
Synthesis and Conformational Analysis of Naphthoxazine-Fused Phenanthrene Derivatives

 , and
, and 
Abstract
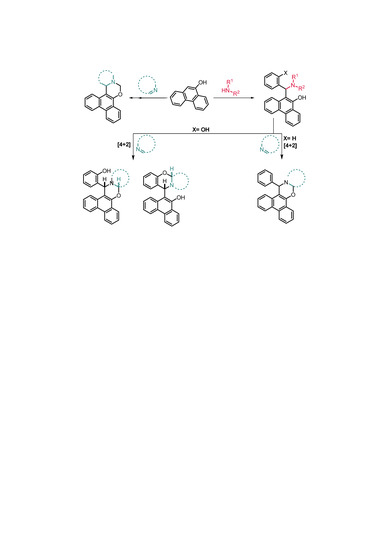
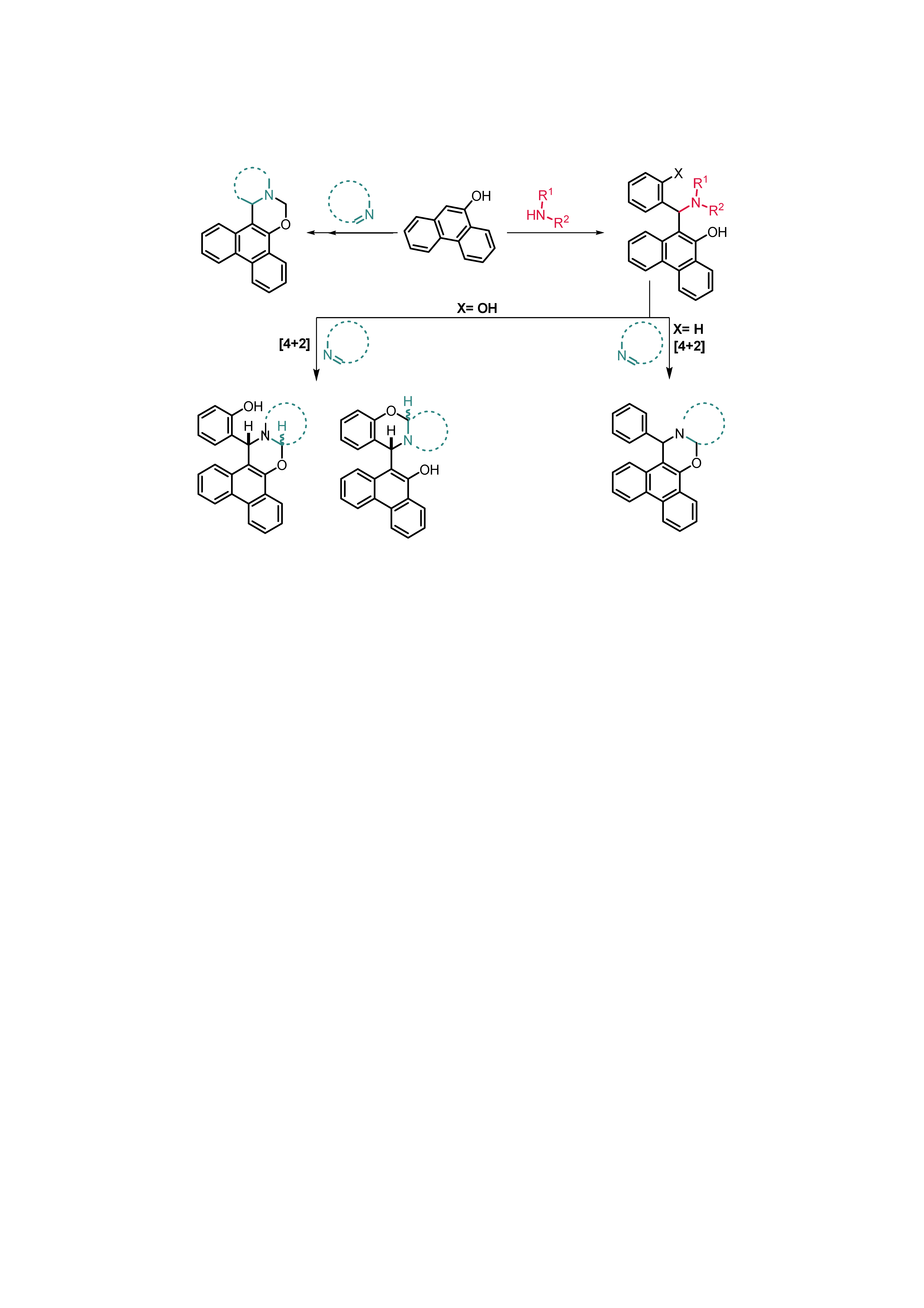
1. Introduction
2. Results
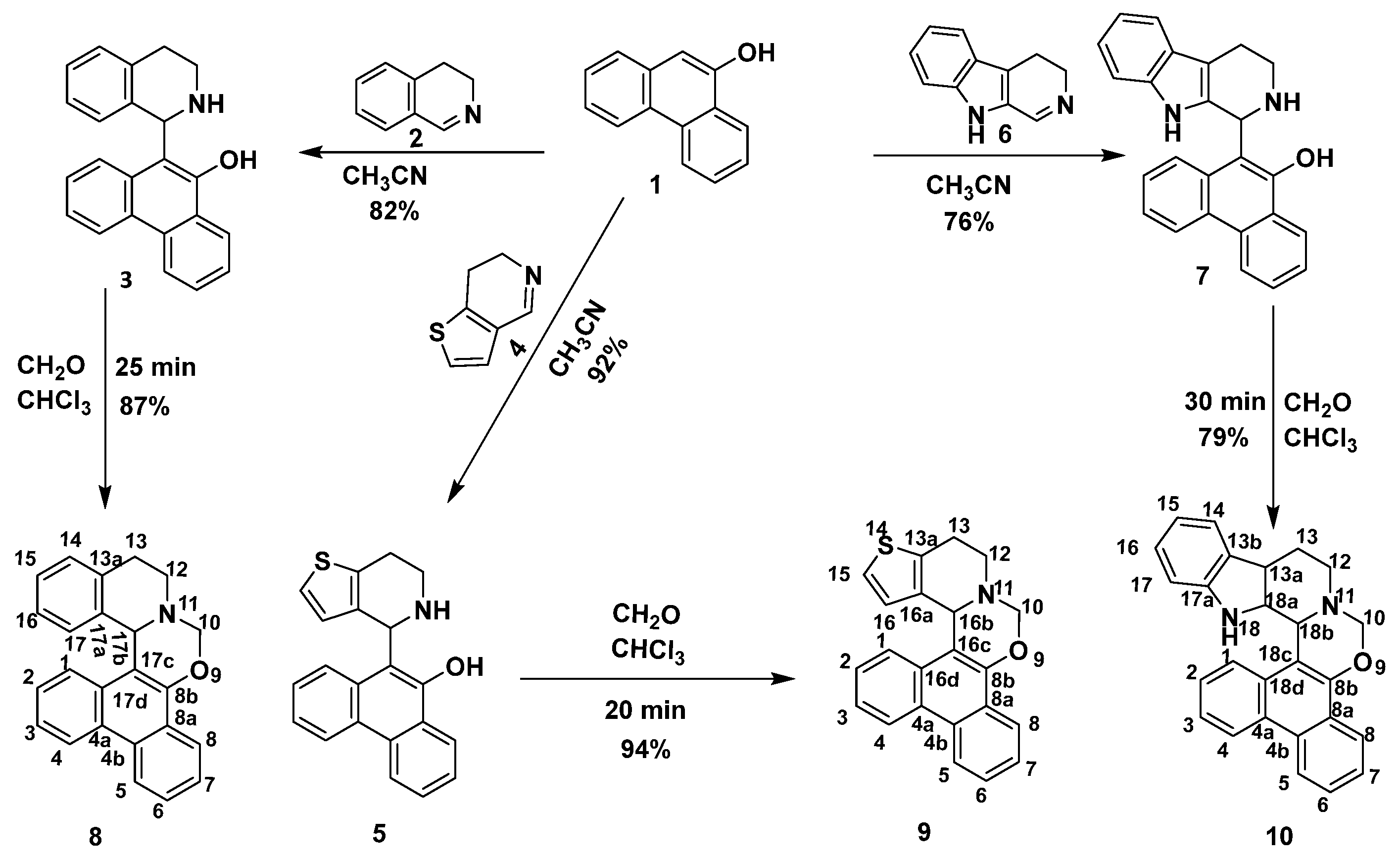
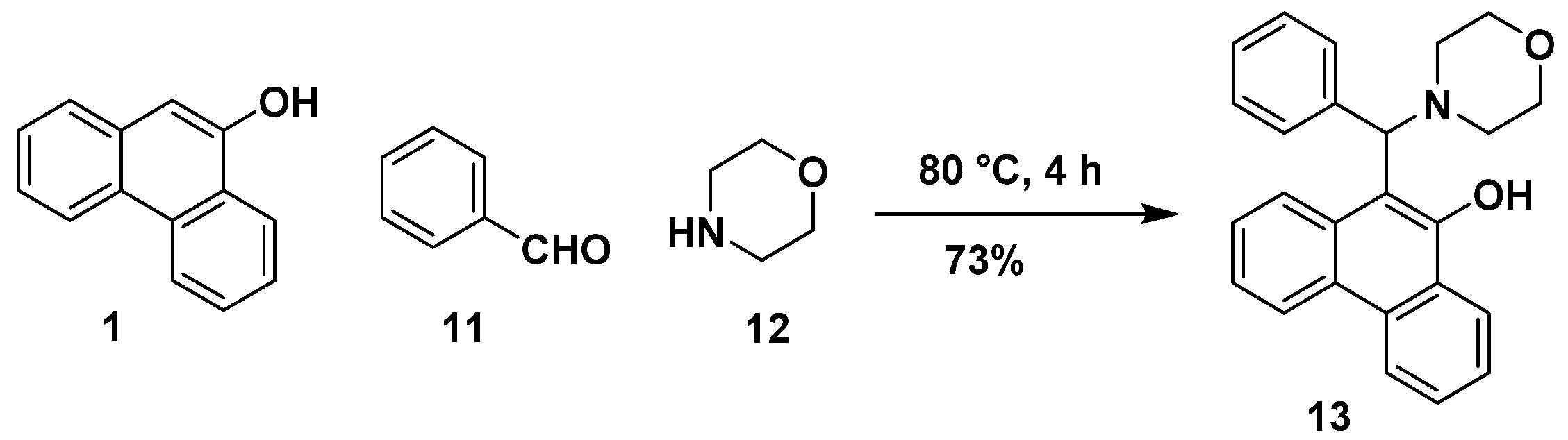
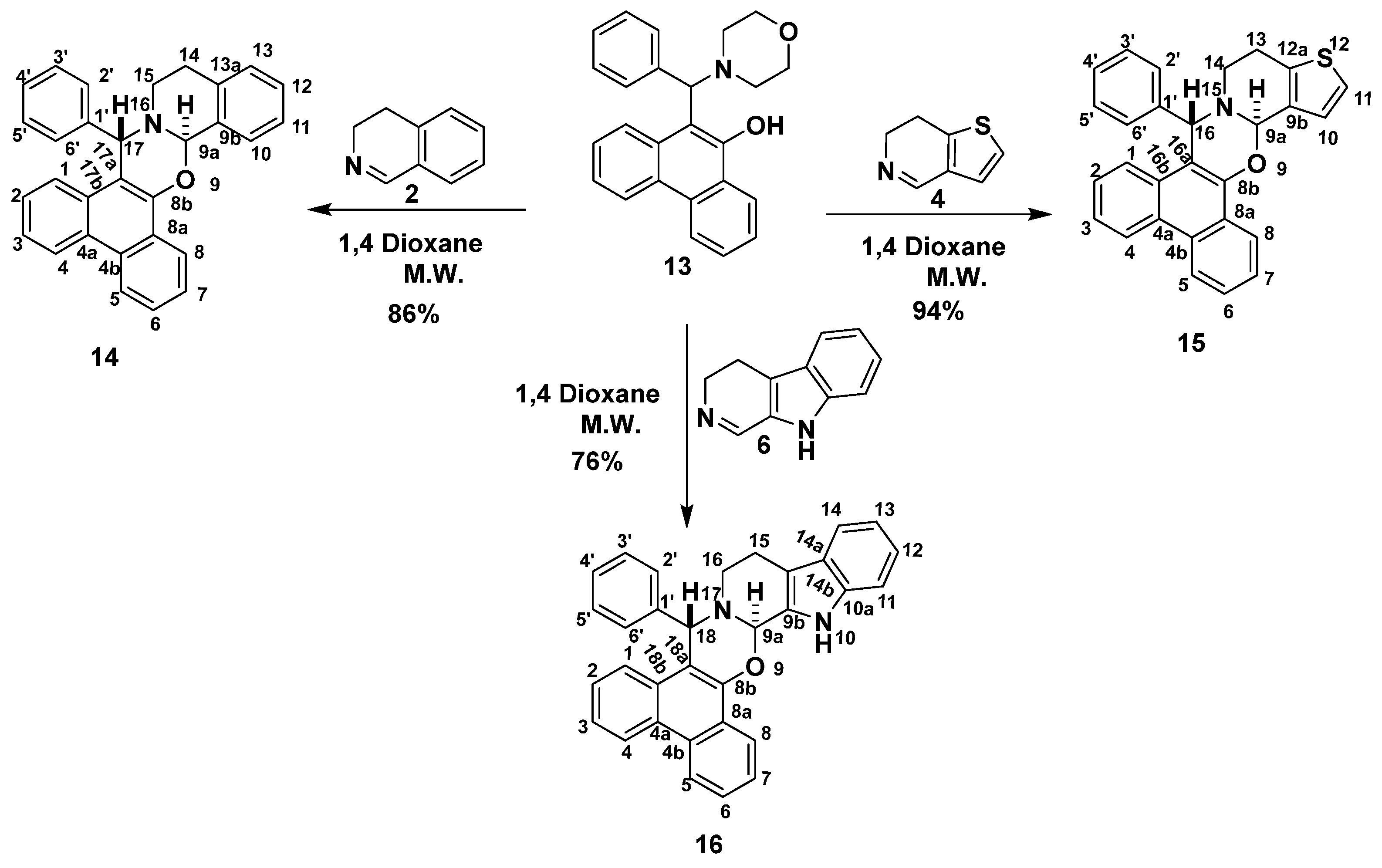
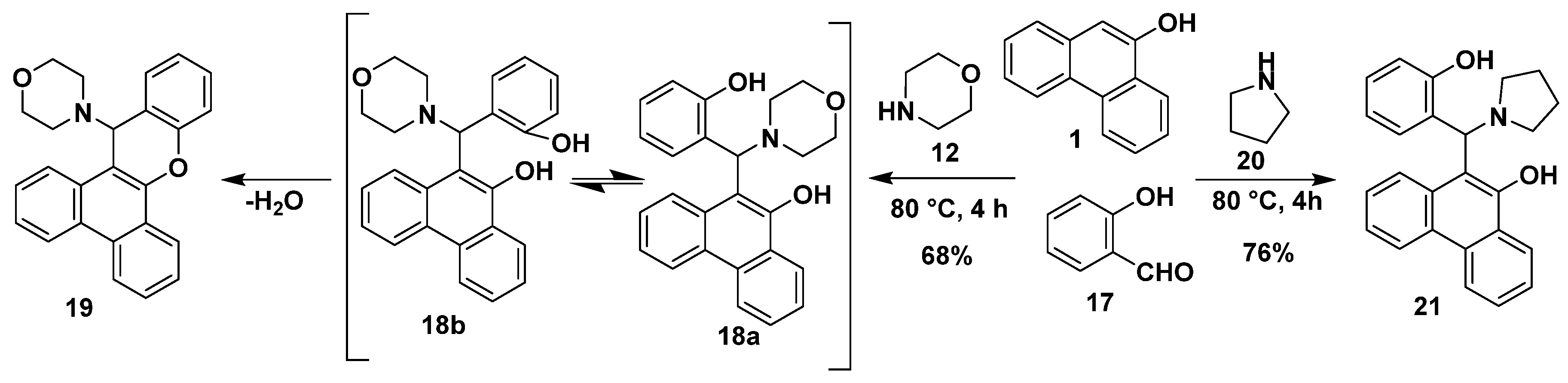
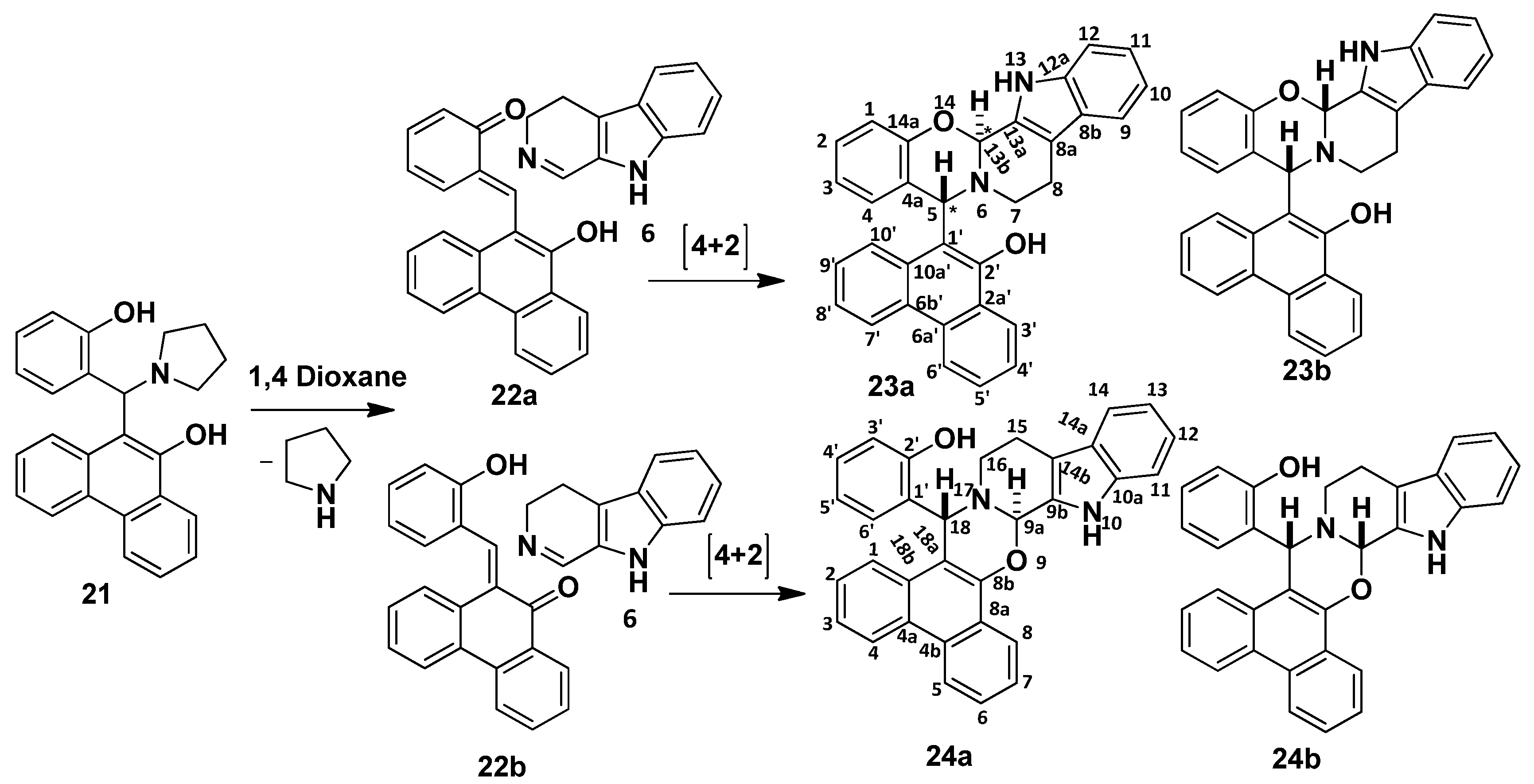
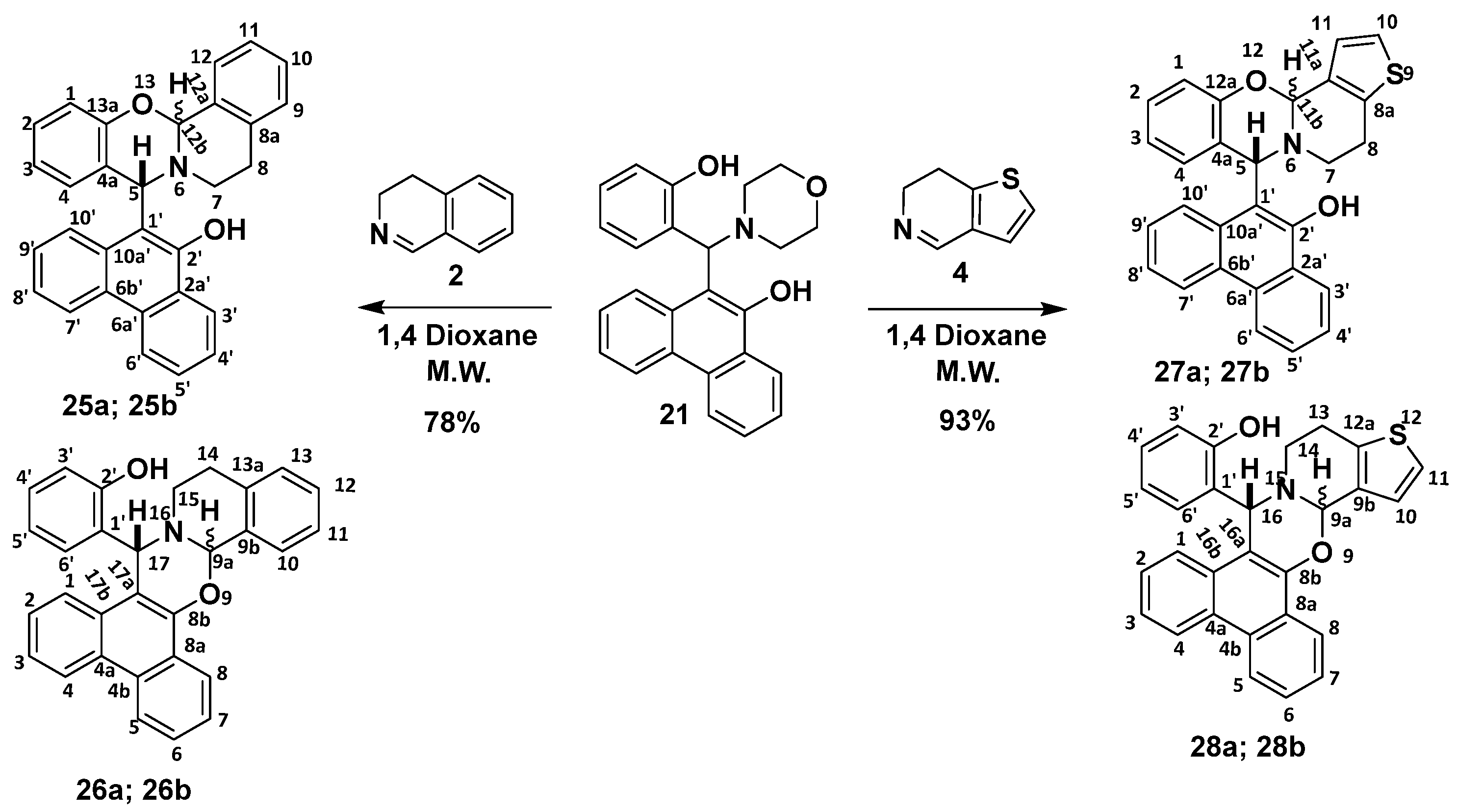
2.1. Synthesis
2.2. Structural and Conformational Analysis
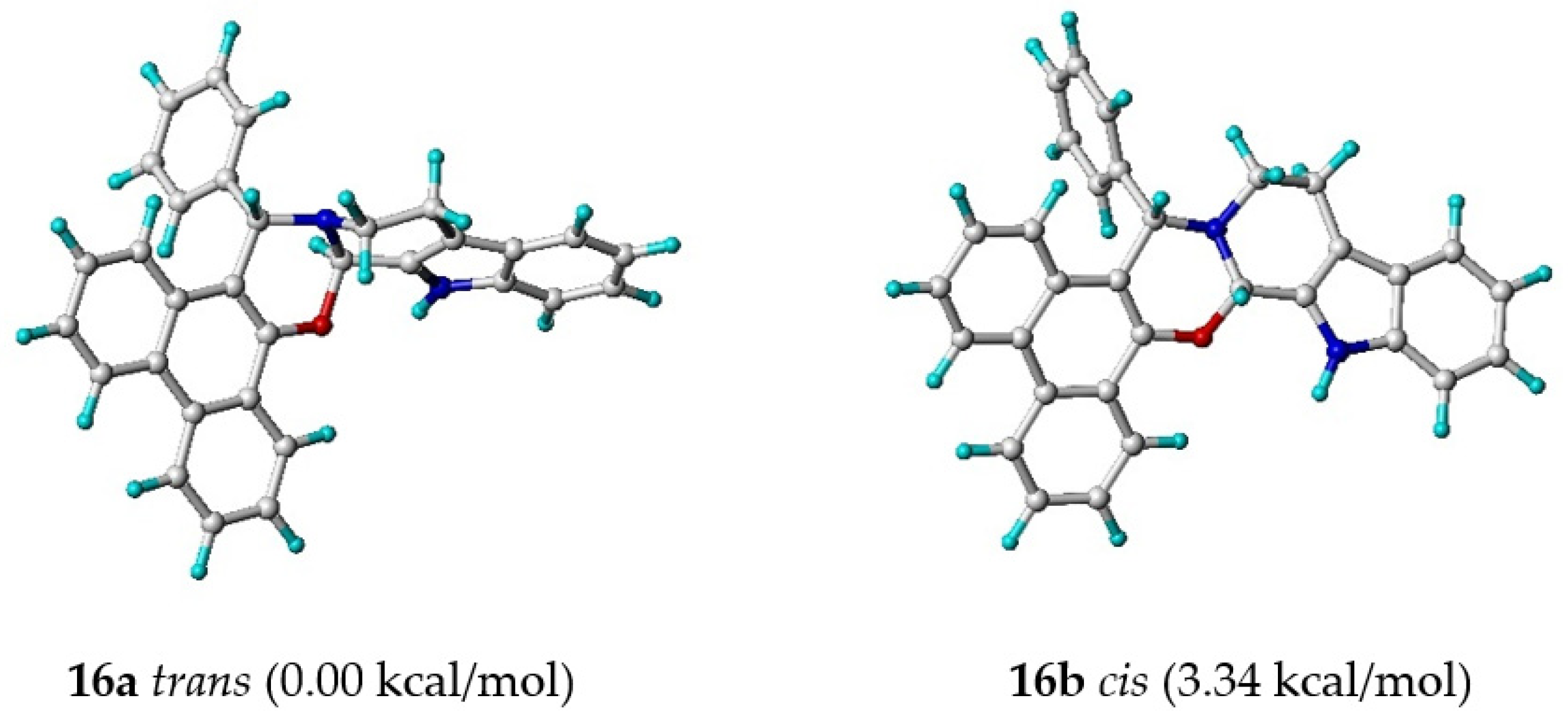
2.3. Detailed NMR Analysis of New Phenanthr[9,10-e]Oxazine 16
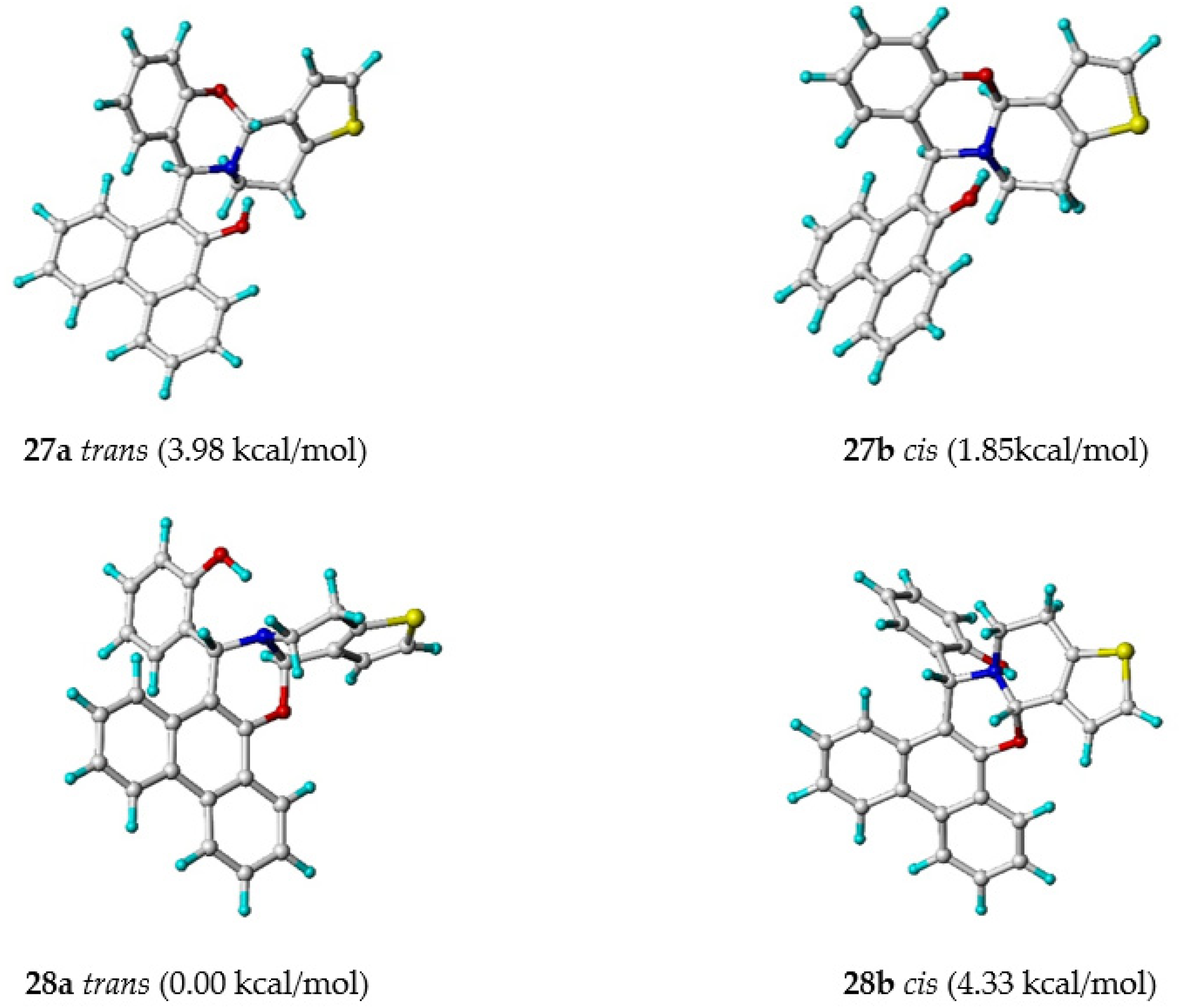
2.4. Detailed NMR Analysis of New Phenanthr[9,10-e]Oxazines 27a,b−28a,b
3. Materials and Methods
3.1. General Methods
3.2. Procedures
3.2.1. General Procedure for the Synthesis of Hydroxyphenanthryl-Isoquinoline, -Thienopyridine and -β-Carboline (3, 5 and 7)
3.2.2. General Procedure for the Synthesis of Phenanthr[9,10-e][1,3]Oxazinoisoquinoline,-Thienopyridine and -β-Carboline (8–10)
3.2.3. General Procedure for the Synthesis of Phenanthr[9,10-e][1,3]Oxazines (14, 15 and 16)
3.2.4. General Procedure for the Synthesis of Phenanthr[9,10-e][1,3]Oxazines (24, 26 and 28)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Grand, T.; Demion, M.; Norez, C.; Mettey, Y.; Launay, P.; Becq, F.; Bois, P.; Guinamard, R. 9-Phenanthrol Inhibits Human TRPM4 but not TRPM5 Cationic Channels. Br. J. Pharmacol. 