
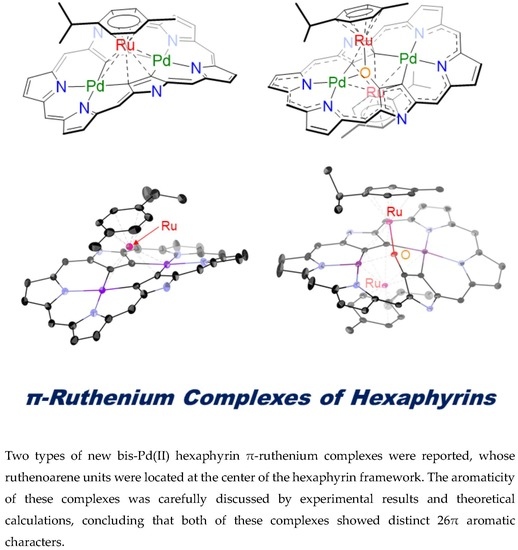
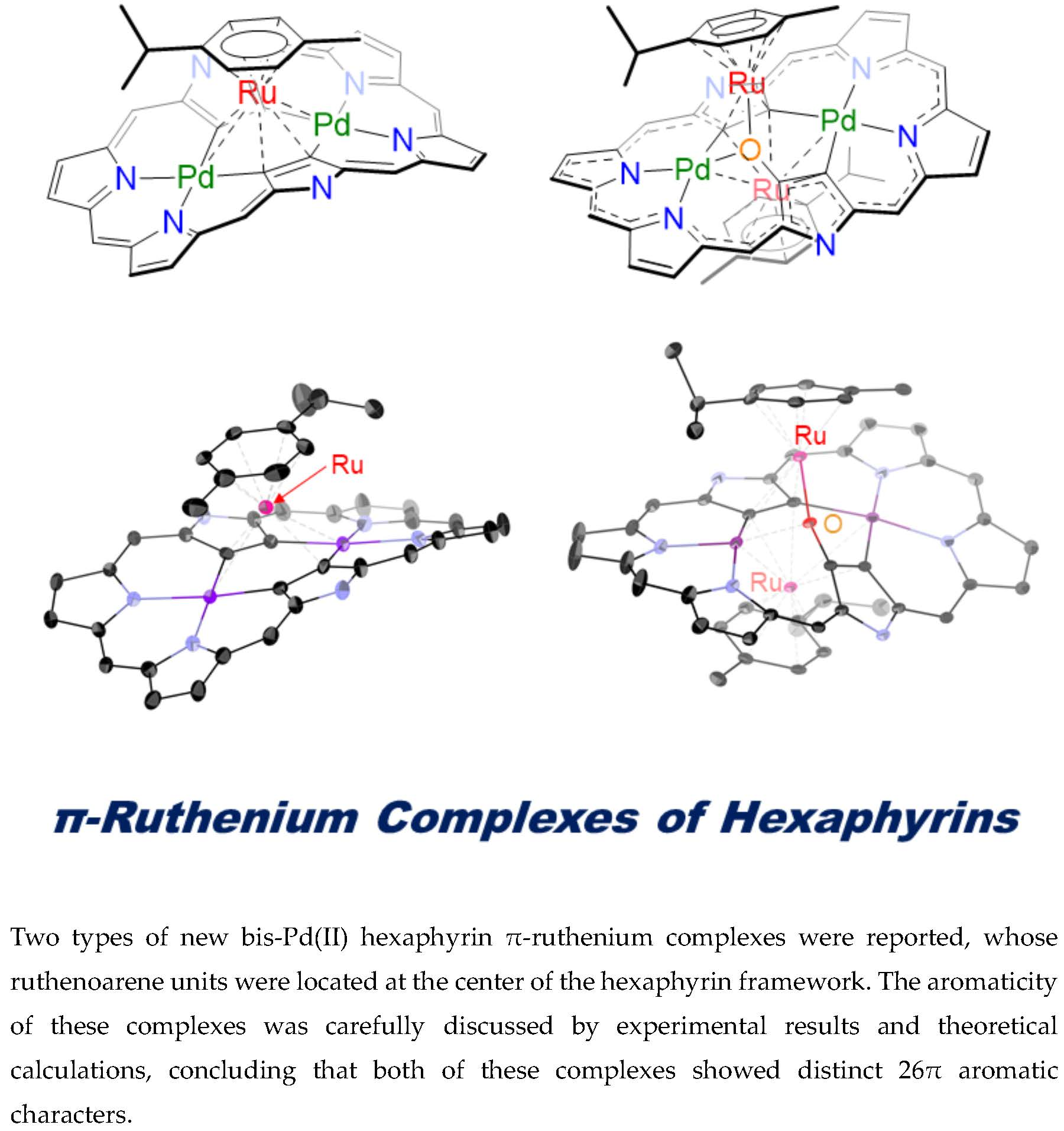
Oxidation-Induced Detachment of Ruthenoarene Units and Oxygen Insertion in Bis-Pd(II) Hexaphyrin π-Ruthenium Complexes


Abstract
:
1. Introduction
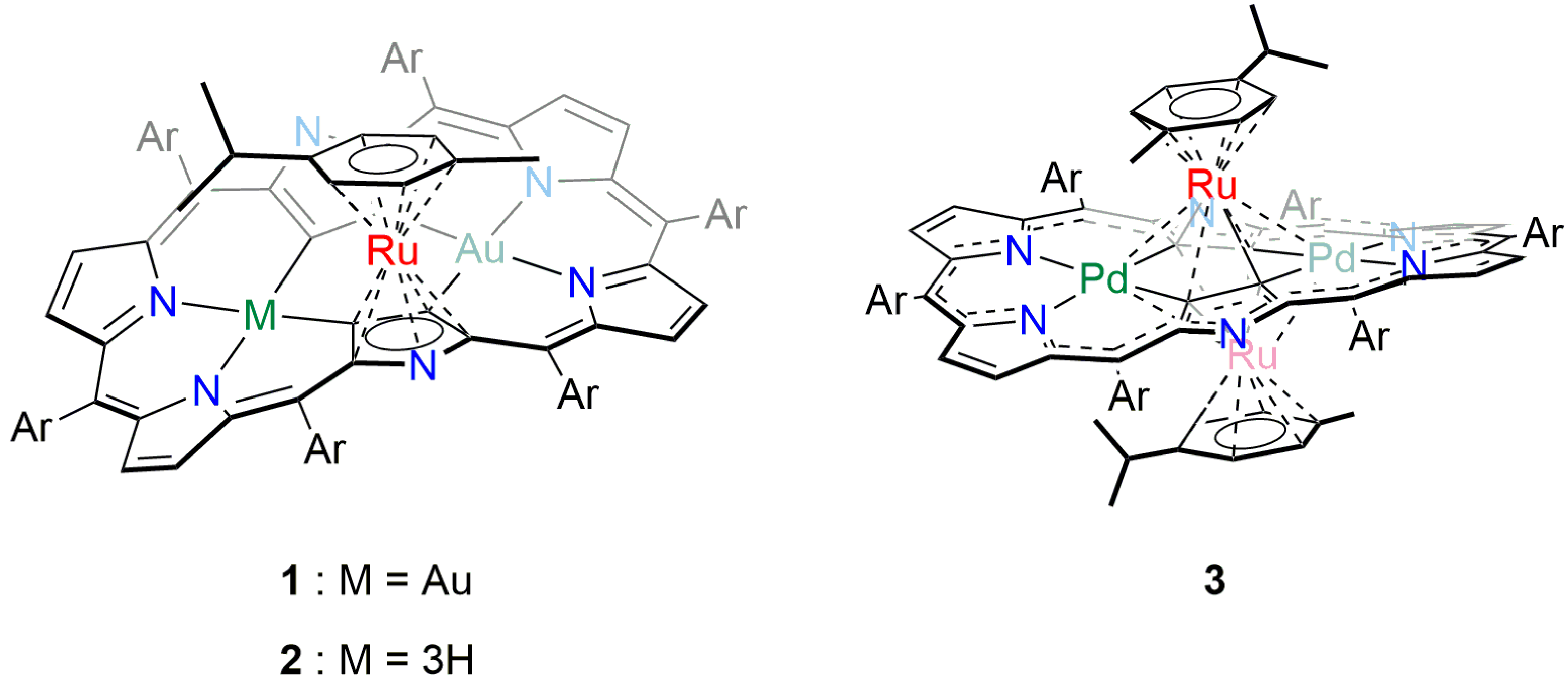
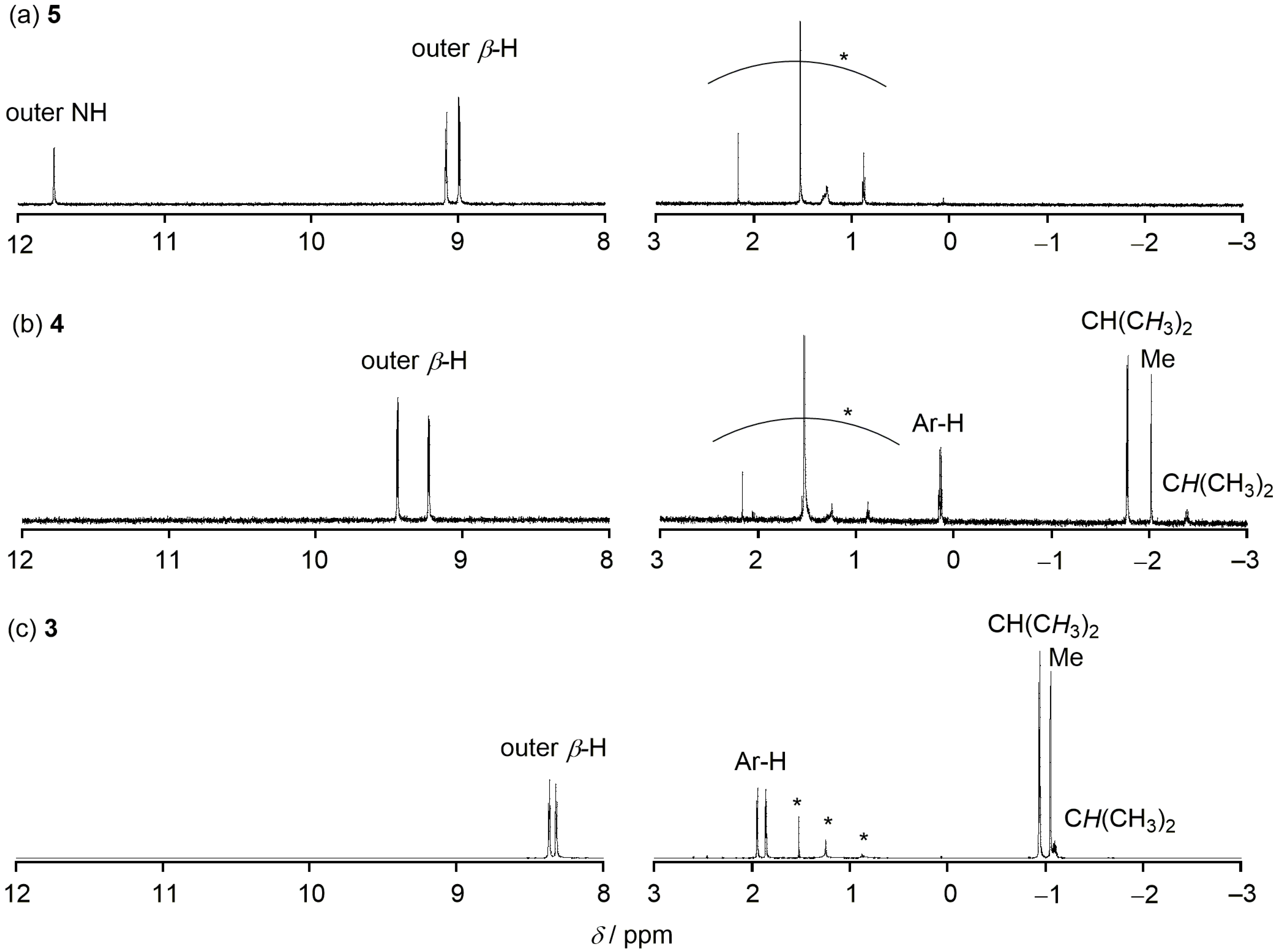
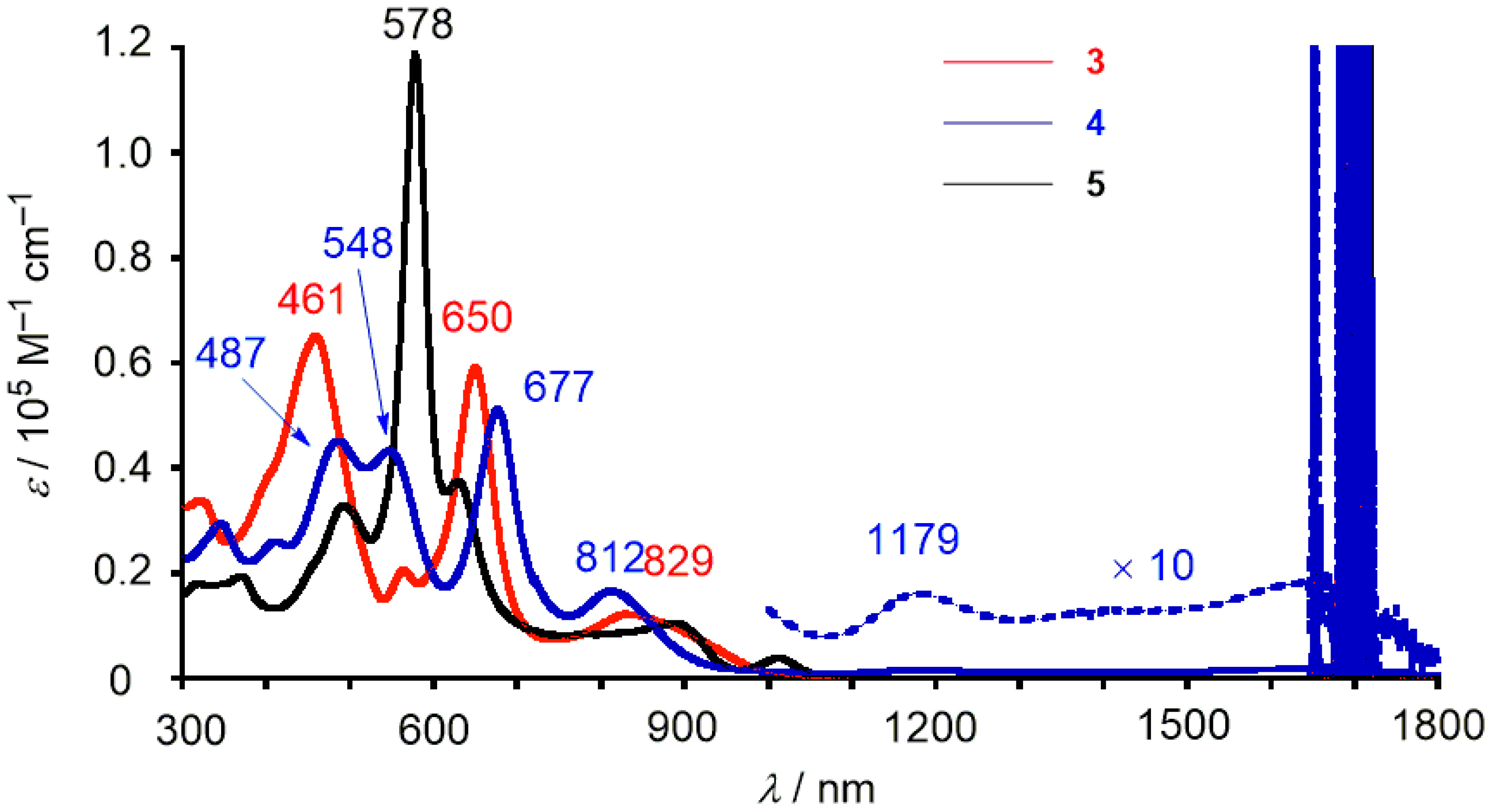
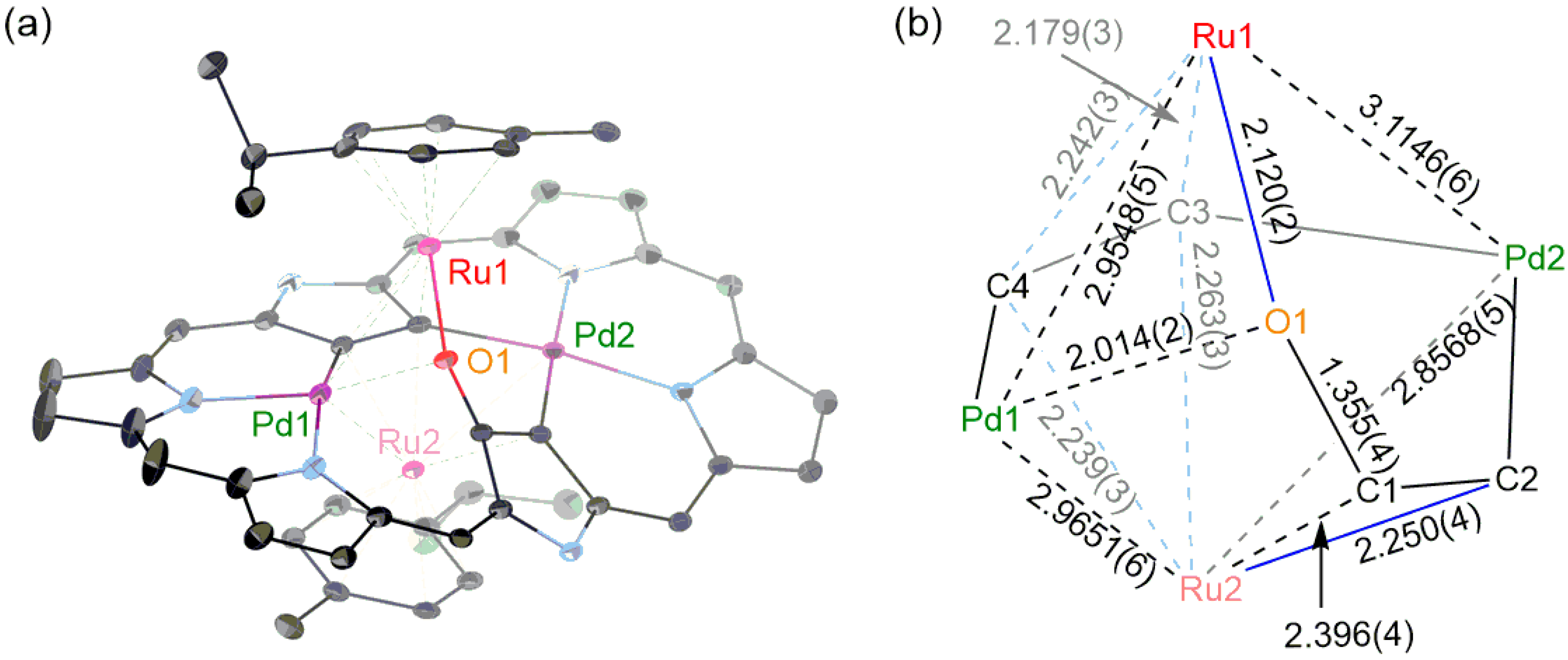
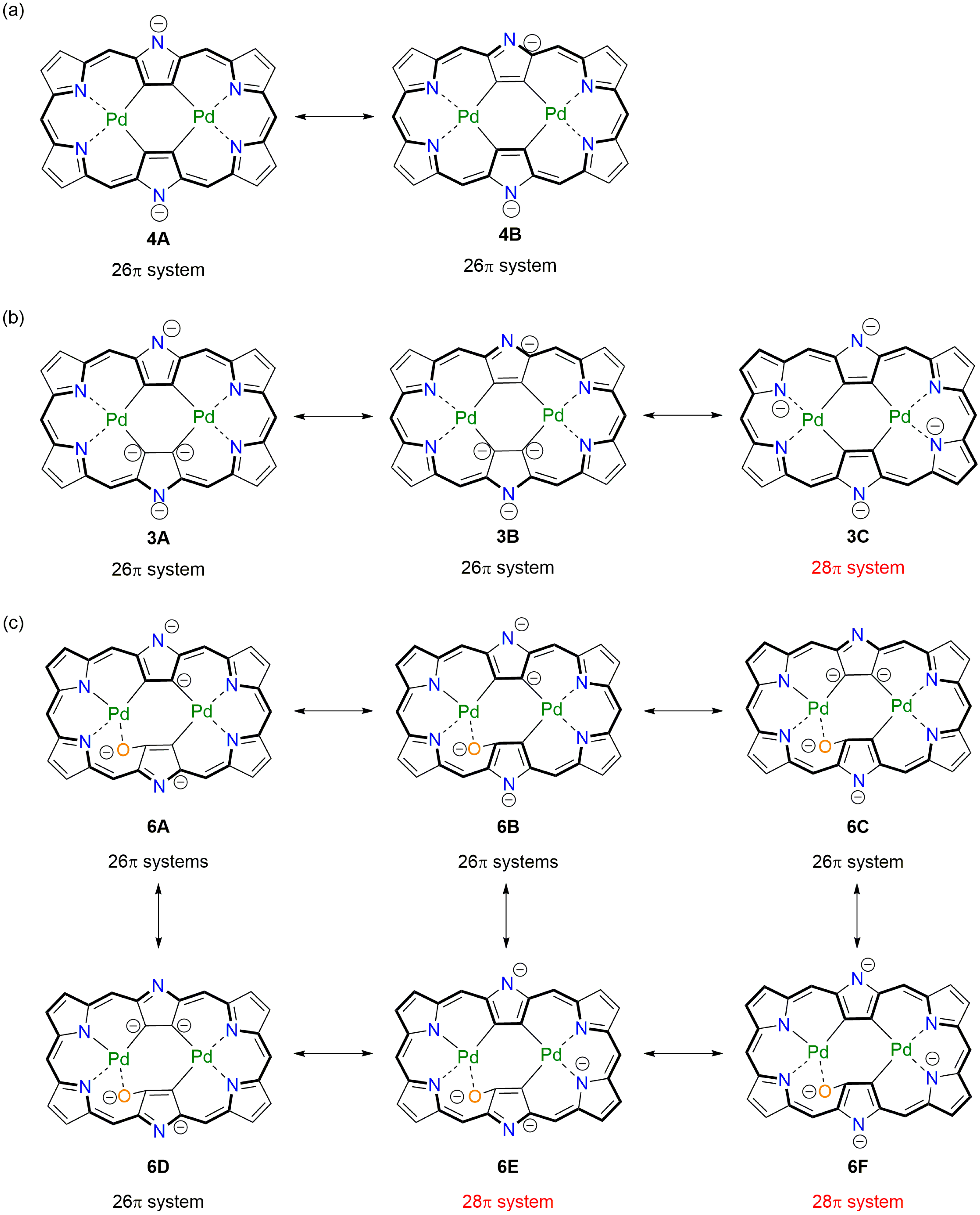
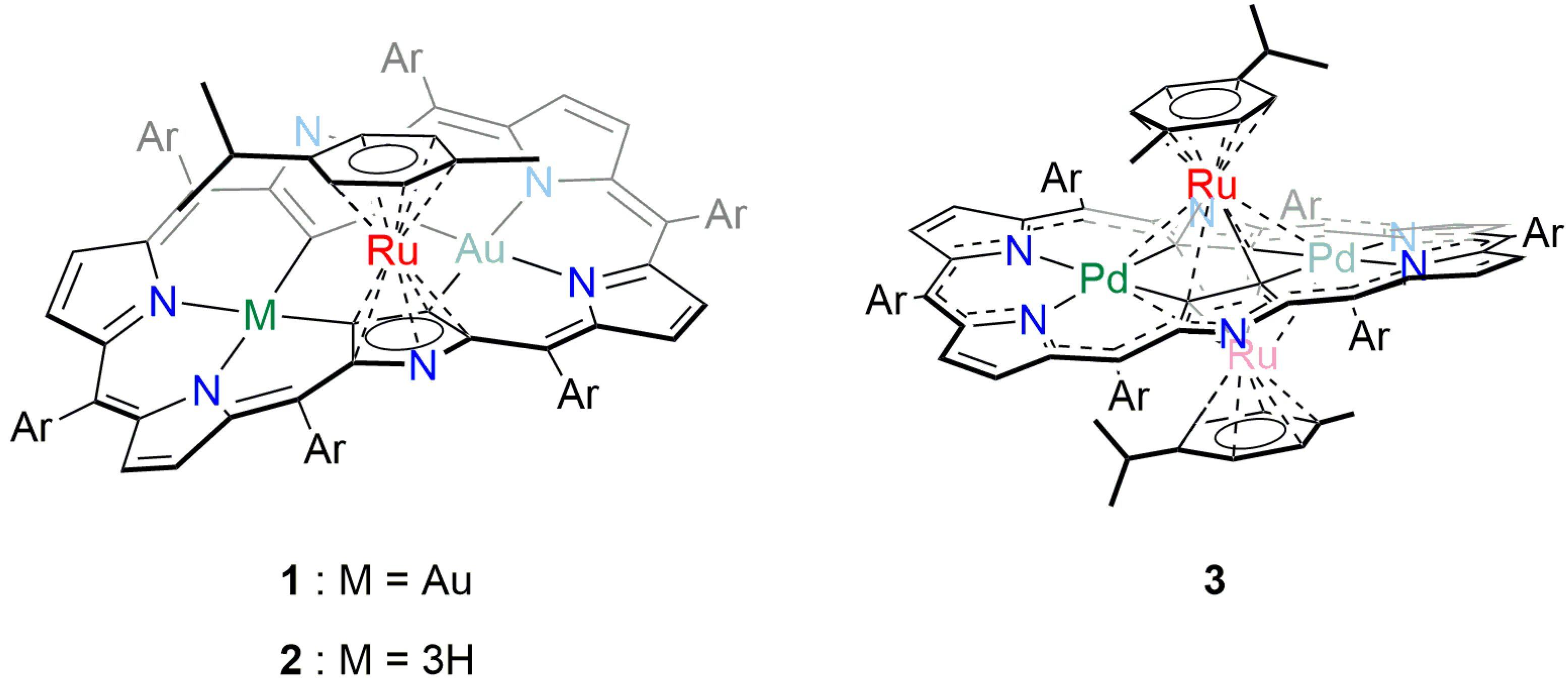
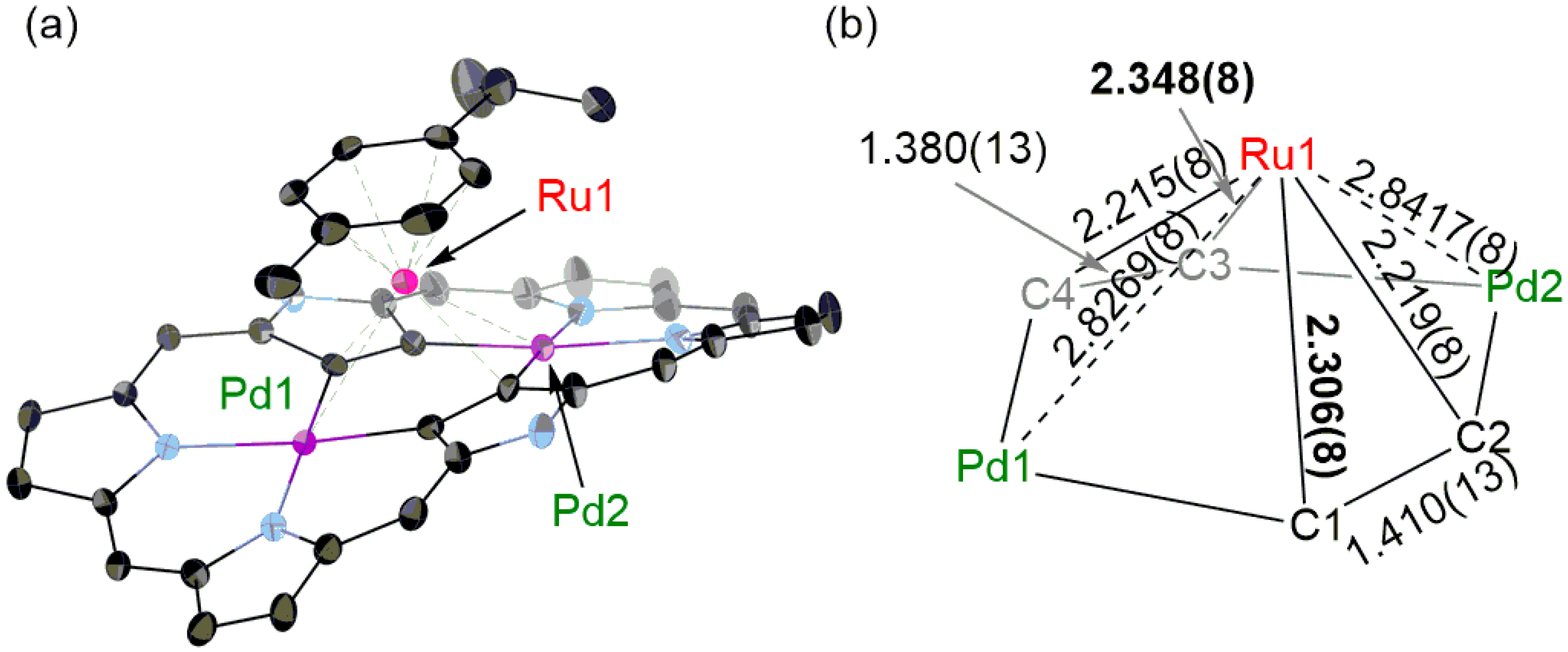
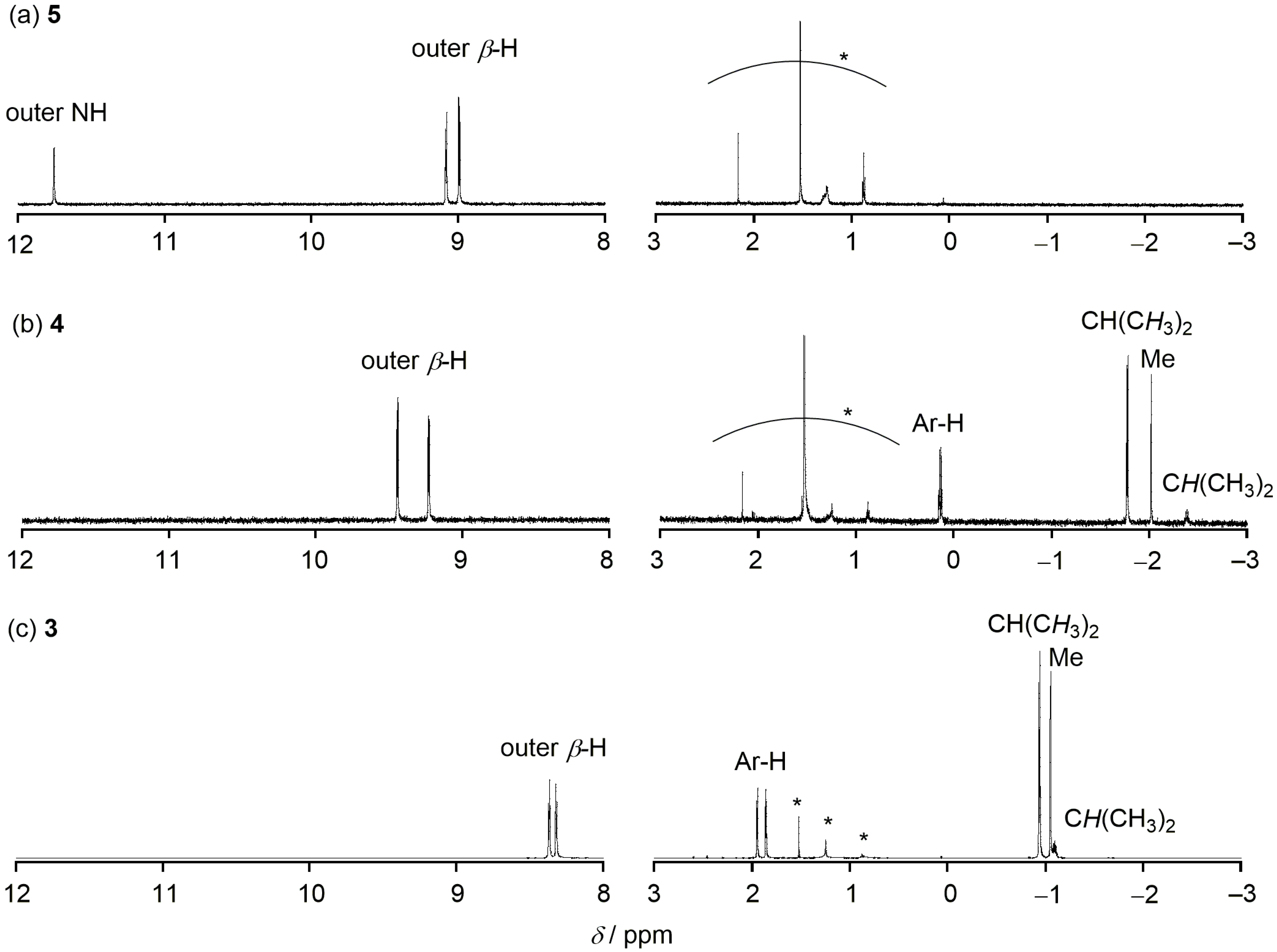
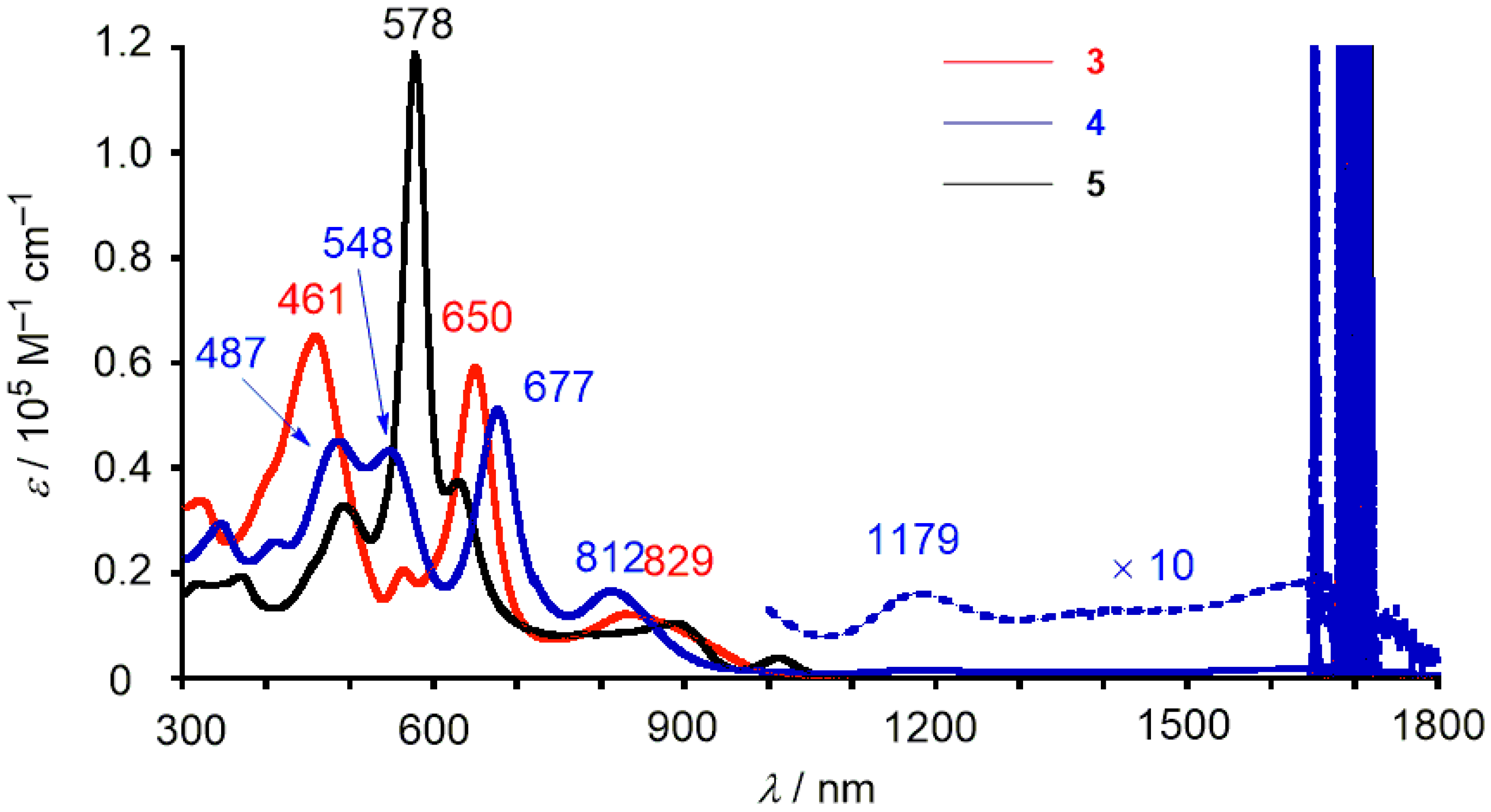
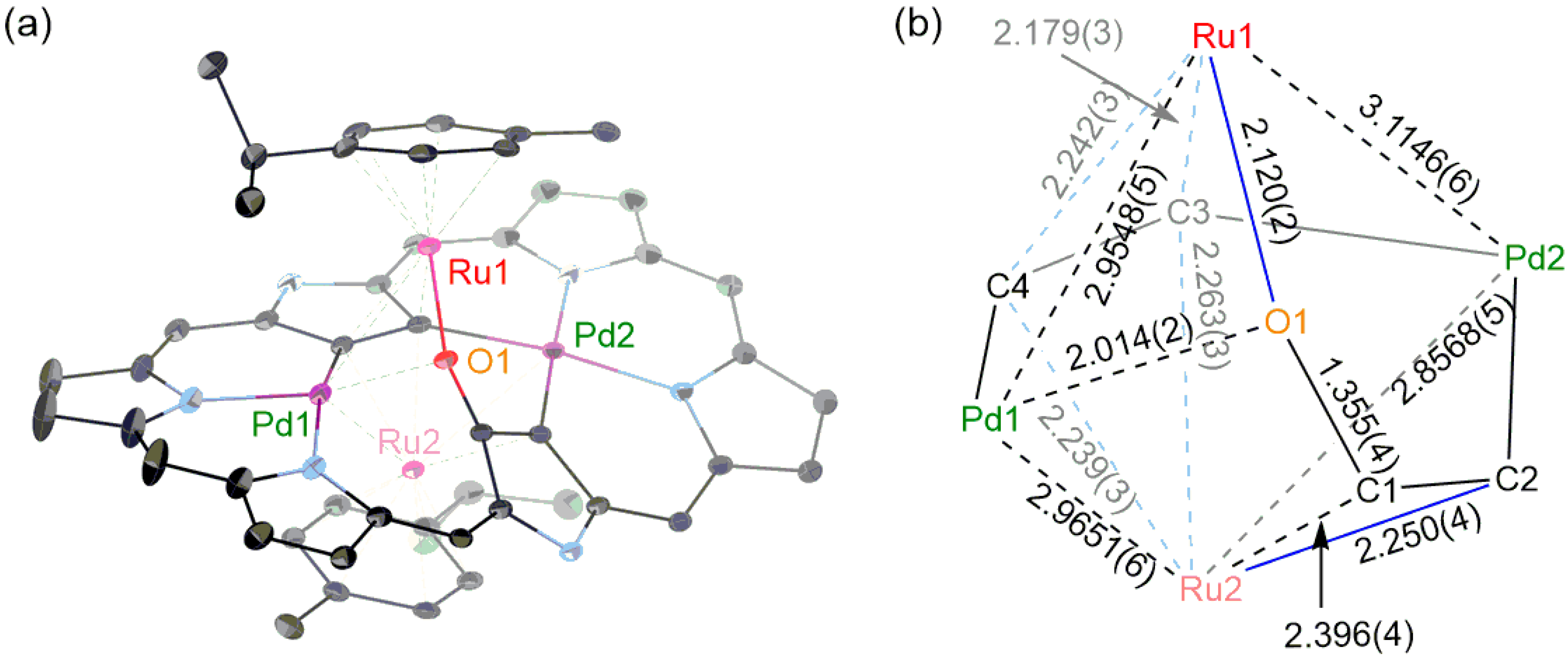
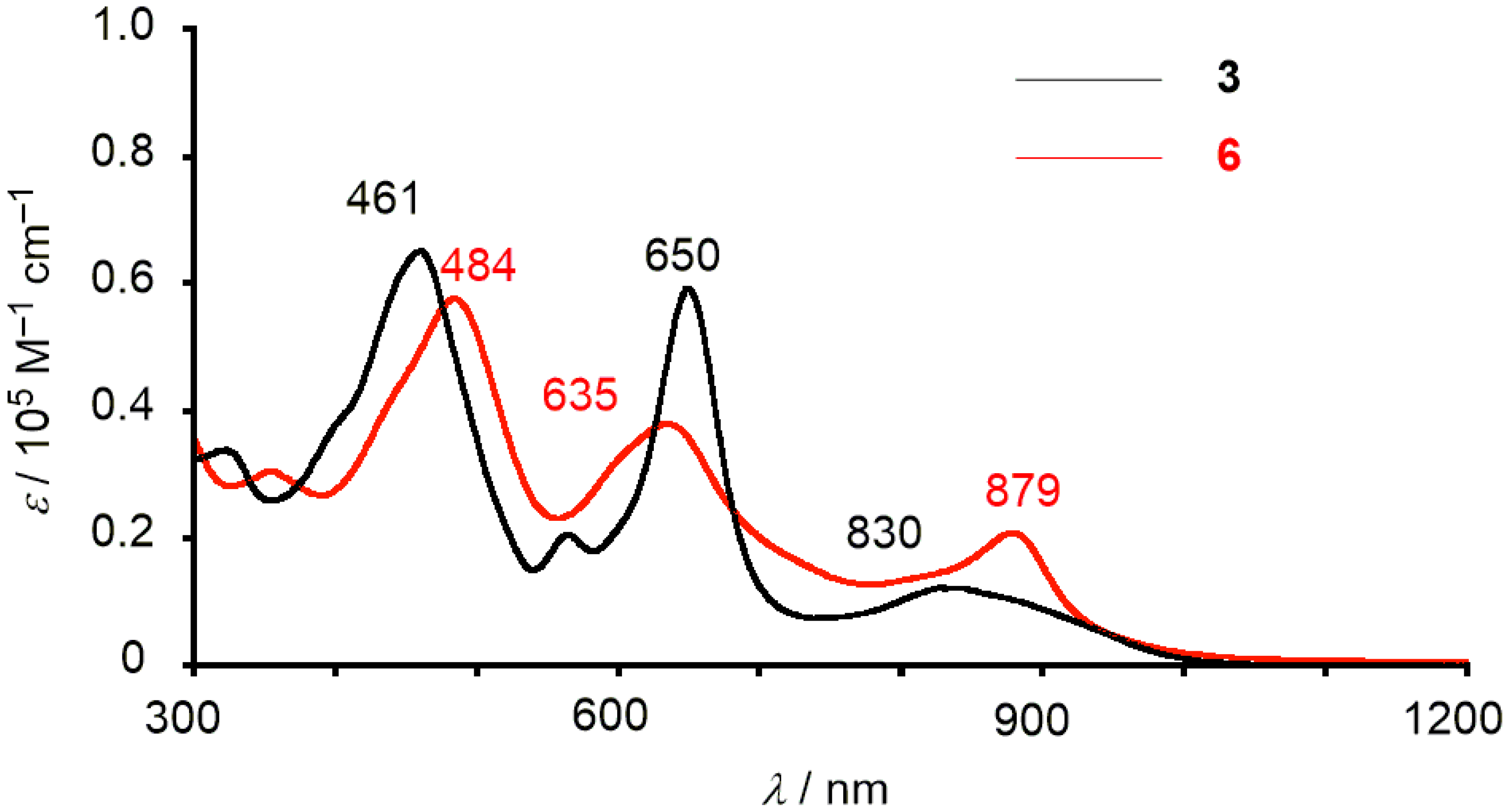
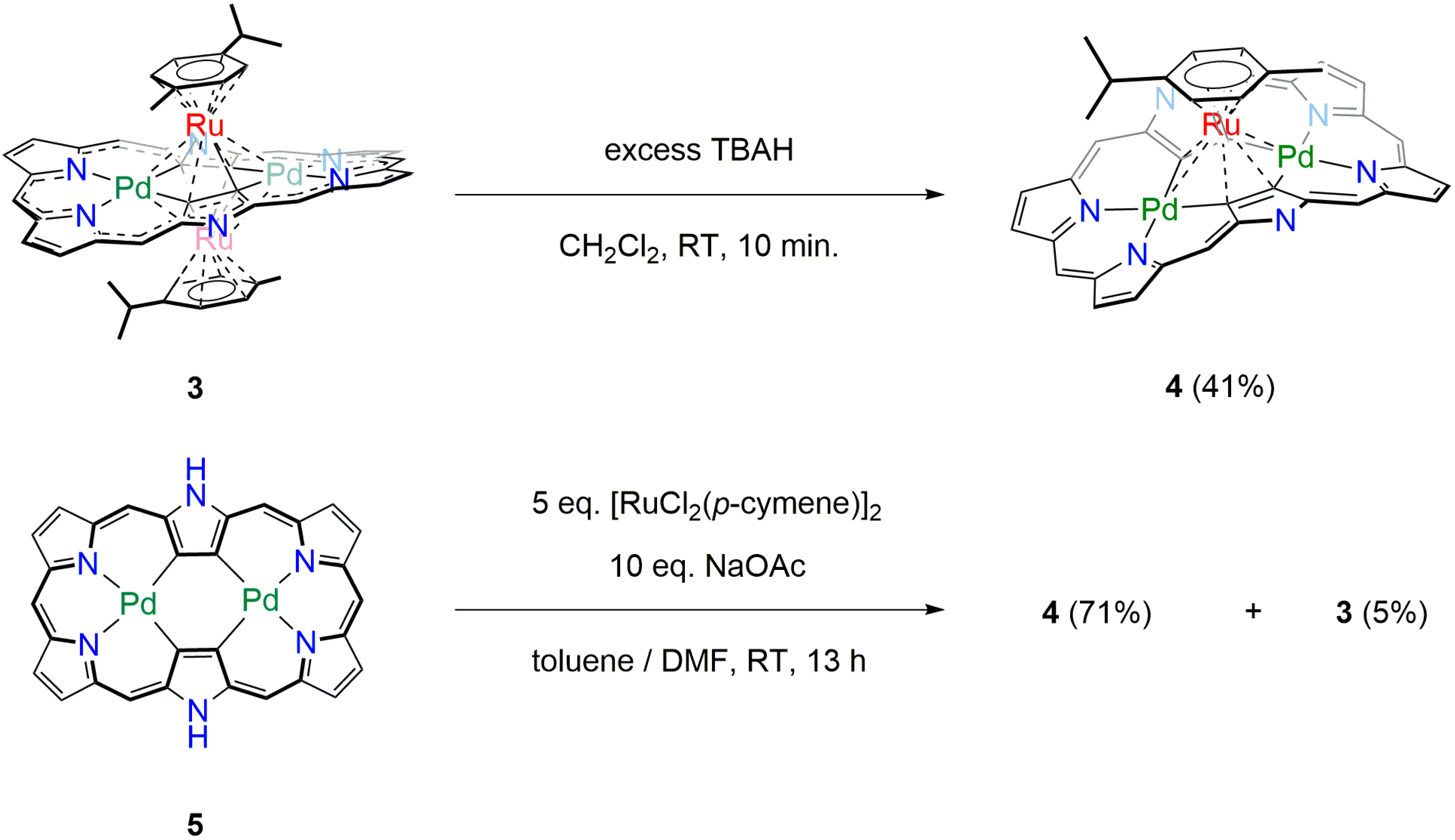
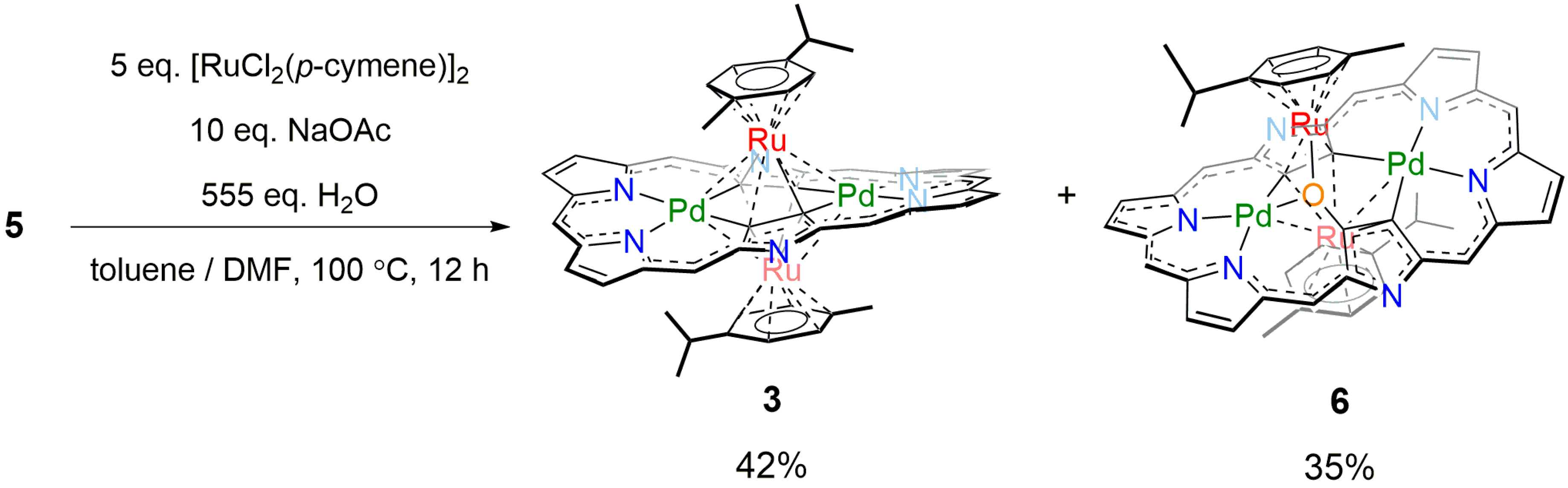
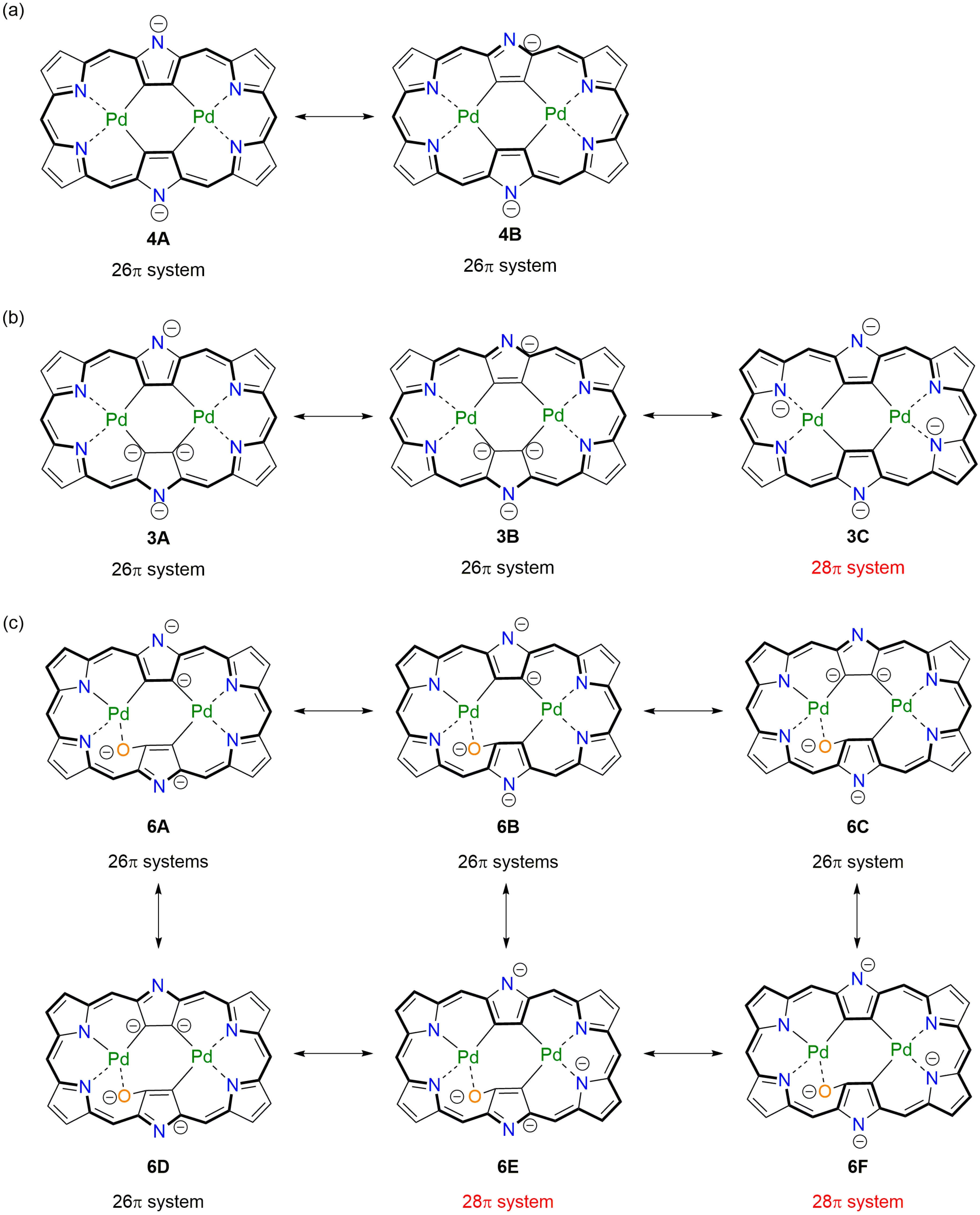
2. Results and Discussion
3. Materials and Methods
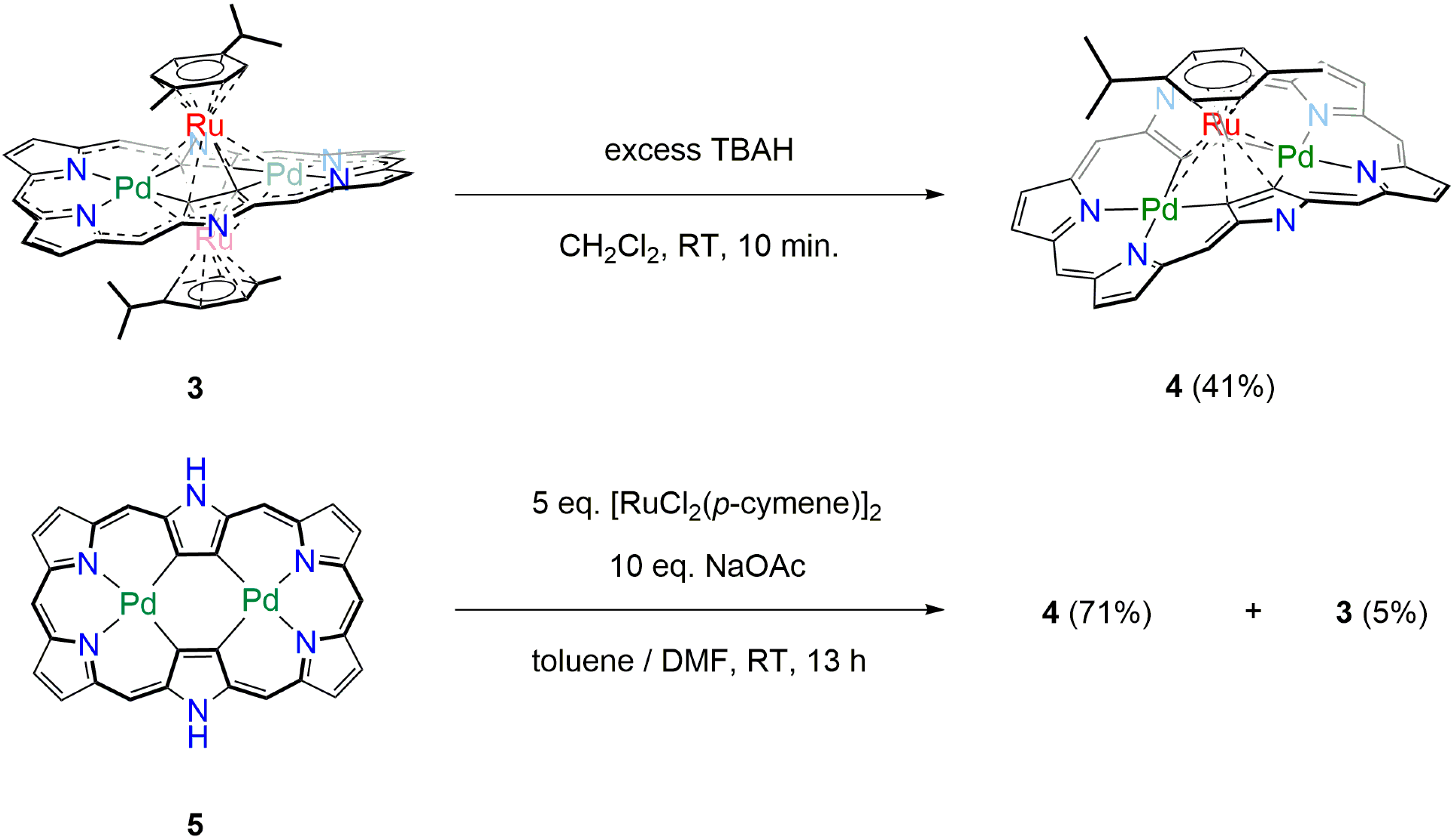
3.1. Oxidation of 3 into [26]Hexaphyrin Bis-Pd(II)-Mono-Ru(II) Complex 4
3.2. Direct Synthesis of [26]Hexaphyrin Bis-Pd(II)-Mono-Ru(II) Complex 4
3.3. [26]Hexaphyrin Bis-Pd(II)-Bis-Ru(II)-Oxygen Inserted Complex 6
