Effect of Mo Dispersion on the Catalytic Properties and Stability of Mo–Fe Catalysts for the Partial Oxidation of Methanol

Abstract
1. Introduction
2. Experimental
2.1. Materials
2.2. Preparation of Catalysts
2.3. Catalytic Test and Characterization
3. Results and Discussion
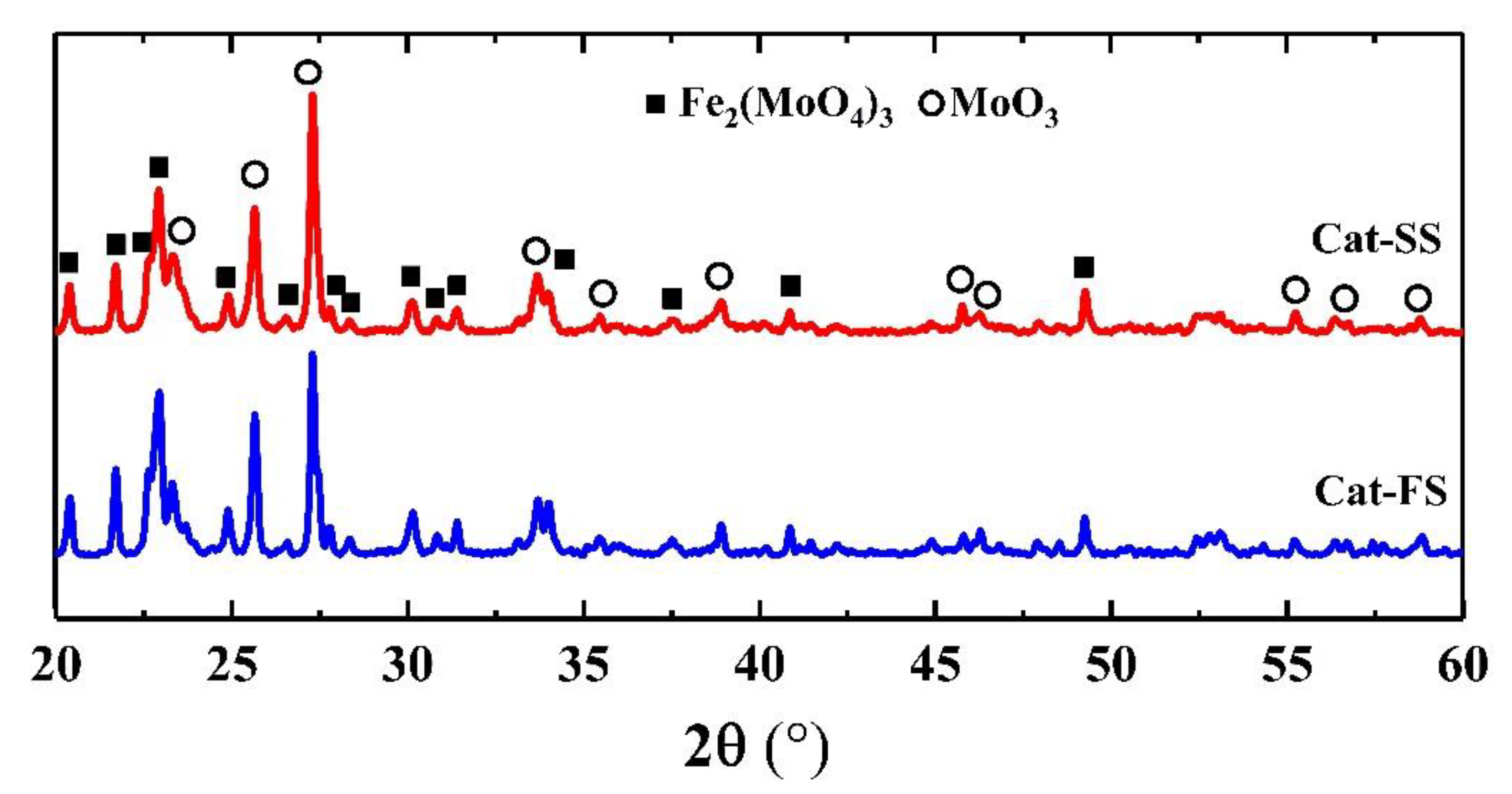
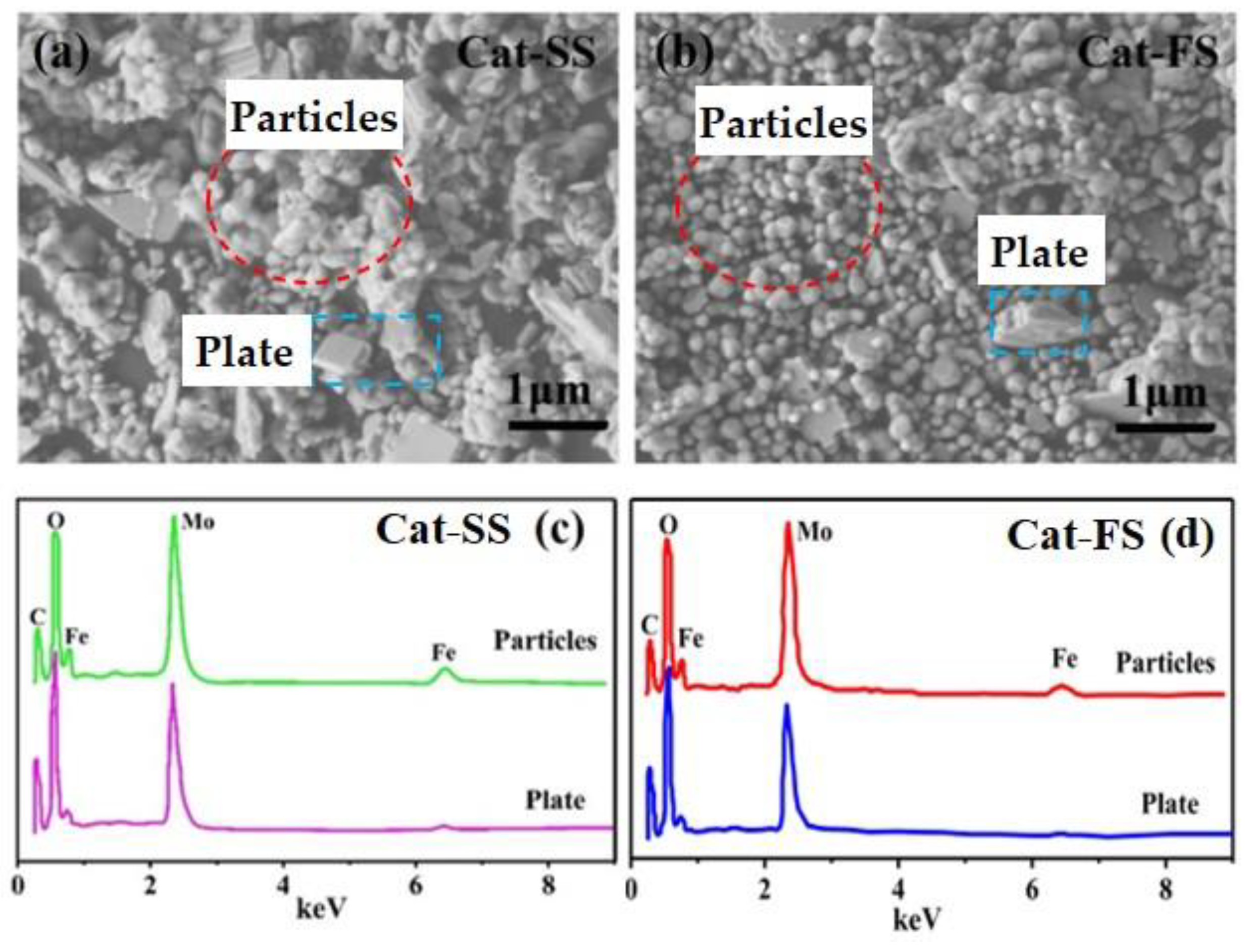
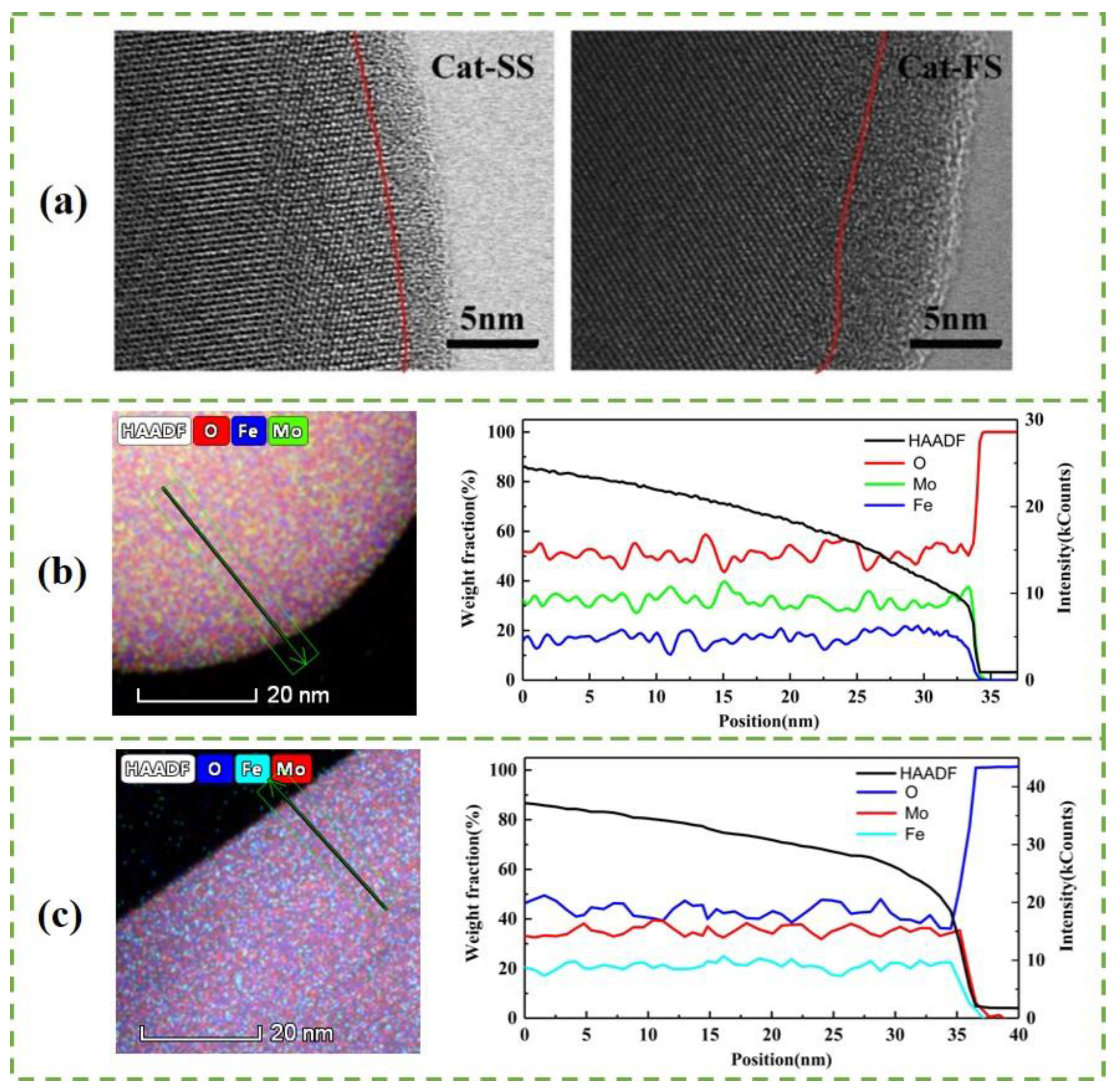
3.1. Characterization of the Catalysts
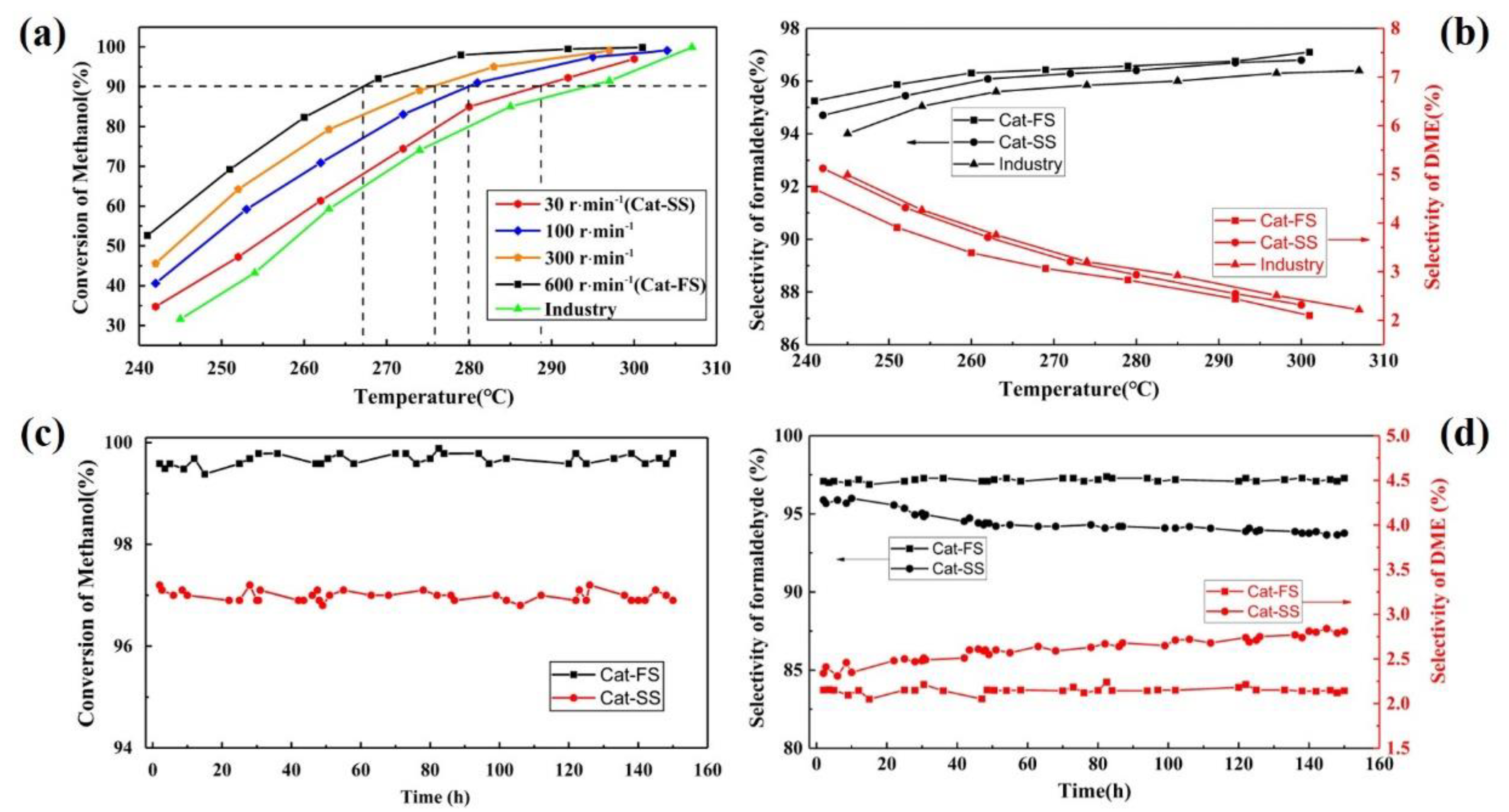
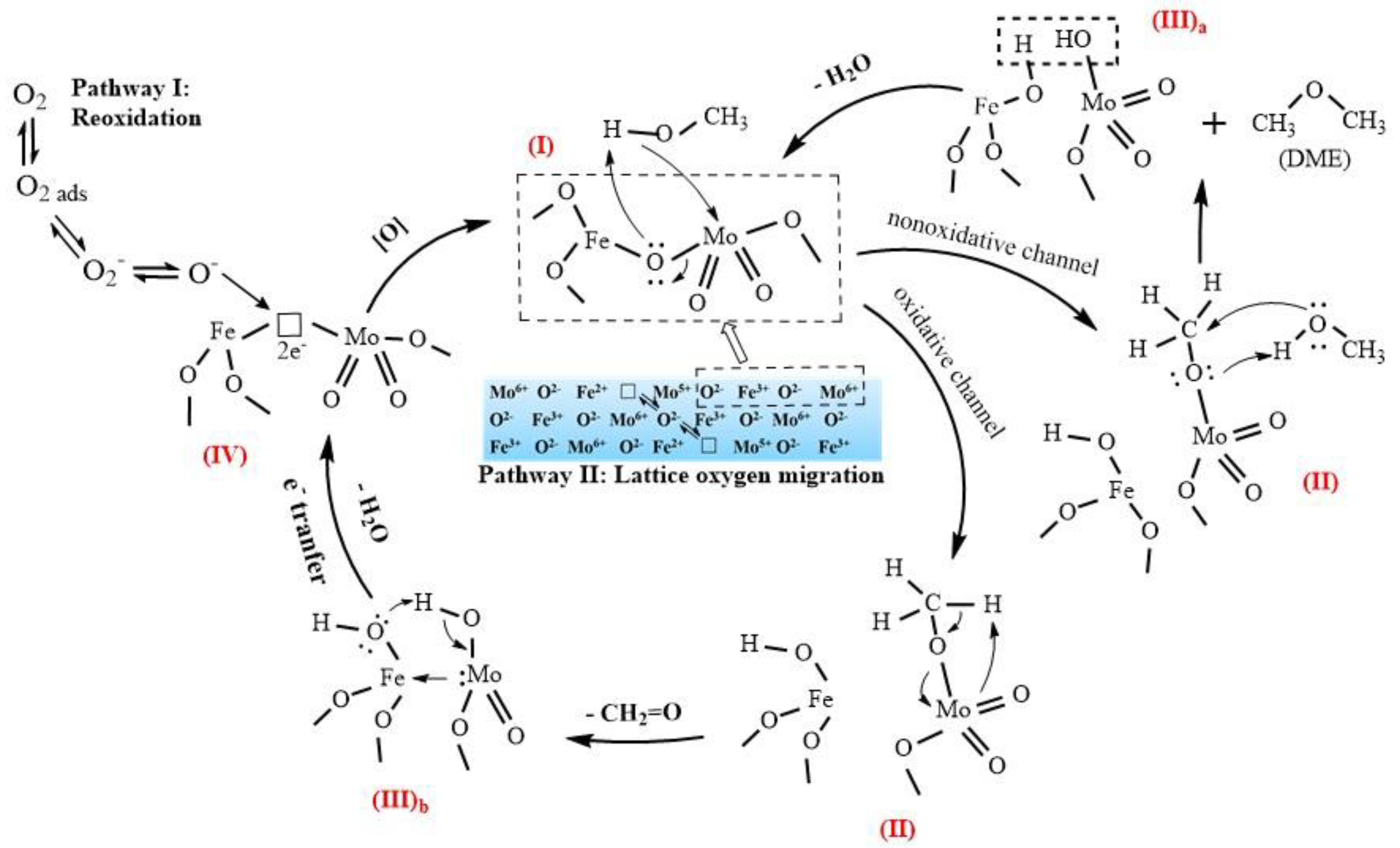
3.2. Activity and Stability of the Catalyst
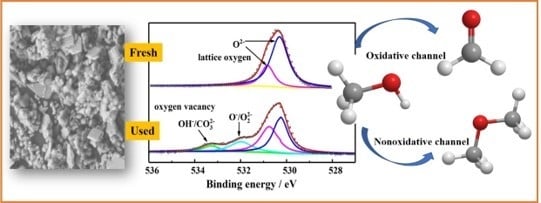
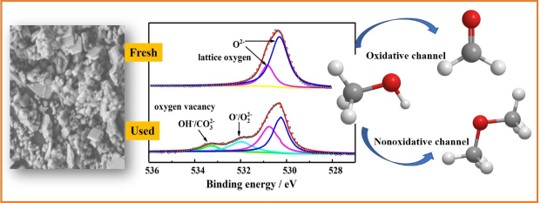
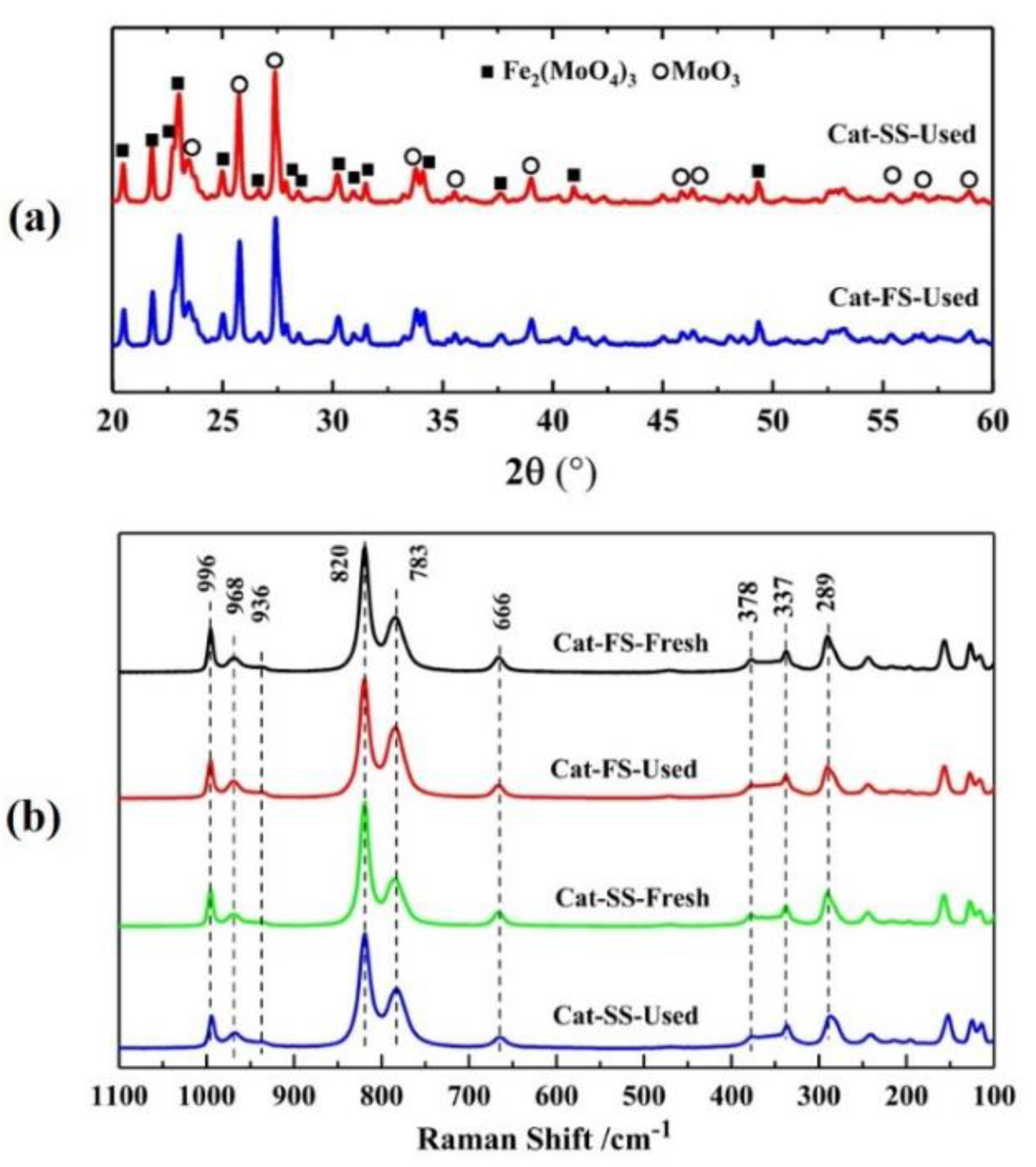
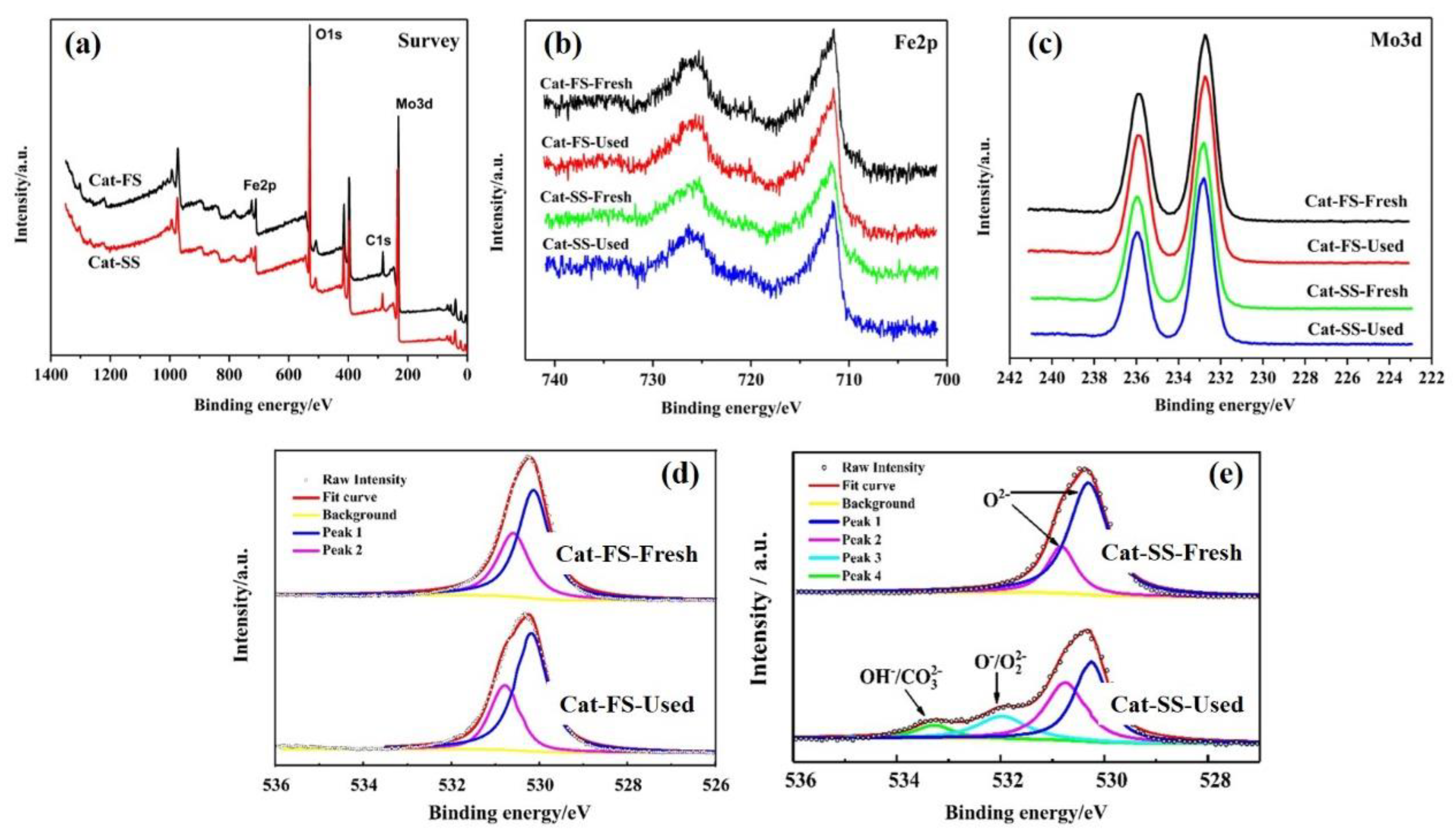
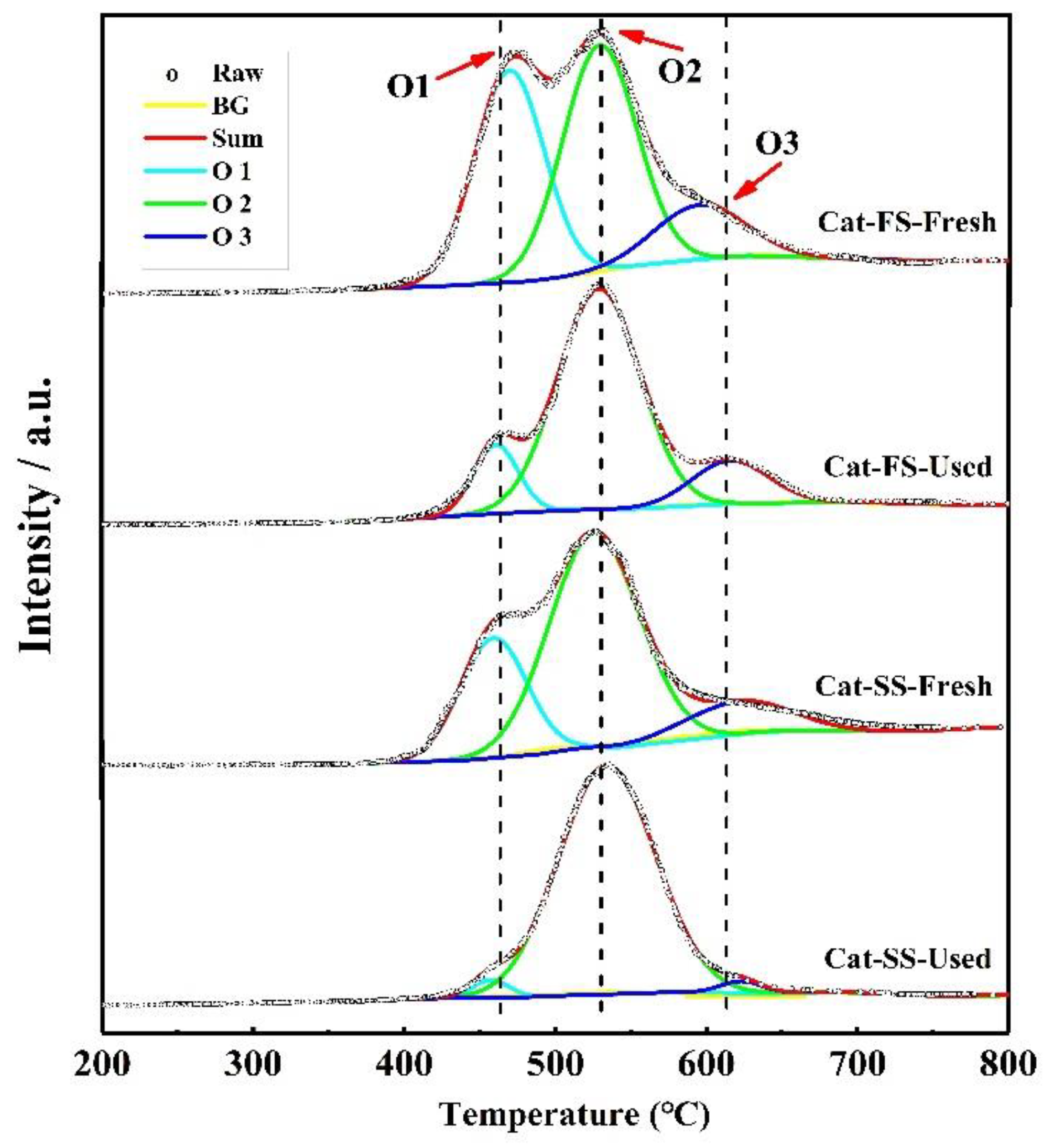
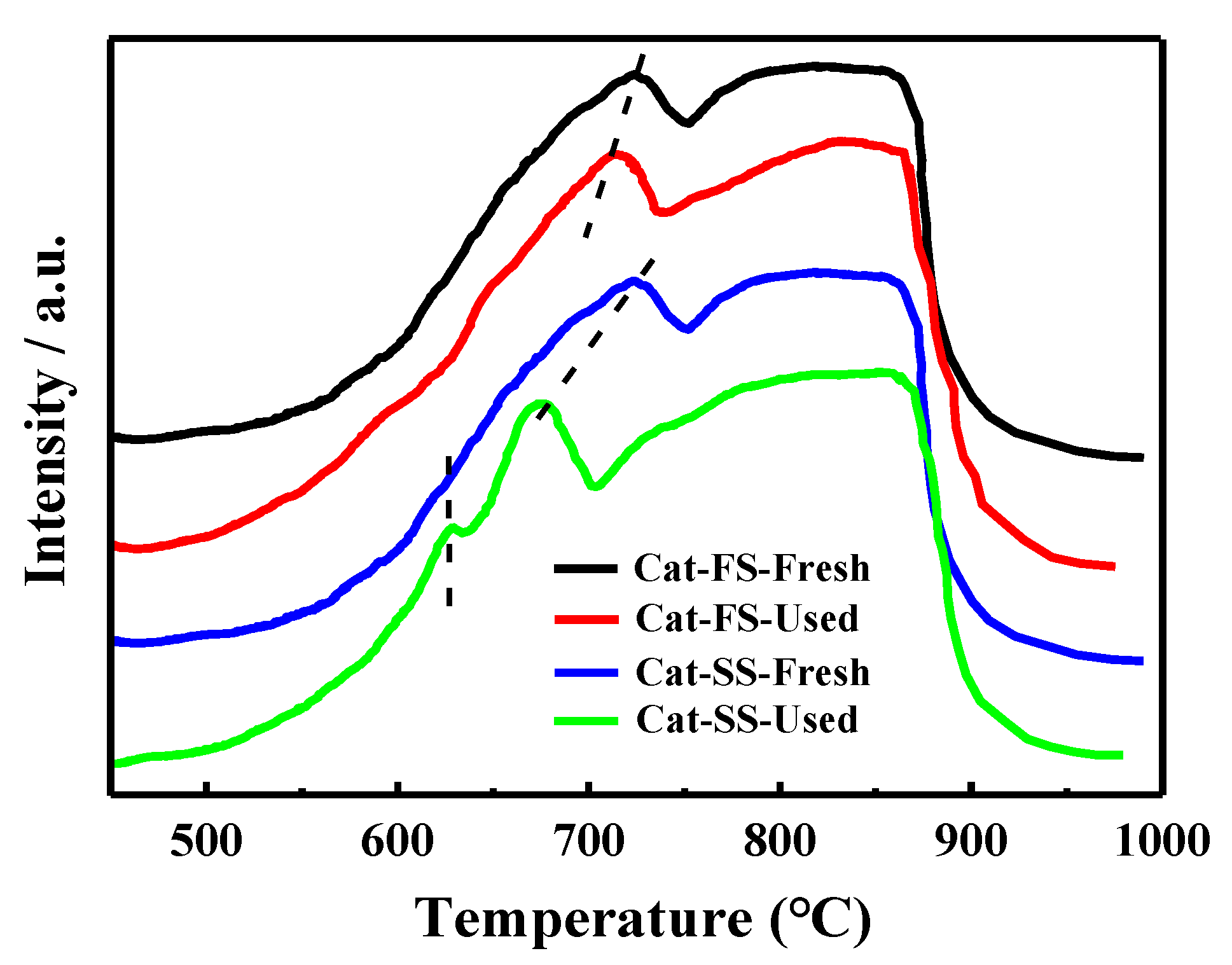
3.3. Analysis of Fresh and Used Catalysts