2008, 153, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Guinamard, R.; Hof, T.; Del Negro, C.A. The TRPM4 Channel Inhibitor 9-Phenanthrol. Br. J. Pharmacol. 2014, 171, 1600–1613. [Google Scholar] [CrossRef] [PubMed]
- Cho, C.H.; Lee, Y.S.; Kim, E.; Hwang, E.M.; Park, J.Y. Physiological Functions of the TRPM4 Channels via Protein Interactions. BMB Reports 2015, 48, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, A.L.; Amberg, G.C.; Earley, S. Ca2+ Release from the Sarcoplasmic Reticulum is Required for Sustained TRPM4 Activity in Cerebral Artery Smooth Muscle Cells. Am. J. Physiol. Cell. Physiol. 2010, 299, C279–C288. [Google Scholar] [CrossRef]
- Parajuli, S.P.; Hristov, K.L.; Sullivan, M.N.; Xin, W.; Smith, A.C.; Earley, S.; Malysz, J.; Petkov, G.V. Control of Urinary Bladder Smooth Muscle Excitability by the TRPM4 Channel Modulator 9-Phenanthrol. Channels 2013, 7, 537–540. [Google Scholar] [CrossRef]
- Becerra, A.; Echeverría, C.; Varela, D.; Sarmiento, D.; Armisén, R.; Nuñez-Villena, F.; Montecinos, M.; Simon, F. Transient Receptor Potential Melastatin 4 Inhibition Prevents Lipopolysaccharide-Induced Endothelial Cell Death. Cardiovasc. Res. 2011, 91, 677–684. [Google Scholar] [CrossRef]