3.4. Synthesis of [26]Hexaphyrin Bis-Pd(II)-Bis-Ru(II)-Oxygen Inserted Complex 6 under Argon Atmosphere
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sessler, J.L.; Seidel, D. Synthetic Expanded Porphyrin Chemistry. Angew. Chem. Int. Ed. 2003, 42, 5134–5175. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, T.K.; Venkatraman, S. Core-Modified Expanded Porphyrins: New Generation Organic Materials. Acc. Chem. Res. 2003, 36, 676–691. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Osuka, A. Metalation Chemistry of meso-Aryl-Substituted Expanded Porphyrins. Eur. J. Inorg. Chem. 2006, 1319–1335. [Google Scholar] [CrossRef]
- Stępień, M.; Sprutta, N.; Latos-Grażyński, L. Figure Eights, Möbius Bands, and More: Conformation and Aromaticity of Porphyrinoids. Angew. Chem. Int. Ed. 2011, 50, 4288–4340. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Osuka, A. Expanded Porphyrins: Intriguing Structures, Electronic Properties, and Reactivities. Angew. Chem. Int. Ed. 2011, 50, 4342–4373. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Osuka, A. Chemistry of meso-Aryl-Substituted Expanded Porphyrins: Aromaticity and Molecular Twist. Chem. Rev. 2017, 117, 2584–2640. [Google Scholar] [CrossRef]