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fajardo, C.A.G.; Niznansky, D.; N’Guyen, Y.; Courson, C.; Roger, A.-C. Methane selective oxidation to formaldehyde with Fe-catalysts supported on silica or incorporated into the support. Catal. Commun. 2008, 9, 864–869. [Google Scholar] [CrossRef]
- Pestryakov, A.N.; Petranovskii, V.P.; Pfänder, N.; Knop-Gericke, A. Supported foam–copper catalysts for methanol selective oxidation. Catal. Commun. 2004, 5, 777–781. [Google Scholar] [CrossRef]
- Soares, A.P.V.; Portela, M.F.; Kiennemann, A. Methanol selective oxidation to formaldehyde over iron-molybdate catalysts. Catal. Rev. 2005, 47, 125–174. [Google Scholar] [CrossRef]
- Jin, G.; Weng, W.; Lin, Z.; Dummer, N.F.; Taylor, S.H.; Kiely, C.J.; Bartley, J.K.; Hutchings, G.J. Fe2(MoO4)3/MoO3 nano-structured catalysts for the oxidation of methanol to formaldehyde. J. Catal. 2012, 296, 55–64. [Google Scholar] [CrossRef]
- Raun, K.V.; Lundegaard, L.F.; Chevallier, J.; Beato, P.; Appel, C.C.; Nielsen, K.; Thorhauge, M.; Jensen, A.D.; Høj, M. Deactivation behavior of an iron-molybdate catalyst during selective oxidation of methanol to formaldehyde. Catal. Sci. Technol. 2018, 8, 4626–4637. [Google Scholar] [CrossRef]
- Pernicone, N. Deactivation of Fe-Mo oxide catalyst in industrial plant and simulation tests on laboratory scale. Catal. Today 1991, 11, 85–91. [Google Scholar] [CrossRef]
- House, M.P.; Carley A., F.; Echeverria-Valda, R.; Bowker, M. Effect of Varying the Cation Ratio within Iron Molybdate Catalysts for the Selective Oxidation of Methanol. J. Phys. Chem. C 2008, 112, 4333–4341. [Google Scholar] [CrossRef]
- Soares, A.P.V.; Portela, M.F.; Kiennemann, A. Iron molybdates for selective oxidation of methanol: Mo excess effects on the deactivation behaviour. Catal. Commun. 2001, 2, 159–164. [Google Scholar] [CrossRef]