- Hou, J.; Fei, Y.; Li, W.; Chen, Y.; Wang, Q.; Xiao, Y.; Wang, Y.; Li, Y. The Transient Receptor Potential Melastatin 4 Channel Inhibitor 9-Phenanthrol Modulates Cardiac Sodium Channel. Br. J. Pharmacol. 2018, 175, 4325–4337. [Google Scholar] [CrossRef]
- Bur, S.K.; Martin, S.F. Vinylogous Mannich Reactions: Selectivity and Synthetic Utility. Tetrahedron 2001, 57, 3221–3242. [Google Scholar] [CrossRef]
- Speckamp, W.N.; Moolenaar, M.J. New Developments in the Chemistry of N-Acyliminium Ions and Related Intermediates. Tetrahedron 2000, 56, 3817–3856. [Google Scholar] [CrossRef]
- Arend, M.; Westermann, B.; Risch, N. Modern Variants of the Mannich Reaction. Angew. Chem. Int. Ed. Engl. 1998, 37, 1045–1070. [Google Scholar] [CrossRef]
- Liras, S.; Davoren, J.E.; Bordner, J. An Approach to the Skeleton of the Securinega Alkaloids. The Total Synthesis of (±)-Securinine. Org. Lett. 2001, 3, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Clark, C.W.; Mortimore, M.; Goh, J.B.; Martin, S.F. Biogenetically Inspired Approach to the Strychnos Alkaloids. Concise Syntheses of (±)-Akuammicine and (±)-Strychnine. J. Am. Chem. Soc. 2001, 123, 8003–8010. [Google Scholar] [CrossRef] [PubMed]
- Szatmári, I.; Fülöp, F. Syntheses and Transformations of 1-(α-Aminobenzyl)-2-Naphthol Derivatives. Curr. Org. Synth. 2004, 1, 155–165. [Google Scholar] [CrossRef]