- Szyszko, B.; Białek, M.J.; Pacholska-Dudziak, E.; Latos-Grażyński, L. Flexible Porphyrinoids. Chem. Rev. 2017, 117, 2839–2909. [Google Scholar] [CrossRef]
- Nakai, A.; Ishida, S.-i.; Soya, T.; Osuka, A. π-Ruthenium Complexes of Hexaphyrins(1.1.1.1.1.1): A Triple-Decker Complex Bearing Two Ruthenoarene Units. Angew. Chem. Int. Ed. 2019, 58, 8197–8200. [Google Scholar] [CrossRef]
- Cuesta, L.; Sessler, J.L. π-Metal complexes of tetrapyrrolic systems. A novel coordination mode in “porphyrin-like” chemistry. Chem. Soc. Rev. 2009, 38, 2716–2729. [Google Scholar] [CrossRef]
- Dailey, K.K.; Yap, G.P.A.; Rheingold, A.L.; Rauchfuss, T.B. Metalloporphyrins as Ligands: Synthesis and Characterization of [(η6-cymene)-Ru{η5-Ni(OEP)}]2+. Angew. Chem. Int. Ed. Engl. 1996, 35, 1833–1835. [Google Scholar] [CrossRef]
- Dailey, K.K.; Rauchfuss, T.B. π-Complexes of metalloporphyrins as model intermediates in hydrodemetallation (HDM) catalysis. Polyhedron 1997, 16, 3129–3138. [Google Scholar] [CrossRef]
