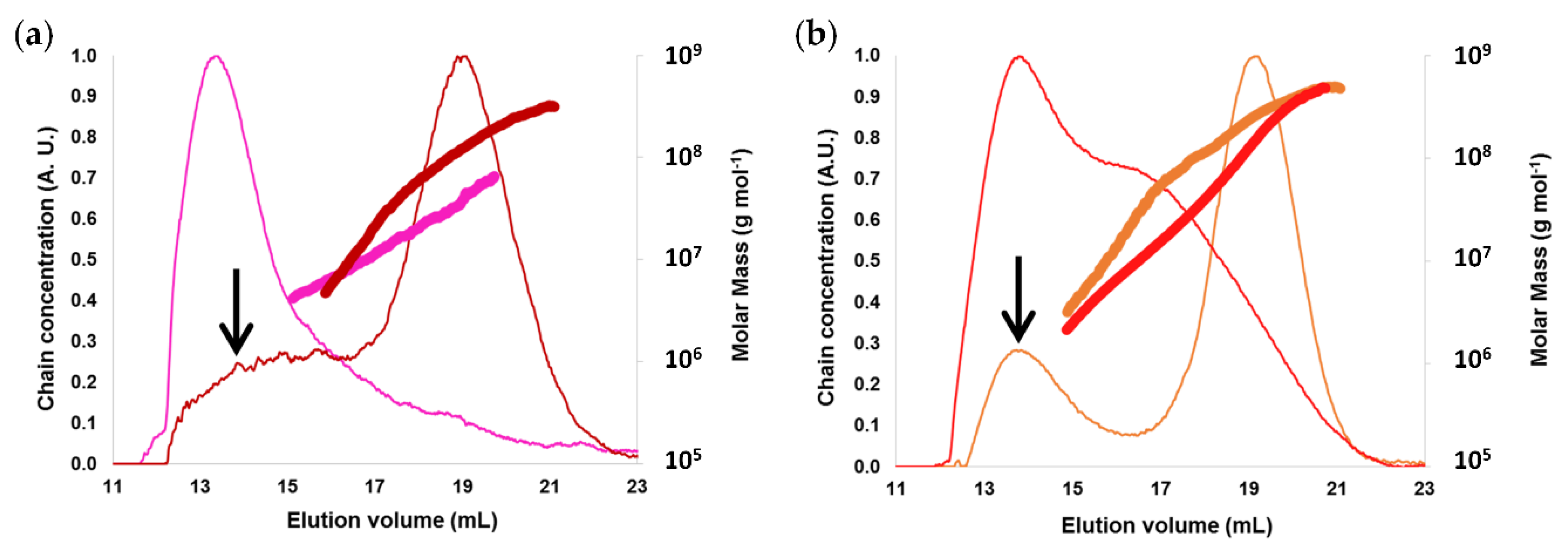
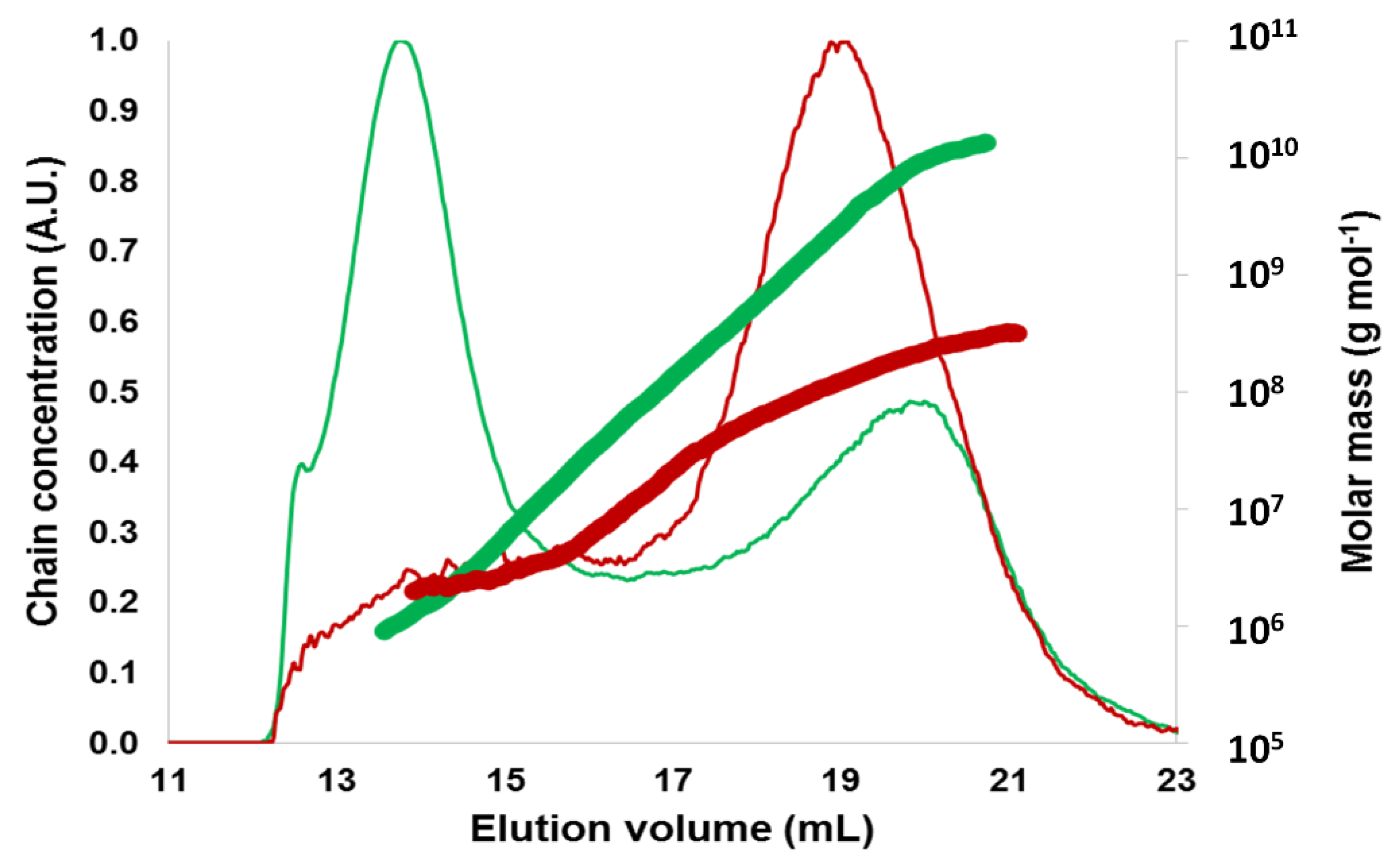
2.1.2. Macromolecular and Molecular Structure
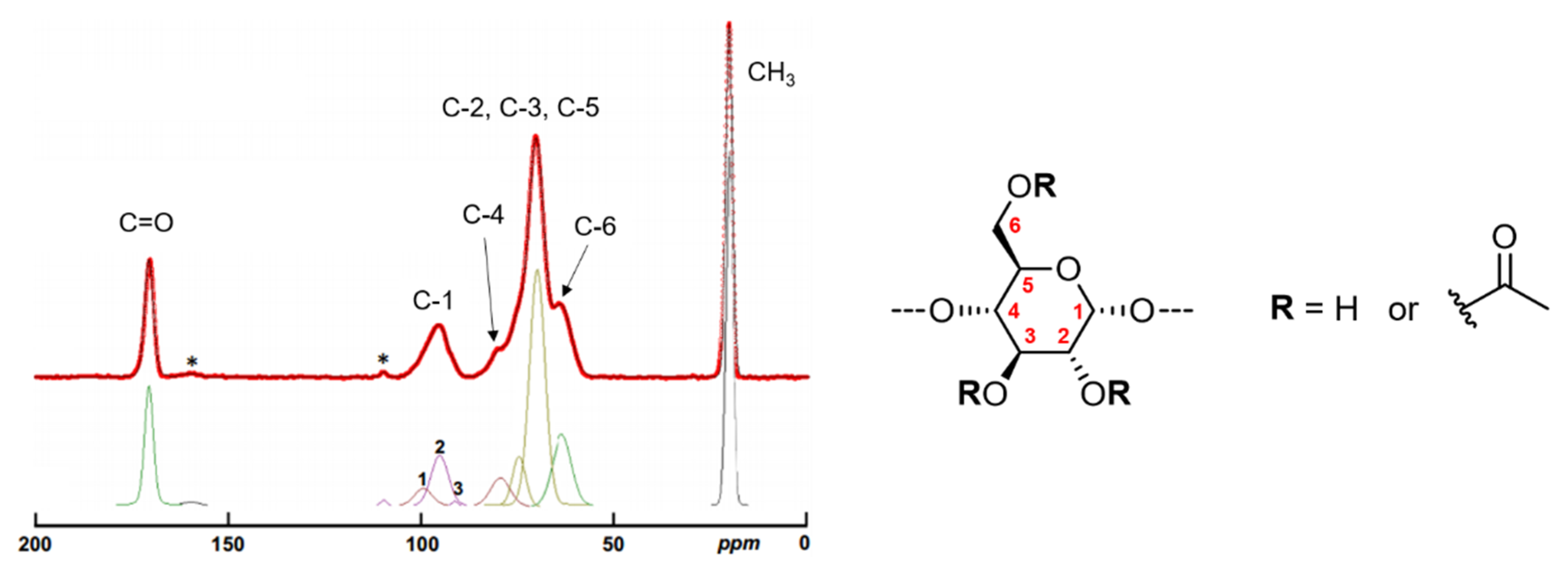
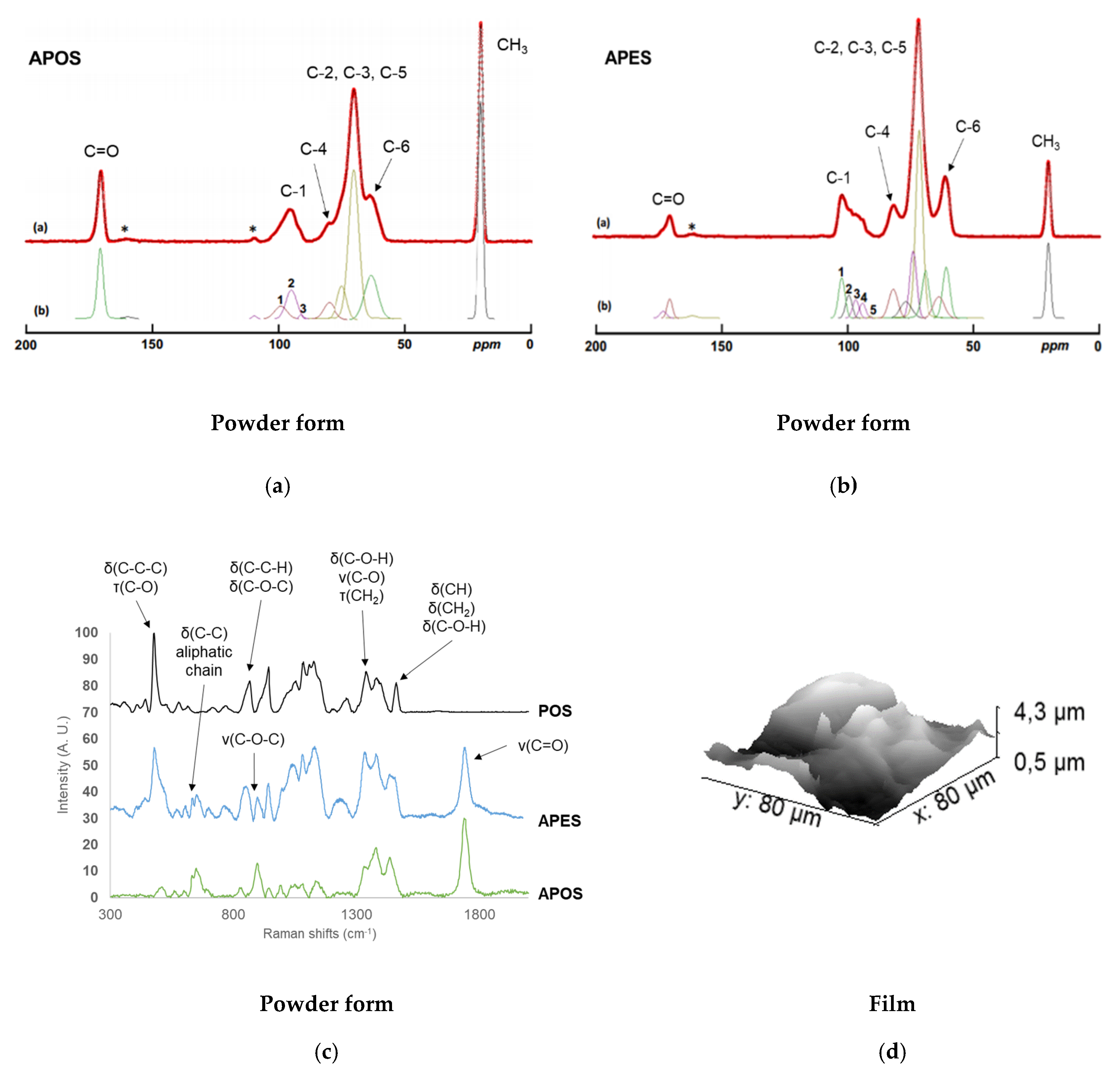
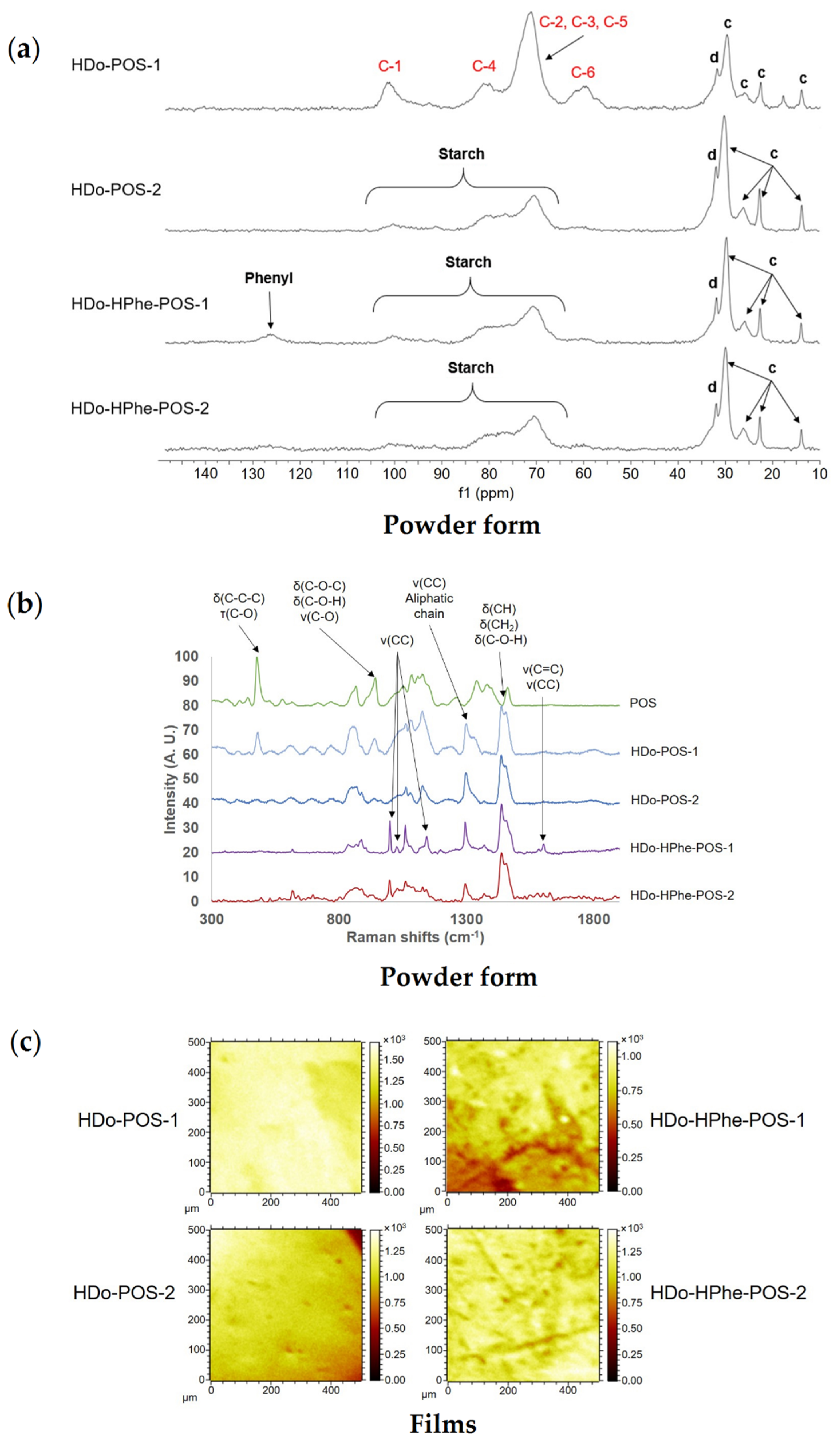
2.1.3. Composition, localization of Acetyl Groups and Supramolecular Structure
2.1.4. Surface Characterization of Materials
2.2. Characterization of Acetylated Starches with Various DS and Amylose Content
2.2.1. Macromolecular Characteristics
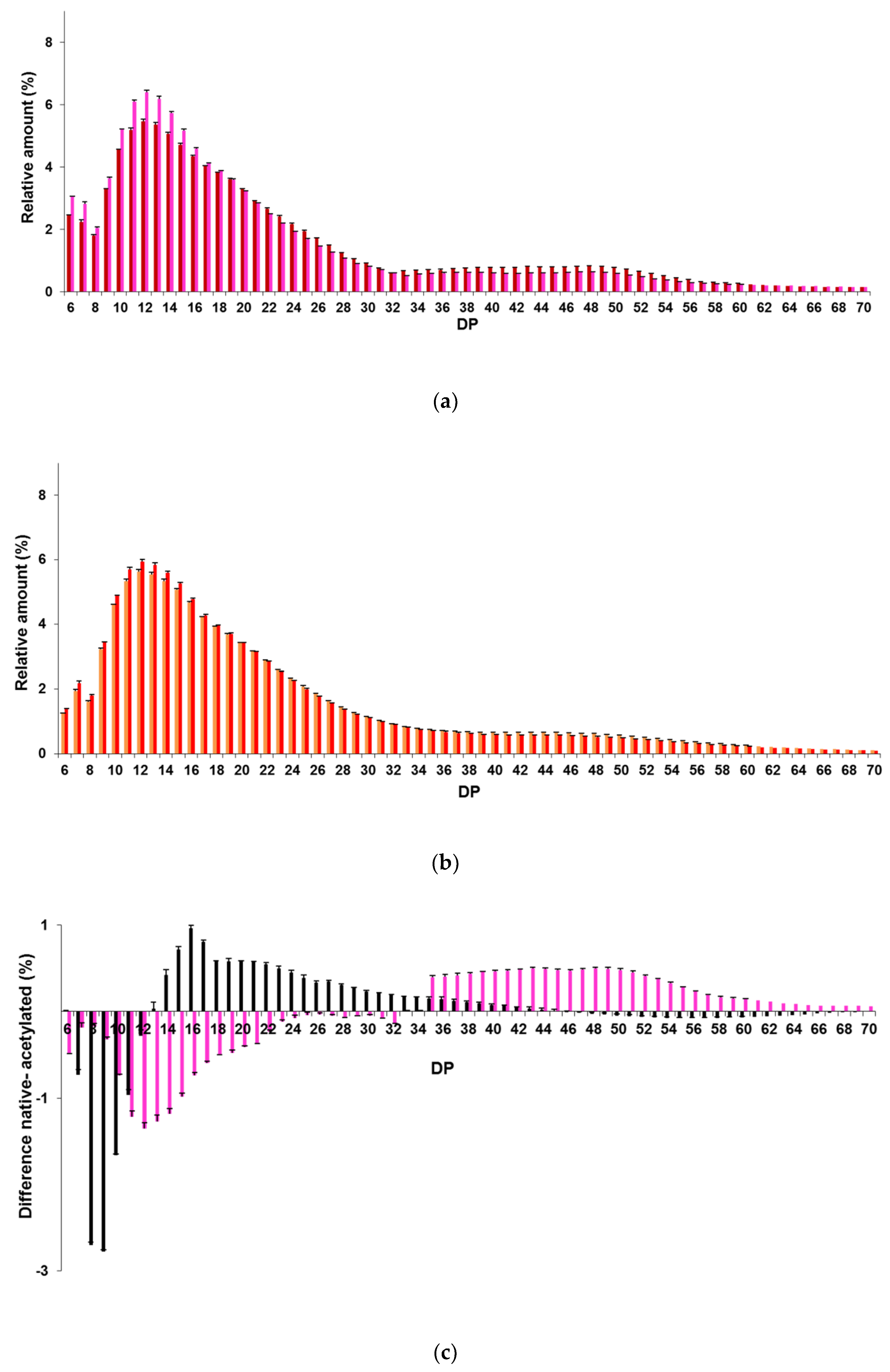
2.2.2. Chain-Length Distributions of Acetylated Starches
2.2.3. Localization of Acetyl Groups, Supramolecular Structure and Surface Characteristics of Acetylated Starches
2.3. Characterization of Etherified Starches
2.3.1. Degree of Substitution
2.3.2. Macromolecular Characteristics
2.3.3. Composition, Localization of Ether Groups and Supramolecular Structure
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Nuclear Magnetic Resonance Measurements
3.2.2. Determination of size, Molar Mass and Chain Length Distributions
- Pretreatment of the samples
- Determination of macromolecular characteristics
- Determination of chain length (CL) distributions
3.2.3. Topology and Surface Chemical Composition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pérez, S.; Bertoft, E. The molecular structures of starch components and their contribution to the architecture of starch granules: A comprehensive review. Starch Stärke 2010, 62, 389–420. [Google Scholar] [CrossRef]
- Avérous, L.; Halley, P. Biocomposites based on plasticized starch. Biofuels Bioprod. Biorefining 2009, 3, 329–343. [Google Scholar] [CrossRef]