- Szatmári, I.; Fülöp, F. Syntheses, Transformations and Applications of Aminonaphthol Derivatives Prepared via Modified Mannich Reactions. Tetrahedron 2013, 69, 1255–1278. [Google Scholar] [CrossRef]
- MacLeod, P.D.; Li, Z.; Feng, J.; Li, C.J. Solvent-Free Direct aza-Friedel–Crafts Reactions between 3,4-Dihydroisoquinoline and 1- or 2-Naphthols. Tetrahedron Lett. 2006, 47, 6791–6794. [Google Scholar] [CrossRef]
- Heydenreich, M.; Koch, A.; Klod, S.; Szatmári, I.; Fülöp, F.; Kleinpeter, E. Synthesis and Conformational Analysis of Naphth[1′,2′:5,6][1,3]oxazino[3,2-c][1,3]benzoxazine and Naphth[1′,2′:5,6][1,3]oxazino[3,4-c][1,3]benzoxazine Derivatives. Tetrahedron 2006, 62, 11081–11089. [Google Scholar] [CrossRef]
- Heydenreich, M.; Koch, A.; Szatmári, I.; Fülöp, F.; Kleinpeter, E. Synthesis and Conformational Analysis of Naphth[1,2-e][1,3]oxazino[4,3-a][1,3]isoquinoline and Naphth[2,1-e][1,3]oxazino[4,3-a]isoquinoline Derivatives. Tetrahedron 2008, 64, 7378–7385. [Google Scholar] [CrossRef]
- Szatmári, I.; Barta, P.; Tóth, G.; Balázs, A.; Halász, J.; Fülöp, F. Synthesis and Conformational Behaviour of Enantiomeric Naphthoxazinoquinoxalinone Derivatives. Eur. J. Org. Chem. 2017, 5537–5545. [Google Scholar]
- Barta, P.; Szatmári, I.; Fülöp, F.; Heydenreich, M.; Koch, A.; Kleinpeter, E. Synthesis and Stereochemistry of New Naphth[1,3]oxazino[3,2-a]benzazepine and Naphth[1,3]oxazino[3,2-e]thienopyridine Derivatives. Tetrahedron. 2016, 72, 2402–2410. [Google Scholar] [CrossRef]