- Cuesta, L.; Karnas, E.; Lynch, V.M.; Sessler, J.L.; Kajonkijya, W.; Zhu, W.; Zhang, M.; Ou, Z.; Kadish, K.M.; Ohkubo, K.; et al. (Pentamethylcyclopentadienyl)ruthenium π-Complexes of Metalloporphyrins: Platforms with Novel Photo- and Electrochemical Properties. Chem. Eur. J. 2008, 14, 10206–10210. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, L.; Karnas, E.; Lynch, V.M.; Chen, P.; Shen, J.; Kadish, K.M.; Ohkubo, K.; Fukuzumi, S.; Sessler, J.L. Metalloporphycenes: Synthesis and Characterization of (Pentamethylcyclopentadienyl)ruthenium Sitting-Atop and π-Complexes. J. Am. Chem. Soc. 2009, 131, 13538–13547. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Zúñigaa, G.I.; Roznyatovskiy, V.V.; Nepomnyaschii, A.; Lynch, V.M.; Sessler, J.L. π-Metal complexes of i-propyldinaphthoporphycene. J. Porphyrins Phthalocyanines 2012, 16, 479–487. [Google Scholar] [CrossRef]
- Caballero, E.; Fernández-Ariza, J.; Lynch, V.M.; Romero-Nieto, C.; Rodríguez-Morgade, M.S.; Sessler, J.L.; Guldi, D.M.; Torres, T. Cyclopentadienylruthenium π Complexes of Subphthalocyanines: A “Drop-Pin” Approach To Modifying the Electronic Features of Aromatic Macrocycles. Angew. Chem. Int. Ed. 2012, 51, 11337–11342. [Google Scholar] [CrossRef]
- Caballero, E.; Romero-Nieto, C.; Strauβ, V.; Rodríguez-Morgade, M.S.; Guldi, D.M.; Sessler, J.L.; Torres, T. Ruthenoarenes versus Phenol Derivatives as Axial Linkers for Subporphyrazine Dimers and Trimers. Chem. Eur. J. 2014, 20, 6518–6525. [Google Scholar] [CrossRef] [PubMed]
- Perekalin, D.S.; Kudinov, A.P. Cyclopentadienyl ruthenium complexes with naphthalene and other polycyclic aromatic ligands. Coord. Chem. Rev. 2014, 276, 153–173. [Google Scholar] [CrossRef]
- Kadish, K.M.; Caemelbecke, E.V.; Royal, G. Electrochemistry of Metalloporphyrins in Nonaqueous Media in The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: Boston, MA, USA, 2000; Volume 8, Chapter 55; pp. 1–260. [Google Scholar]
- Yoneda, T.; Osuka, A. Synthesis of a [26]Hexaphyrin Bis-PdII Complex with a Characteristic Aromatic Circuit. Chem. Eur. J. 2013, 19, 7314–7318. [Google Scholar] [CrossRef]