- Xie, F.; Luckman, P.; Milne, J.; McDonald, L.; Young, C.; Tu, C.Y.; Di Pasquale, T.; Faveere, R.; Halley, P.; Xie, F. Thermoplastic Starch. J. Renew. Mater. 2014, 2, 95–106. [Google Scholar] [CrossRef]
- Tomasik, P.; Schilling, C.H. CHEMICAL MODIFICATION OF STARCH. In Advances in Carbohydrate Chemistry and Biochemistry; Elsevier BV: Amsterdam, The Netherlands, 2004; Volume 59, pp. 175–403. [Google Scholar]
- Ashogbon, A.O.; Akintayo, E.T. Recent trend in the physical and chemical modification of starches from different botanical sources: A review. Starch Stärke 2013, 66, 41–57. [Google Scholar] [CrossRef]
- Miao, M.; Li, R.; Jiang, B.; Cui, S.W.; Zhang, T.; Jin, Z. Structure and physicochemical properties of octenyl succinic esters of sugary maize soluble starch and waxy maize starch. Food Chem. 2014, 151, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Simsek, S.; Ovando-Martínez, M.; Whitney, K.; Bello-Pérez, L.A. Effect of acetylation, oxidation and annealing on physicochemical properties of bean starch. Food Chem. 2012, 134, 1796–1803. [Google Scholar] [CrossRef]
- Sun, S.; Ganwei, Z.; Ma, C. Preparation, physicochemical characterization and application of acetylated lotus rhizome starches. Carbohydr. Polym. 2016, 135, 10–17. [Google Scholar] [CrossRef]
- Lee, S.; Kim, S.T.; Pant, B.R.; Kwen, H.D.; Song, H.H.; Lee, S.K.; Nehete, S.V. Carboxymethylation of corn starch and characterization using asymmetrical flow field-flow fractionation coupled with multiangle light scattering. J. Chromatogr. A 2010, 1217, 4623–4628. [Google Scholar] [CrossRef]
