
The Nature of Triel Bonds, a Case of B and Al Centres Bonded with Electron Rich Sites


Abstract
1. Introduction
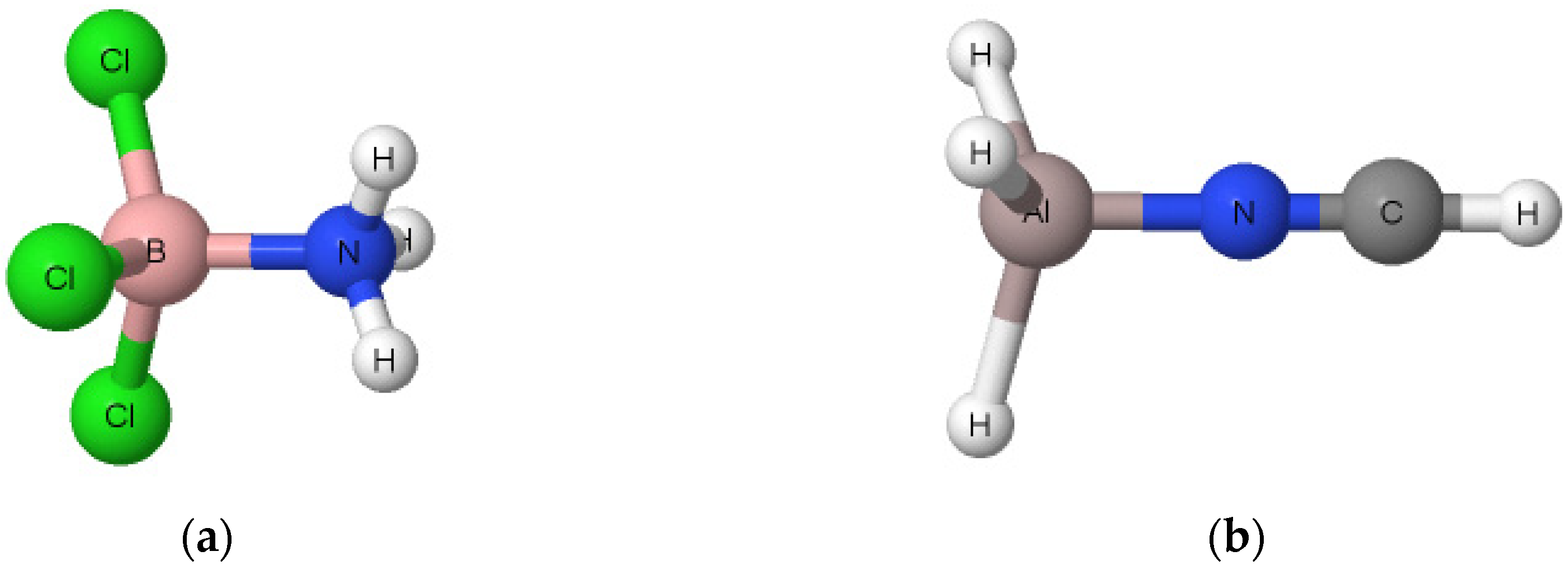
2. Results and Discussion
2.1. The Strength of Triel Bonds
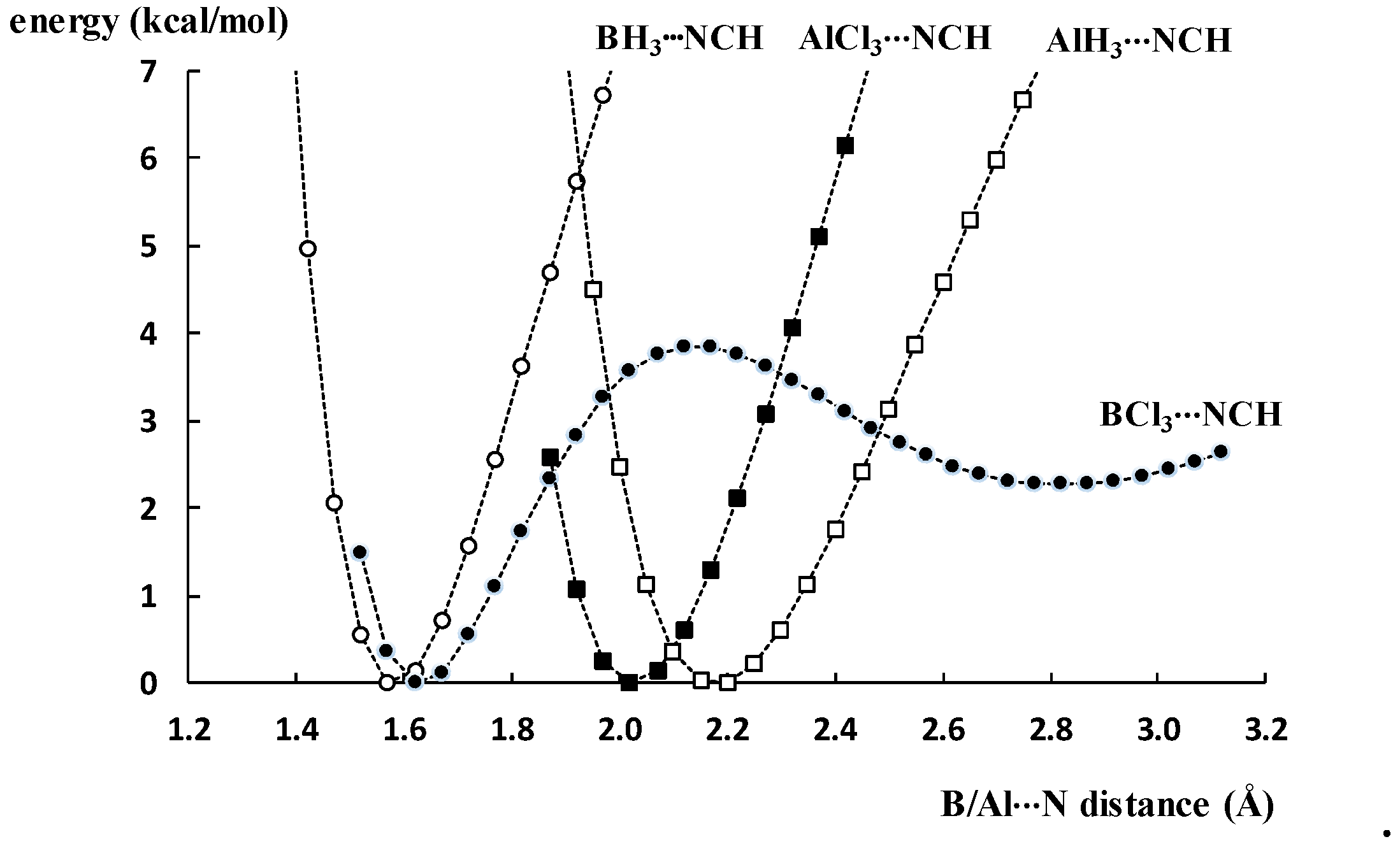
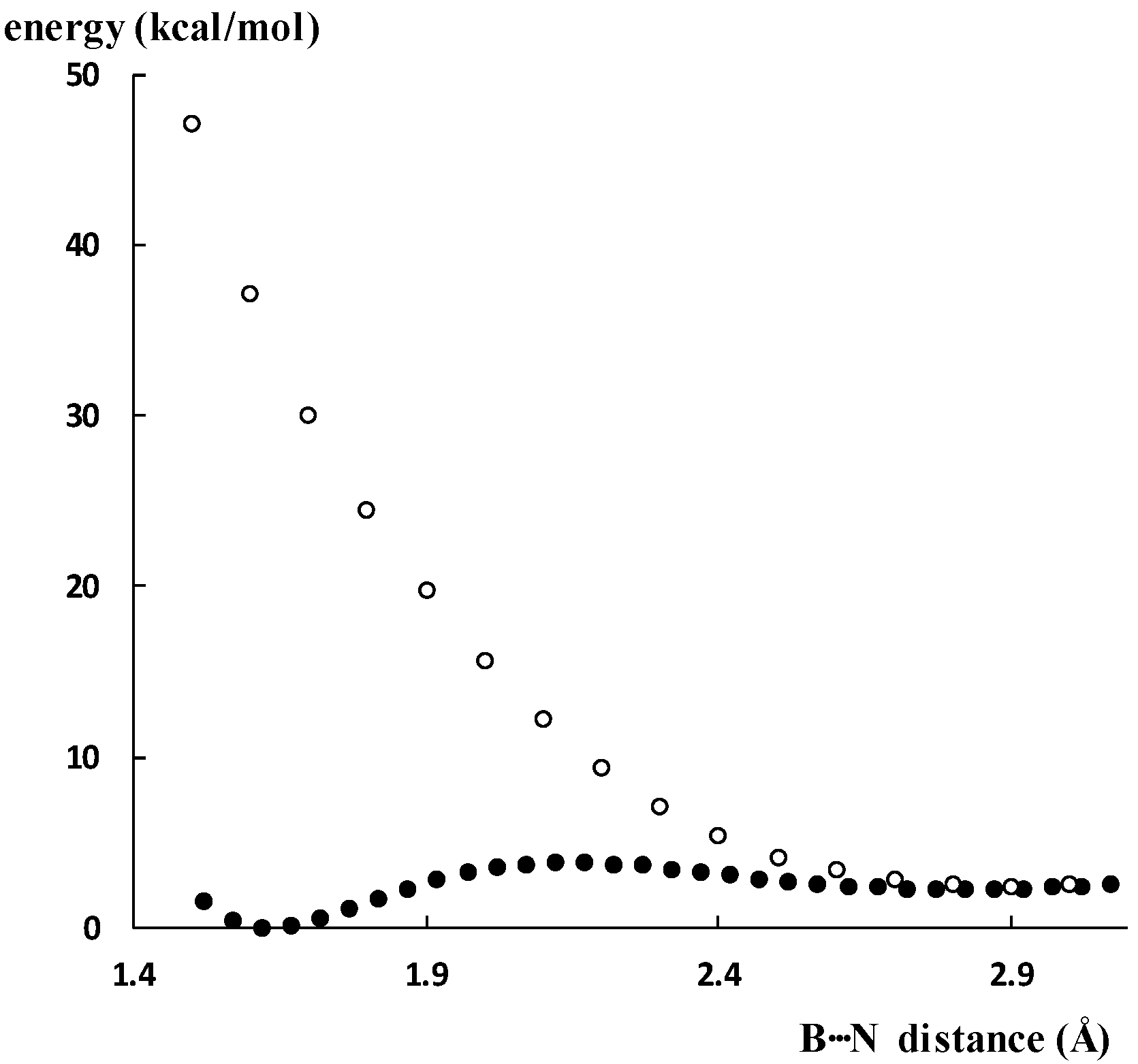
2.2. Double Minima for Triel–Lewis Base Potential Energy Curves
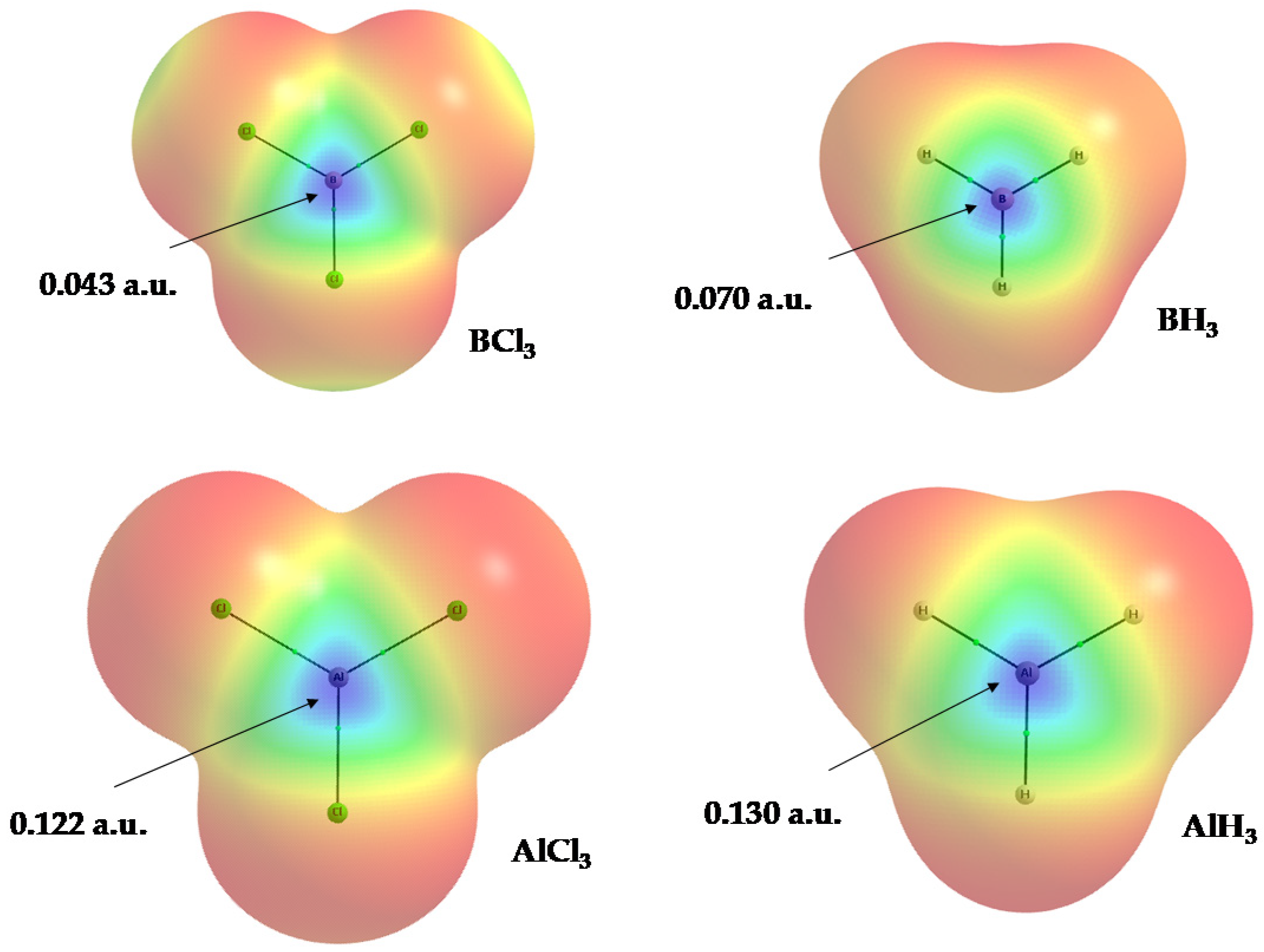
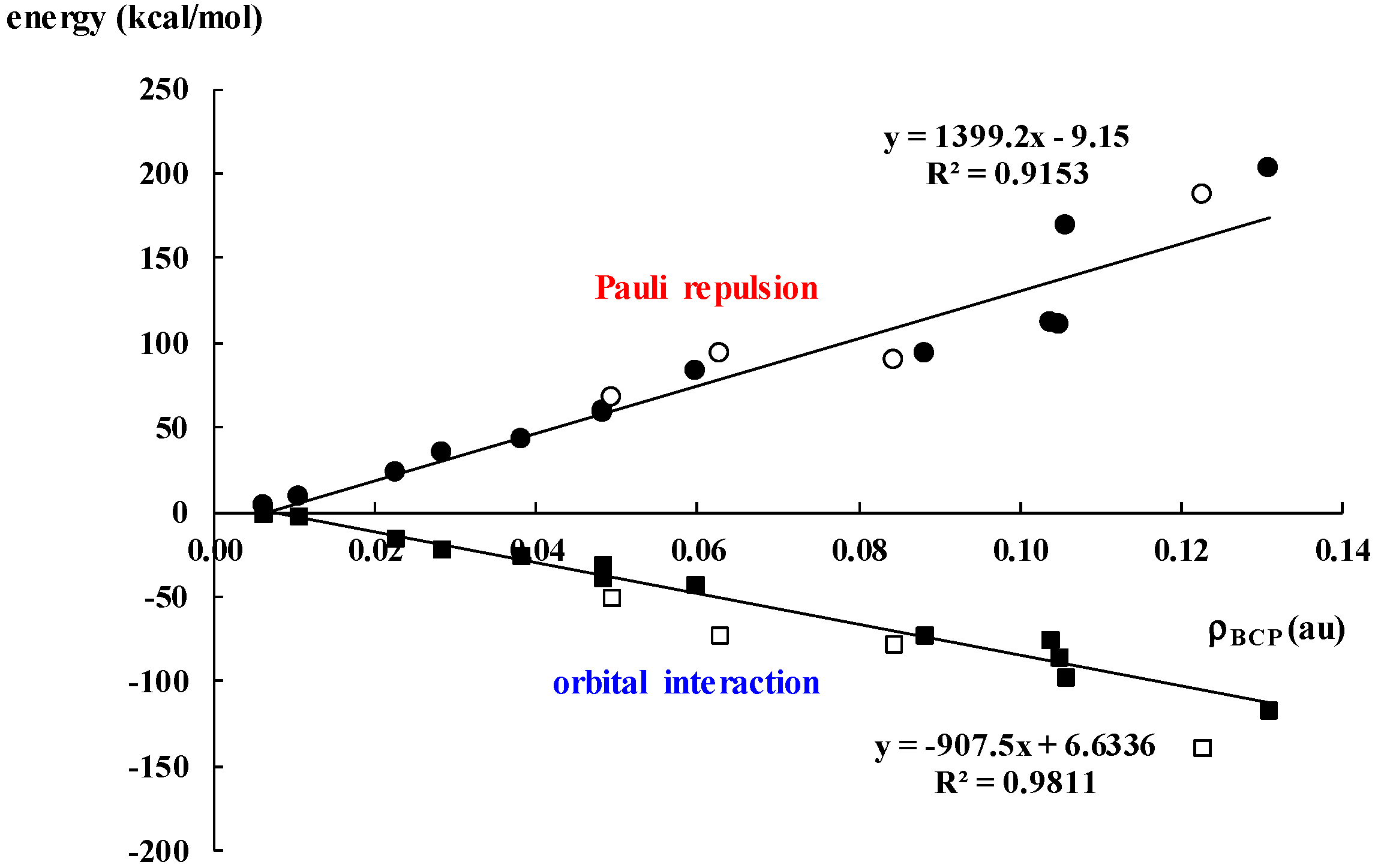
2.3. Covalency of Triel Bonds
3. Conclusions
4. Materials and Methods
Computational Details
Funding
Acknowledgments
Conflicts of Interest
References
- Novoa, J.J. Intermolecular Interactions in Crystals: Fundamentals of Crystal Engineering; The Royal Society of Chemistry: London, UK, 2018. [Google Scholar]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7758. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Detailed Comparison of the Pnicogen Bond with Chalcogen, Halogen, and Hydrogen Bonds. Int. J. Quantum Chem. 2013, 113, 1609–1620. [Google Scholar] [CrossRef]
- Scheiner, S. The Pnicogen Bond: Its Relation to Hydrogen, Halogen, and Other Noncovalent Bonds. Acc. Chem. Res. 2013, 46, 280. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Frontera, A. Aerogen Bonding Interaction: A New Supramolecular Force. Angew. Chem. Int. Ed. 2015, 54, 7340–7343. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen bonds, and σ-hole and π-hole bonds – mechanisms protecting doublet and octet electron structures. Phys. Chem. Chem. Phys. 2017, 19, 29742–29759. [Google Scholar] [CrossRef]