- Barta, P.; Fülöp, F.; Szatmári, I. Mannich Base-Connected Syntheses Mediated by Ortho-Quinone Methides. Beilstein J. Org. Chem. 2018, 14, 560–575. [Google Scholar] [CrossRef]
- Osyanin, V.A.; Osipov, D.V.; Klimochkin, Y.N. Convenient One-Step Synthesis of 4-Unsubstituted 2-Amino-4H-chromene-2-carbonitriles and 5-Unsubstituted 5H-chromeno[2,3-b]pyridine-3-carbonitriles From Quaternary Ammonium salts. Tetrahedron 2012, 68, 5612–5618. [Google Scholar] [CrossRef]
- Pettigrew, J.D.; Freeman, R.P.; Wilson, P.D. Total Synthesis of ()-Xyloketal D and its Enantiomer Confirmation of Absolute Stereochemistry. Can. J. Chem. 2004, 82, 1640–1648. [Google Scholar] [CrossRef]
- Szatmári, I.; Fülöp, F. Simple Access to Pentacyclic Oxazinoisoquinolines via an Unexpected Transformation of Aminomethylnaphthols. Tetrahedron Lett. 2011, 52, 4440–4442. [Google Scholar] [CrossRef]
- Szatmári, I.; Barta, P.; Csámpai, A.; Fülöp, F. Synthesis and Detailed Conformational Analysis of New Naphthoxazino[2,3-a]benz[c]azepine and Naphthoxazino[2,3-a]thieno[3,2-c]pyridine Derivatives. Tetrahedron 2017, 73, 4790–4804. [Google Scholar] [CrossRef]
- Szatmári, I.; Belasri, K.; Heydenreich, M.; Koch, A.; Kleinpeter, E.; Fülöp, F. Ortho-Quinone Methide Driven Synthesis of New O,N- or N,N-Heterocycles. ChemistryOpen. 2019, 8, 961–971. [Google Scholar] [CrossRef]