- Modig, G.; Nilsson, L.; Bergenståhl, B.; Wahlund, K.-G. Homogenization-induced degradation of hydrophobically modified starch determined by asymmetrical flow field-flow fractionation and multi-angle light scattering. Food Hydrocoll. 2006, 20, 1087–1095. [Google Scholar] [CrossRef]
- Nilsson, L.; Leeman, M.; Wahlund, K.-G.; Bergenståhl, B. Mechanical Degradation and Changes in Conformation of Hydrophobically Modified Starch. Biomacromolecules 2006, 7, 2671–2679. [Google Scholar] [CrossRef]
- Heins, D.; Kulicke, W.-M.; Käuper, P.; Thielking, H. Characterization of Acetyl Starch by Means of NMR Spectroscopy and SEC/MALLS in Comparison with Hydroxyethyl Starch. Starch Stärke 1998, 50, 431–437. [Google Scholar] [CrossRef]
- Chen, Z.; A Schols, H.; Voragen, A.G. Differently sized granules from acetylated potato and sweet potato starches differ in the acetyl substitution pattern of their amylose populations. Carbohydr. Polym. 2004, 56, 219–226. [Google Scholar] [CrossRef]
- Huang, J.; Schols, H.A.; Klaver, R.; Jin, Z.; Voragen, A.G. Acetyl substitution patterns of amylose and amylopectin populations in cowpea starch modified with acetic anhydride and vinyl acetate. Carbohydr. Polym. 2007, 67, 542–550. [Google Scholar] [CrossRef]
- Richardson, S.; Gorton, L. Characterisation of the substituent distribution in starch and cellulose derivatives. Anal. Chim. Acta 2003, 497, 27–65. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Wang, L. Characterization of Acetylated Waxy Maize Starches Prepared under Catalysis by Different Alkali and Alkaline-Earth Hydroxides. Starch Stärke 2002, 54, 25–30. [Google Scholar] [CrossRef]
- Chen, Z.; Huang, J.; Suurs, P.; Schols, H.A.; Voragen, A.G. Granule size affects the acetyl substitution on amylopectin populations in potato and sweet potato starches. Carbohydr. Polym. 2005, 62, 333–337. [Google Scholar] [CrossRef]