- Batsanov, S.S. Van der Waals Radii of Elements. Inorg. Mater. 2001, 37, 871–885. [Google Scholar] [CrossRef]
- DFT Calculations Were Performed at the B3LYP/6-31G(d) (C, H, O, N, F) + LANL2DZ (Pd, Ru) Level, Using the Gaussian 16 Program; Gaussian Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Suzuki, M.; Osuka, A. Reversible caterpillar-motion like isomerization in a N,N′-dimethyl hexaphyrin(1.1.1.1.1.1) induced by two-electron oxidation or reduction. Chem. Commun. 2005, 3685–3687. [Google Scholar] [CrossRef]
- Cha, W.-Y.; Lim, J.M.; Yoon, M.-C.; Sung, Y.M.; Lee, B.S.; Katsumata, S.; Suzuki, M.; Mori, H.; Ikawa, H.; Furuta, H.; et al. Deprotonation-Induced Aromaticity Enhancement and New Conjugated Networks in meso-Hexakis(pentafluorophenyl)[26]hexaphyrin. Chem. Eur. J. 2012, 18, 15838–15844. [Google Scholar] [CrossRef] [PubMed]
- Krygowski, T.M.; Szatylowicz, H.; Stasyuk, O.A.; Dominikowska, J.; Palusiak, M. Aromaticity from the Viewpoint of Molecular Geometry: Application to Planar Systems. Chem. Rev. 2014, 114, 6383–6422. [Google Scholar] [CrossRef]