- Routray, K.; Zhou, W.; Kiely, C.J.; Grünert, W.; Wachs, I.E. Origin of the synergistic interaction between MoO3 and iron molybdate for the selective oxidation of methanol to formaldehyde. J. Catal. 2010, 275, 84–98. [Google Scholar] [CrossRef]
- Allison, J.N.; Goddard, W.A. Oxidative dehydrogenation of methanol to formaldehyde. J. Catal. 1985, 92, 127–135. [Google Scholar] [CrossRef]
- Kim, T.H.; Ramachandra, B.; Choi, J.S.; Saidutta, M.; Choo, K.Y.; Song, S.-D.; Rhee, Y.-W. Selective oxidation of methanol to formaldehyde using modified iron-molybdate catalysts. Catal. Lett. 2004, 98, 161–165. [Google Scholar] [CrossRef]
- Brookes, C.; Wells, P.P.; Dimitratos, N.; Jones, W.; Gibson, E.K.; Morgan, D.J.; Cibin, G.; Nicklin, C.; Mora-Fonz, D.; Scanlon, D.O.; et al. The Nature of the Molybdenum Surface in Iron Molybdate. The Active Phase in Selective Methanol Oxidation. J. Phys. Chem. C 2014, 118, 26155–26161. [Google Scholar] [CrossRef]