- Volkert, B.; Lehmann, A.; Greco, T.; Nejad, M.H. A comparison of different synthesis routes for starch acetates and the resulting mechanical properties. Carbohydr. Polym. 2010, 79, 571–577. [Google Scholar] [CrossRef]
- Bello-Pérez, L.A.; Agama-Acevedo, E.; Zamudio-Flores, P.B.; Mendez-Montealvo, G.; Rodriguez-Ambriz, S.L. Effect of low and high acetylation degree in the morphological, physicochemical and structural characteristics of barley starch. LWT 2010, 43, 1434–1440. [Google Scholar] [CrossRef]
- Cave, R.A.; Seabrook, S.A.; Gidley, M.J.; Gilbert, R.G. Characterization of Starch by Size-Exclusion Chromatography: The Limitations Imposed by Shear Scission. Biomacromolecules 2009, 10, 2245–2253. [Google Scholar] [CrossRef] [PubMed]
- Rolland-Sabaté, A.; Guilois, S.; Jaillais, B.; Colonna, P. Molecular size and mass distributions of native starches using complementary separation methods: Asymmetrical Flow Field Flow Fractionation (A4F) and Hydrodynamic and Size Exclusion Chromatography (HDC-SEC). Anal. Bioanal. Chem. 2010, 399, 1493–1505. [Google Scholar] [CrossRef]
- Colussi, R.; Pinto, V.Z.; El Halal, S.L.M.; Vanier, N.L.; Villanova, F.A.; Silva, R.; Zavareze, E.D.R.; Dias, A.R.G. Structural, morphological, and physicochemical properties of acetylated high-, medium-, and low-amylose rice starches. Carbohydr. Polym. 2014, 103, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Elomaa, M. Determination of the degree of substitution of acetylated starch by hydrolysis, 1H NMR and TGA/IR. Carbohydr. Polym. 2004, 57, 261–267. [Google Scholar] [CrossRef]