- Szatmári, I.; Sas, I.; Fülöp, F. Catalyst-Free Coupling of Indole Derivatives with 3,4-Dihydroisoquinoline and Related Compounds. Tetrahedron Lett. 2013, 54, 5069–5071. [Google Scholar] [CrossRef]
- Herz, W.; Tsai, L. Sulfur analogs of isoquinolines. IV. The Pictet-Gams Reaction and Attempts to Prepare Analogs of Papaverine1,2. J. Chem. Soc. 1955, 77, 3529–3533. [Google Scholar] [CrossRef]
- Chen, Z.; Hu, G.; Li, D.; Chen, J.; Li, Y.; Zhou, H.; Xie, Y. Synthesis and Vasodilator Effects of Rutaecarpine Analogues which Might be Involved Transient Receptor Potential Vanilloid Subfamily, Member 1 (TRPV1). Bioorg. Med. Chem. 2009, 17, 2351–2359. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; (Revision A.02); Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D.A. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Hehre, W.J.; Radom, L.; von Rague Schleyer, P.; Pople, J. Ab Initio Molecular Orbital Theory; Wiley: New York, NY, USA, 1986. [Google Scholar]
- Becke, A.D. A New Mixing of Hartree–Fock and Local Density-Functional Theories. J. Chem. Phys. 1993, 98, 1372–1492. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-Consistent Perturbation Theory of Diamagnetism. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Cheeseman, G.G.W.; Trucks, T.A.; Keith, M.J. A Comparison of Models for Calculating Nuclear Magnetic Resonance Shielding Tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Products | Type of Heating | Solvent | Reaction Time | Temperature | Yield (%) |