- Uhlrich, J.J.; Sainio, J.; Lei, Y.; Edwards, D.; Davies, R.; Bowker, M.; Shaikhutdinov, S.; Freund, H.-J. Preparation and characterization of iron–molybdate thin films. Surf. Sci. 2011, 605, 1550–1555. [Google Scholar] [CrossRef]
- Yeo, B.R.; Pudge, G.J.; Bugler, K.G.; Rushby, A.V.; Kondrat, S.; Bartley, J.; Golunski, S.; Taylor, S.H.; Gibson, E.; Wells, P.P. The surface of iron molybdate catalysts used for the selective oxidation of methanol. Surf. Sci. 2016, 648, 163–169. [Google Scholar] [CrossRef]
- Trifiro, F. The chemistry of oxidation catalysts based on mixed oxides. Catal. Today 1998, 41, 21–35. [Google Scholar] [CrossRef]
- Alessandrini, G.; Cairati, L.; Forzatti, P.; Villa, P.; Trifirò, F. Chemical, structural and catalytic modifications of pure and doped iron (III) molybdate. J. Less Common Met. 1977, 54, 373–386. [Google Scholar] [CrossRef]
- Bowker, M.; Holroyd, R.; House, M.; Bracey, R.; Bamroongwongdee, C.; Shannon, M.; Carley, A. The Selective Oxidation of Methanol on Iron Molybdate Catalysts. Top. Catal. 2008, 48, 158–165. [Google Scholar] [CrossRef]
- Sutthiumporn, K.; Kawi, S. Promotional effect of alkaline earth over Ni–La2O3 catalyst for CO2 reforming of CH4: Role of surface oxygen species on H2 production and carbon suppression. Int. J. Hydrogen Energy 2011, 36, 14435–14446. [Google Scholar] [CrossRef]
- Wang, J.; Xia, Y.; Dong, Y.; Chen, R.; Xiang, L.; Komarneni, S. Defect-rich ZnO nanosheets of high surface area as an efficient visible-light photocatalyst. Appl. Catal. B Environ. 2016, 192, 8–16. [Google Scholar] [CrossRef]