- Rolland-Sabaté, A.; Colonna, P.; Mendez-Montealvo, M.G.; Planchot, V. Branching Features of Amylopectins and Glycogen Determined by Asymmetrical Flow Field Flow Fractionation Coupled with Multiangle Laser Light Scattering. Biomacromolecules 2007, 8, 2520–2532. [Google Scholar] [CrossRef]
- Rolland-Sabaté, A.; Sánchez, T.; Buleon, A.; Colonna, P.; Jaillais, B.; Ceballos, H.; Dufour, D. Structural characterization of novel cassava starches with low and high-amylose contents in comparison with other commercial sources. Food Hydrocoll. 2012, 27, 161–174. [Google Scholar] [CrossRef]
- Gidley, M.J.; Bociek, S.M. Molecular organization in starches: A carbon 13 CP/MAS NMR study. J. Am. Chem. Soc. 1985, 107, 7040–7044. [Google Scholar] [CrossRef]
- Paris, M.; Bizot, H.; Emery, J.; Buzaré, J.; Buleon, A. Crystallinity and structuring role of water in native and recrystallized starches by 13C CP-MAS NMR spectroscopy. Carbohydr. Polym. 1999, 39, 327–339. [Google Scholar] [CrossRef]
- Chevigny, C.; Foucat, L.; Rolland-Sabaté, A.; Buleon, A.; Lourdin, D. Shape-memory effect in amorphous potato starch: The influence of local orders and paracrystallinity. Carbohydr. Polym. 2016, 146, 411–419. [Google Scholar] [CrossRef]
- Paris, M.; Bizot, H.; Emery, J.; Buzaré, J.; Buléon, A. NMR local range investigations in amorphous starchy substrates I. Structural heterogeneity probed by (13)C CP-MAS NMR. Int. J. Boil. Macromol. 2001, 29, 127–136. [Google Scholar] [CrossRef]
- David, A. Etude de dérivés d’amidon: Relation entre la structure et le comportement thermomécanique. Ph.D. Thesis, Université de Lille, Lille, France, 2017. [Google Scholar]