- Tanaka, T.; Aratani, N.; Lim, J.M.; Kim, K.S.; Kim, D.; Osuka, A. Porphyrin-hexaphyrin hybrid tapes. Chem. Sci. 2011, 2, 1414–1418. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M.; Schneider, T.R. SHELXL: High-resolution refinement. Methods Enzymol. 1997, 277, 319–343. [Google Scholar] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Herges, R.; Geuenich, D. Delocalization of Electrons in Molecules. J. Phys. Chem. A 2001, 105, 3214–3220. [Google Scholar] [CrossRef]
- Geuenich, D.; Hess, K.; Köhler, F.; Herges, R. Anisotropy of the Induced Current Density (ACID), a General Method To Quantify and Visualize Electronic Delocalization. Chem. Rev. 2005, 105, 3758–3772. [Google Scholar] [CrossRef]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO Version 3.1; Gaussian Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
Sample Availability: Samples of the compounds 4 and 6 are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Eox.2 [V] | Eox.1 [V] | Ered.1 [V] | Ered.2 [V] | ΔE [eV] | |
|---|---|---|---|---|---|
| 3 [8] | 0.66 | 0.22 | −1.29 | 1.51 | |
| 4 | 0.81 | 0.56 | −0.43 | −1.01 | 0.99 |
| 5 [8] | 0.71 | −0.50 | −0.93 | 1.21 | |
| 6 | 0.56 | 0.17 | −1.08 | −1.62 | 1.25 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakai, A.; Tanaka, T.; Osuka, A. Oxidation-Induced Detachment of Ruthenoarene Units and Oxygen Insertion in Bis-Pd(II) Hexaphyrin π-Ruthenium Complexes. Molecules 2020, 25, 2753. https://doi.org/10.3390/molecules25122753
Nakai A, Tanaka T, Osuka A. Oxidation-Induced Detachment of Ruthenoarene Units and Oxygen Insertion in Bis-Pd(II) Hexaphyrin π-Ruthenium Complexes. Molecules. 2020; 25(12):2753. https://doi.org/10.3390/molecules25122753
Chicago/Turabian StyleNakai, Akito, Takayuki Tanaka, and Atsuhiro Osuka. 2020. "Oxidation-Induced Detachment of Ruthenoarene Units and Oxygen Insertion in Bis-Pd(II) Hexaphyrin π-Ruthenium Complexes" Molecules 25, no. 12: 2753. https://doi.org/10.3390/molecules25122753
APA StyleNakai, A., Tanaka, T., & Osuka, A. (2020). Oxidation-Induced Detachment of Ruthenoarene Units and Oxygen Insertion in Bis-Pd(II) Hexaphyrin π-Ruthenium Complexes. Molecules, 25(12), 2753. https://doi.org/10.3390/molecules25122753