- Grabowski, S.J. Boron and other Triel Lewis Acid Centers: From Hypovalency to Hypervalency. ChemPhysChem 2014, 15, 2985–2993. [Google Scholar] [CrossRef]
- Grabowski, S.J. π-Hole Bonds: Boron and Aluminum Lewis Acid Centers. ChemPhysChem 2015, 16, 1470–1479. [Google Scholar] [CrossRef]
- Grabowski, S.J. Triel Bonds, π-Hole-π-Electrons Interactions in Complexes of Boron and Aluminium Trihalides and Trihydrides with Acetylene and Ethylene. Molecules 2015, 20, 11297–11316. [Google Scholar] [CrossRef]
- Beckett, M.A.; Strickland, G.C.; Holland, J.R.; Varma, K.S. A convenient n.m.r. method for the measurement of Lewis acidity at boron centres: correlation of reaction rates of Lewis acid initiated epoxide polymerizations with Lewis acidity. Polymer 1996, 37, 4629–4631. [Google Scholar] [CrossRef]
- Hiberty, P.C.; Ohanessian, G. Comparison of minimal and extended basis sets in terms of resonant formulas. Application to 1,3 dipoles. J. Am. Chem. Soc. 1982, 104, 66–70. [Google Scholar] [CrossRef]
- Brinck, T.; Murray, J.S.; Politzer, P. A computational analysis of the bonding in boron trifluoride and boron trichloride and their complexes with ammonia. Inorg. Chem. 1993, 32, 2622–2625. [Google Scholar] [CrossRef]
- Kutzelnigg, W. Chemical Bonding in Higher Main Group Elements. Angew. Chem. Int. Ed. 1984, 23, 272–295. [Google Scholar] [CrossRef]