|---|---|---|---|---|---|
| 3 | Oil bath | - | 120 min | 80 °C | 10 |
| Oil bath | - | 60 min | 100 °C | 19 | |
| Oil bath | Acetonitrile | 60 min | 90 °C | 49 | |
| MW | Acetonitrile | 10 min | 100 °C | 67 | |
| MW | Acetonitrile | 20 min | 100 °C | 82 | |
| 5 | MW | Acetonitrile | 20 min | 100 °C | 72 |
| MW | Acetonitrile | 35 min | 100 °C | 92 | |
| 7 | MW | Acetonitrile | 20 min | 100 °C | 63 |
| MW | Acetonitrile | 40 min | 100 °C | 76 |
| Product | Reaction Time | Temperature (°C) | Yield (%) |
|---|---|---|---|
| 14 | 15 min | 60 | 47 |
| 80 | 86 | ||
| 100 | 29 | ||
| 15 | 15 min | 60 | 52 |
| 80 | 94 | ||
| 100 | 37 | ||
| 16 | 15 min | 60 | 21 |
| 80 | 76 | ||
| 100 | 32 |
| Products | Reaction Time | Temperature (°C) | Yield (%) |
|---|---|---|---|
| 23a,b 24a,b | 15 min | 60 | 48 |
| 80 | 85 | ||
| 100 | 33 | ||
| 25a,b 26a,b | 15 min | 60 | 28 |
| 80 | 78 | ||
| 100 | 19 | ||
| 27a,b 28a,b | 15 min | 60 | 51 |
| 80 | 93 | ||
| 100 | 38 |
| Positions | 1/18 | 9a/18 | 18/16 (ř-eq) | 9a/16 (ř-eq) | 9a/16 (ř-ax) | 9a/10 | 14/15 (ř-ax) | 14/15 (ř-eq) |
|---|---|---|---|---|---|---|---|---|
| Measured NOE | strong | medium | strong | weak | weak | medium | weak | medium |