- Kizil, R.; Irudayaraj, J.; Seetharaman, K. Characterization of Irradiated Starches by Using FT-Raman and FTIR Spectroscopy. J. Agric. Food Chem. 2002, 50, 3912–3918. [Google Scholar] [CrossRef]
- Phillips, D.L.; Liu, H.; Pan, D.; Corke, H. General Application of Raman Spectroscopy for the Determination of Level of Acetylation in Modified Starches. Cereal Chem. J. 1999, 76, 439–443. [Google Scholar] [CrossRef]
- Chung, H.-J.; Liu, Q. Impact of molecular structure of amylopectin and amylose on amylose chain association during cooling. Carbohydr. Polym. 2009, 77, 807–815. [Google Scholar] [CrossRef]
- Jane, J.; Chen, Y.Y.; Lee, L.F.; McPherson, A.E.; Wong, K.S.; Radosavljević, M.; Kasemsuwan, T. Effects of Amylopectin Branch Chain Length and Amylose Content on the Gelatinization and Pasting Properties of Starch. Cereal Chem. J. 1999, 76, 629–637. [Google Scholar] [CrossRef]
- Ratnayake, W.; Hoover, R.; Shahidi, F.; Perera, C.; Jane, J. Composition, molecular structure, and physicochemical properties of starches from four field pea (Pisum sativum L.) cultivars. Food Chem. 2001, 74, 189–202. [Google Scholar] [CrossRef]
- McPherson, A. Comparison of waxy potato with other root and tuber starches. Carbohydr. Polym. 1999, 40, 57–70. [Google Scholar] [CrossRef]
- Xu, Y.; Miladinov, V.; Hanna, M. Synthesis and Characterization of Starch Acetates with High Substitution. Cereal Chem. J. 2004, 81, 735–740. [Google Scholar] [CrossRef]
- Shogren, R.L. Scandium triflate catalyzed acetylation of starch at low to moderate temperatures. Carbohydr. Polym. 2008, 72, 439–443. [Google Scholar] [CrossRef]