- Rowsell, B.D.; Gillespie, R.J.; Heard, G.L. Ligand Close-Packing and the Lewis Acidity of BF3 and BCl3. Inorg. Chem. 1999, 38, 4659–4662. [Google Scholar] [CrossRef] [PubMed]
- Bessac, F.; Frenking, G. Why Is BCl3 a Stronger Lewis Acid with Respect to String Bases than BF3? Inorg. Chem. 2003, 42, 7990–7994. [Google Scholar] [CrossRef] [PubMed]
- Jonas, V.; Frenking, G.; Reetz, M.T. Comparative Theoretical Study of Lewis Acid-Base Complexes of BH3, BF3, BCl3, AlCl3, and SO2. J. Am. Chem. Soc. 1994, 116, 8741–8753. [Google Scholar] [CrossRef]
- Van der Veken, B.J.; Sluyts, E.J. Reversed Lewis Acidity of Mixed Boron Halides: An Infrared Study of the Van der Waals Complexes of BFxCly with CH3F in Cryosolution. J. Am. Chem. Soc. 1997, 119, 11516–11522. [Google Scholar] [CrossRef]
- Phillips, J.A. Structural and energetic properties of nitrile–BX3 complexes: substituent effects and their impact on condensed-phase sensitivity. Theor. Chem. Acc. 2017, 136, 16. [Google Scholar] [CrossRef]
- Giesen, D.J.; Phillips, J.A. Structure, Bonding, and Vibrational Frequencies of CH3CN-BF3: New Insight into Medium Effects and the Discrepancy between the Experimental and Theoretical Geometries. J. Phys. Chem. A 2003, 107, 4009–4018. [Google Scholar] [CrossRef]
- Phillips, J.A.; Cramer, C.J. B-N Distance Potential of CH3CN-BF3 Revisited: Resolving the Experiment-Theory Structure Discrepancy and Modeling the Effects of Low-Dielectric Environments. J. Phys. Chem. B 2007, 111, 1408–1415. [Google Scholar] [CrossRef] [PubMed]