| Calculated distances d [Ĺ] | 2.2 2.2(cis) | 3.6 2.7(cis) | 2.2 2.7(cis) | 4.1 3.8(cis) | 3.8 2.7(cis) | 2.8 3.0(cis) | 4.3 3.4(cis) | 2.9 2.9(cis) |
| NOE-estimated distances d [Ĺ] | 2.2 | 3.4 | 2.3 | 4.4 | 4.3 | 2.8 | 3.5 | 3.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belasri, K.; Topal, L.; Heydenreich, M.; Koch, A.; Kleinpeter, E.; Fülöp, F.; Szatmári, I. Synthesis and Conformational Analysis of Naphthoxazine-Fused Phenanthrene Derivatives. Molecules 2020, 25, 2524. https://doi.org/10.3390/molecules25112524
Belasri K, Topal L, Heydenreich M, Koch A, Kleinpeter E, Fülöp F, Szatmári I. Synthesis and Conformational Analysis of Naphthoxazine-Fused Phenanthrene Derivatives. Molecules. 2020; 25(11):2524. https://doi.org/10.3390/molecules25112524
Chicago/Turabian StyleBelasri, Khadija, Leila Topal, Matthias Heydenreich, Andreas Koch, Erich Kleinpeter, Ferenc Fülöp, and István Szatmári. 2020. "Synthesis and Conformational Analysis of Naphthoxazine-Fused Phenanthrene Derivatives" Molecules 25, no. 11: 2524. https://doi.org/10.3390/molecules25112524
APA StyleBelasri, K., Topal, L., Heydenreich, M., Koch, A., Kleinpeter, E., Fülöp, F., & Szatmári, I. (2020). Synthesis and Conformational Analysis of Naphthoxazine-Fused Phenanthrene Derivatives. Molecules, 25(11), 2524. https://doi.org/10.3390/molecules25112524