- Hong, J.; Zeng, X.; Buckow, R.; Han, Z.; Wang, M.-S. Nanostructure, morphology and functionality of cassava starch after pulsed electric fields assisted acetylation. Food Hydrocoll. 2016, 54, 139–150. [Google Scholar] [CrossRef]
- Gilet, A. Développement de résines thermoplastiques pour fonctionnalisation de matières premières amyglacées. Ph.D. Thesis, Université d’Artois, Arras, France, 2016. [Google Scholar]
- Quettier, C. Procédé de préparation de dérivés acétylés de matière amylacée. International Patent Application No. PCT/FR2010/052147, 11 October 2010. [Google Scholar]
- Bien, F.; Wiege, B.; Warwel, S. Hydrophobic Modification of Starch by Alkali-Catalyzed Addition of 1,2-Epoxyalkanes. Starch Stärke 2001, 53, 555–559. [Google Scholar] [CrossRef]
- Funke, U.; Lindhauer, M.G. Effect of Reaction Conditions and Alkyl Chain Lengths on the Properties of Hydroxyalkyl Starch Ethers. Starch Stärke 2001, 53, 547. [Google Scholar] [CrossRef]
- Gilet, A.; Quettier, C.; Wiatz, V.; Bricout, H.; Ferreira, M.; Rousseau, C.; Monflier, E.; Tilloy, S. Synthesis of 2-Hydroxydodecyl Starch Ethers: Importance of the Purification Process. Ind. Eng. Chem. Res. 2018, 58, 2437–2444. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | |||
|---|---|---|---|
| WMS | 60.0 | 282 | 1.12 |
| AWMS | 52.0 | 283 | 1.16 |
| Reference | Solubilization Recovery (%) | Elution Recovery (%) | |||
|---|---|---|---|---|---|
| POS | 100 | 70 | 10.0 | 179 | 4.79 |
| PES | 89 | 100 | 20.4 | 219 | 3.49 |
| APOS | 100 | 40 | 1.5 | 104 | 1.35 |
| APES | 100 | 85 | 5.1 | 181 | 5.71 |
| Starch Ethers | DSHDo.liqNMR a (DMSO-d6) | DSHDo.EA(C) b | DSHDo.EA(H) c | DSHDo.liqNMR d (CDCl3 and THF-d8) | DSHPhe.liqNMR e (CDCl3 and THF-d8) |