- Wrass, J.P.; Sadowsky, D.; Bloomgren, K.M.; Cramer, C.J.; Phillips, J.A. Quantum chemical and matrix-IR characterization of CH3CN-BCl3: a complex with two distinct minima along the B-N bond potential. Phys. Chem. Chem. Phys. 2014, 16, 16480–16491. [Google Scholar] [CrossRef]
- Helminiak, H.M.; Knauf, R.R.; Danforth, S.J.; Phillips, J.A. Structural and Energetic Properties of Acetonitrile-Group IV (A & B) Halide Complexes. J. Phys. Chem. A 2014, 118, 4266–4277. [Google Scholar]
- Grabowski, S.J. Triel bond and coordination of triel centres – Comparison with hydrogen bond interaction. Coord. Chem. Rev. 2020, 407, 213171. [Google Scholar] [CrossRef]
- Grabowski, S.J. Triel bonds-complexes of boron and aluminum trihalides and trihydrides with benzene. Struct. Chem. 2017, 28, 1163–1171. [Google Scholar] [CrossRef]
- Grabowski, S.J. Bifurcated Triel Bonds—Hydrides and Halides of 1,2-Bis(Dichloroboryl)Benzene and 1,8-Bis(Dichloroboryl)Naphthalene. Crystals 2019, 9, 503. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Acta Cryst. 2016, B72, 171–179. [Google Scholar]
- Wong, R.; Allen, F.H.; Willett, P. The scientific impact of the Cambridge Structural Database: A citation-based study. J. Appl. Cryst. 2010, 43, 811–824. [Google Scholar] [CrossRef]
- Piela, L. Ideas of Quantum Chemistry; Elsevier Science Publishers: Amsterdam, The Netherlands, 2007; pp. 684–691. [Google Scholar]
- Grabowski, S.J.; Sokalski, W.A. Different types of hydrogen bonds: correlation analysis of interaction energy components. J. Phys. Org. Chem. 2005, 18, 779–784. [Google Scholar] [CrossRef]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–561. [Google Scholar] [CrossRef]
- Grabowski, S.J. Lewis Acid Properties of Tetrel Tetrafluorides—The Coincidence of the σ-Hole Concept with the QTAIM Approach. Crystals 2017, 7, 43. [Google Scholar] [CrossRef]
- Reed, E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C. Valency and Bonding, A Natural Bond Orbital Donor—Acceptor Perspective; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Mantina, M.; Chamberlin, A.C.; Valero, R.; Cramer, C.J.; Truhlar, D.G. Consistent van der Waals Radii for the Whole Main Group. J. Phys. Chem. A 2009, 113, 5806–5812. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen bonds with BF4- anion as a proton acceptor. Crystals 2020, 10, 460. [Google Scholar] [CrossRef]
- Gillespie, R.J.; Hargittai, I. The VSEPR Model of Molecular Geometry; Dover Publications, Inc.: Mineola, NY, USA, 2012. [Google Scholar]
- Grabowski, S.J. Two Faces of Triel Bonds in Boron Trihalide Complexes. J. Comput. Chem. 2018, 39, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules, A Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Matta, C.; Boyd, R.J. (Eds.) Quantum Theory of Atoms in Molecules: Recent Progress in Theory and Application; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Jenkins, S.; Morrison, I. The chemical character of the intermolecular bonds of seven phases of ice as revealed by AB initio calculation of electron densities. Chem. Phys. Lett. 2000, 317, 97–102. [Google Scholar] [CrossRef]
- Arnold, W.D.; Oldfield, E. The Chemical Nature of Hydrogen Bonding in Proteins via NMR: J-Couplings, Chemical Shifts, and AIM Theory. J. Am. Chem. Soc. 2000, 122, 12835–12841. [Google Scholar] [CrossRef]
- Grabowski, S.J. What is the Covalency of Hydrogen Bonding? Chem. Rev. 2011, 11, 2597–2625. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, R.J.; Popelier, P.L.A. Chemical Bonding and Molecular Geometry; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H., Jr. Gaussian Basis Sets for Use in Correlated Molecular Calculations. III. The second row atoms, Al-Ar. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll, Version 13.05.06; TK Gristmill Software: Overland Park, KS, USA, 2013. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Van Lenthe, E.; Baerends, E.J. Optimized Slater-type basis sets for the elements 1–118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. NBO, 6.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2013; Available online: http://nbo6.chem.wisc.edu/ (accessed on 18 May 2020)(this web-side refers nowadays to NBO 7.0 version, all pre-NBO7 versions are cease).