|---|---|---|---|---|---|
| HDo-POS-1 | 0.40 ± 0.05 | 0.39 ± 0.05 | 0.41 ± 0.06 | ND f | ND f |
| HDo-POS-2 | ND g | 1.60 ± 0.09 | 1.58 ± 0.11 | ND f | ND f |
| HDo-HPhe-POS-1 | ND g | - | - | 1.45 ± 0.07 | 0.10 ± 0.02 |
| HDo-HPhe-POS-2 | ND g | - | - | 1.67 ± 0.04 | 0.19 ± 0.03 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Volant, C.; Gilet, A.; Beddiaf, F.; Collinet-Fressancourt, M.; Falourd, X.; Descamps, N.; Wiatz, V.; Bricout, H.; Tilloy, S.; Monflier, E.; et al. Multiscale Structure of Starches Grafted with Hydrophobic Groups: A New Analytical Strategy. Molecules 2020, 25, 2827. https://doi.org/10.3390/molecules25122827
Volant C, Gilet A, Beddiaf F, Collinet-Fressancourt M, Falourd X, Descamps N, Wiatz V, Bricout H, Tilloy S, Monflier E, et al. Multiscale Structure of Starches Grafted with Hydrophobic Groups: A New Analytical Strategy. Molecules. 2020; 25(12):2827. https://doi.org/10.3390/molecules25122827
Chicago/Turabian StyleVolant, Chloé, Alexandre Gilet, Fatima Beddiaf, Marion Collinet-Fressancourt, Xavier Falourd, Nicolas Descamps, Vincent Wiatz, Hervé Bricout, Sébastien Tilloy, Eric Monflier, and et al. 2020. "Multiscale Structure of Starches Grafted with Hydrophobic Groups: A New Analytical Strategy" Molecules 25, no. 12: 2827. https://doi.org/10.3390/molecules25122827
APA StyleVolant, C., Gilet, A., Beddiaf, F., Collinet-Fressancourt, M., Falourd, X., Descamps, N., Wiatz, V., Bricout, H., Tilloy, S., Monflier, E., Quettier, C., Mazzah, A., & Rolland-Sabaté, A. (2020). Multiscale Structure of Starches Grafted with Hydrophobic Groups: A New Analytical Strategy. Molecules, 25(12), 2827. https://doi.org/10.3390/molecules25122827