- Ziegler, T.; Rauk, A. CO, CS, N2, PF3, and CNCH3 as σ Donors and π Acceptors. A Theoretical Study by the Hartree-Fock-Slater Transition-State Method. Inorg. Chem. 1979, 18, 1755–1759. [Google Scholar] [CrossRef]
- Velde, G.T.E.; Bickelhaupt, F.M.; Baerends, E.J.; Guerra, C.F.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- ADF2017, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. Available online: http://www.scm.com (accessed on 18 May 2020).
Sample Availability: Samples of the compounds are not available from the author. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Ebin | Eint | BSSE | Edef |
|---|---|---|---|---|
| BCl3⋅⋅⋅NCH a | −3.2 | −3.5 | 0.8 | 0.3 |
| BCl3⋅⋅⋅NCH b | −3.8 | −23.6 | 2.5 | 19.8 |
| BCl3⋅⋅⋅N2 a | −1.5 | −1.5 | 0.6 | 0.0 |
| BCl3⋅⋅⋅NH3 b | −26.7 | −50.4 | 2.7 | 23.6 |
| BCl4− b | −45.6 | −84.9 | 2.8 | 39.2 |
| BH3⋅⋅⋅NCH b | −18.6 | −30.4 | 1.2 | 11.8 |
| BH3⋅⋅⋅N2 b | −6.1 | −14.2 | 1.3 | 8.1 |
| BH3⋅⋅⋅NH3 b | −30.8 | −44.0 | 1.1 | 13.2 |
| BH3Cl− b | −34.1 | −50.9 | 1.7 | 16.8 |
| AlCl3⋅⋅⋅NCH b | −23.8 | −29.5 | 1.5 | 5.7 |
| AlCl3⋅⋅⋅N2 a | −7.0 | −9.0 | 1.3 | 2.0 |
| AlCl3⋅⋅⋅NH3 b | −38.8 | −45.7 | 1.6 | 6.9 |
| AlCl4− b | −76.0 | −98.5 | 1.7 | 22.5 |
| AlH3⋅⋅⋅NCH b | −17.7 | −20.6 | 0.7 | 2.9 |
| AlH3⋅⋅⋅N2 a | −5.8 | −6.5 | 0.7 | 0.7 |
| AlH3⋅⋅⋅NH3 b | −28.7 | −32.8 | 0.6 | 4.1 |
| AlH3Cl− b | −52.2 | −66.6 | 1.1 | 14.4 |
| Complex | B/Al⋅⋅⋅N/Cl | α-angle | Ch-shift | B/Al | Pol-B/Al |
|---|---|---|---|---|---|
| BCl3⋅⋅⋅NCH a | 2.817 | 91.7 | −0.012 | 0.344 | - |
| BCl3⋅⋅⋅NCH b | 1.628 | 104.0 | −0.292 | 0.265 | 22.1 |
| BCl3⋅⋅⋅N2 a | 3.083 | 90.4 | −0.005 | 0.320 | - |
| BCl3⋅⋅⋅NH3 b | 1.614 | 105.0 | −0.411 | 0.312 | 22.8 |
| BCl4− b | 1.862 | 109.5 | −0.690 | 0.238 | 30.4 |
| BH3⋅⋅⋅NCH b | 1.582 | 103.9 | −0.260 | −0.275 | 77.5 |
| BH3⋅⋅⋅N2 b | 1.622 | 101.6 | −0.226 | −0.287 | 21.1 |
| BH3⋅⋅⋅NH3 b | 1.652 | 104.7 | −0.396 | −0.234 | 79.4 |
| BH3Cl− b | 1.968 | 106.5 | −0.549 | −0.289 | 74.5 |
| AlCl3⋅⋅⋅NCH b | 2.024 | 100.0 | −0.126 | 1.327 | 90.9 |
| AlCl3⋅⋅⋅N2 a | 2.213 | 96.1 | −0.099 | 1.297 | 92.5 |
| AlCl3⋅⋅⋅NH3 b | 1.999 | 100.8 | −0.186 | 1.325 | 90.4 |
| AlCl4− b | 2.164 | 109.5 | −0.440 | 1.240 | 83.5 |
| AlH3⋅⋅⋅NCH b | 2.099 | 97.7 | −0.107 | 0.903 | 91.5 |
| AlH3⋅⋅⋅N2 a | 2.290 | 93.8 | −0.114 | 0.916 | 93.5 |
| AlH3⋅⋅⋅NH3 b | 2.071 | 99.2 | −0.179 | 0.899 | 8.9 |
| AlH3Cl− b | 2.255 | 107.1 | −0.376 | 0.822 | 15.1 |
| Complex | ρBCP | ∇2ρBCP | HBCP |
|---|---|---|---|
| BCl3⋅⋅⋅NCH a | 0.011 | 0.035 | 0.001 |
| BCl3⋅⋅⋅NCH b | 0.106 | 0.403 | −0.070 |
| BCl3⋅⋅⋅N2 a | 0.006 | 0.022 | 0.001 |
| BCl3⋅⋅⋅NH3 b | 0.131 | 0.256 | −0.111 |
| BCl4− b | 0.123 | −0.055 | −0.116 |
| BH3⋅⋅⋅NCH b | 0.105 | 0.662 | −0.056 |
| BH3⋅⋅⋅N2 b | 0.088 | 0.652 | −0.038 |
| BH3⋅⋅⋅NH3 b | 0.104 | 0.444 | −0.069 |
| BH3Cl− b | 0.084 | 0.132 | −0.065 |
| AlCl3⋅⋅⋅NCH b | 0.048 | 0.310 | 0.002 |
| AlCl3⋅⋅⋅N2 a | 0.028 | 0.164 | 0.003 |
| AlCl3⋅⋅⋅NH3 b | 0.060 | 0.340 | −0.005 |
| AlCl4− b | 0.063 | 0.292 | −0.011 |
| AlH3⋅⋅⋅NCH b | 0.038 | 0.244 | 0.004 |
| AlH3⋅⋅⋅N2 a | 0.023 | 0.125 | 0.003 |
| AlH3⋅⋅⋅NH3 b | 0.048 | 0.274 | 0.000 |
| AlH3Cl− b | 0.050 | 0.226 | −0.006 |
| Complex | ΔEint | ΔEPauli | ΔEelstat | ΔEorb | ΔEdisp |
|---|---|---|---|---|---|
| BCl3⋅⋅⋅NCH a | −1.9 | 8.5 | −5.8 | −2.6 | −2.1 |
| BCl3⋅⋅⋅NCH b | −18.5 | 168.9 | −86.9 | −98.7 | −1.9 |
| BCl3⋅⋅⋅N2 a | −1.0 | 3.7 | −1.7 | −1.3 | −1.7 |
| BCl3⋅⋅⋅NH3 b | −43.8 | 202.8 | −125.8 | −117.7 | −3.1 |
| BCl4− b | −80.9 | 186.8 | −126.6 | −140.2 | −0.9 |
| BH3⋅⋅⋅NCH b | −36.7 | 109.9 | −59.3 | −86.2 | −1.1 |
| BH3⋅⋅⋅N2 b | −23.6 | 93.6 | −42.3 | −73.8 | −1.1 |
| BH3⋅⋅⋅NH3 b | −45.9 | 110.7 | −78.1 | −76.7 | −1.9 |
| BH3Cl− b | −53.8 | 88.9 | −63.1 | −78.8 | −0.9 |
| AlCl3⋅⋅⋅NCH b | −32.7 | 57.4 | −47.6 | −40.0 | −2.6 |
| AlCl3⋅⋅⋅N2 a | −10.1 | 35.2 | −20.5 | −22.4 | −2.3 |
| AlCl3⋅⋅⋅NH3 b | −43.7 | 82.1 | −78.7 | −43.6 | −3.6 |
| AlCl4− b | −95.1 | 93.6 | −113.2 | −73.8 | −1.6 |
| AlH3⋅⋅⋅NCH b | −21.0 | 41.9 | −34.3 | −27.2 | −1.5 |
| AlH3⋅⋅⋅N2 a | −8.5 | 22.3 | −13.7 | −16.0 | −1.2 |
| AlH3⋅⋅⋅NH3 b | −32.7 | 59.5 | −58.6 | −31.2 | −2.3 |
| AlH3Cl− b | −65.5 | 67.1 | −81.2 | −50.8 | −0.7 |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grabowski, S.J. The Nature of Triel Bonds, a Case of B and Al Centres Bonded with Electron Rich Sites. Molecules 2020, 25, 2703. https://doi.org/10.3390/molecules25112703
Grabowski SJ. The Nature of Triel Bonds, a Case of B and Al Centres Bonded with Electron Rich Sites. Molecules. 2020; 25(11):2703. https://doi.org/10.3390/molecules25112703
Chicago/Turabian StyleGrabowski, Sławomir J. 2020. "The Nature of Triel Bonds, a Case of B and Al Centres Bonded with Electron Rich Sites" Molecules 25, no. 11: 2703. https://doi.org/10.3390/molecules25112703
APA StyleGrabowski, S. J. (2020). The Nature of Triel Bonds, a Case of B and Al Centres Bonded with Electron Rich Sites. Molecules, 25(11), 2703. https://doi.org/10.3390/molecules25112703