- Han, Z.-S.; Pan, W.; Pan, W.-X.; Li, J.-I.; Zhu, Q.-M.; Tin, K.-C.; Wong, N.-B. Preparation and effect of Mo-V-Cr-Bi-Si oxide catalysts on controlled oxidation of methane to methanol and formaldehyde. Korean J. Chem. Eng. 1998, 15, 496–499. [Google Scholar] [CrossRef]
- Yang, T.L.; Feng, L.B.; Shen, S.K. Oxygen Species on the Surface of La2O3/CaO and Its Role in the Oxidative Coupling of Methane. J. Catal. 1994, 145, 384–389. [Google Scholar] [CrossRef]
- Bielański, A.; Haber, J. Oxygen in Catalysis on Transition Metal Oxides. Catal. Rev. 1979, 19, 1–41. [Google Scholar] [CrossRef]
- Hu, X.; Zhang, Z.; Zhang, Y.; Sun, L.; Tian, H.; Yang, X. Synthesis of a Highly Active and Stable Pt/Co3O4 Catalyst and Its Application for the Catalytic Combustion of Toluene. Eur. J. Inorg. Chem. 2019, 2019, 2933–2939. [Google Scholar] [CrossRef]
- Luo, L.; Shao, G.; Duan, Z. Catalytic Oxidation Properties and Characterization of LaSrCo0.9 B′0.1 O4 (B′= Mn, Fe, Ni, Cu) Mixed Oxides. Turk. J. Chem. 2006, 29, 597–605. [Google Scholar]
- Zhang, H.; Shen, J.; Ge, X. The Reduction Behavior of Fe-Mo-O Catalysts Studied by Temperature-Programmed Reduction Combined with in Situ Mössbauer Spectroscopy and X-Ray Diffraction. J. Solid State Chem. 1995, 117, 127–135. [Google Scholar] [CrossRef]
- Farneth, W.E.; Ohuchi, F.; Staley, R.H.; Chowdhry, U.; Sleight, A.W. Mechanism of partial oxidation of methanol over molybdenum (VI) oxide as studied by temperature-programmed desorption. J. Phys. Chem. 1985, 89, 2493–2497. [Google Scholar] [CrossRef]
- Machiels, C.; Sleight, A. Kinetic isotope effect in the selective oxidation of methanol to formaldehyde over some molybdate catalysts. J. Catal. 1982, 76, 1976–1989. [Google Scholar] [CrossRef]
- Bruckman, K.; TATIBouÈT, J.-M.; Che, M.; Serwicka, E.; Haber, J. Catalytic Behavior of Unsupported and Heteropolysalt-Supported H3+nPVnMo12−nO40 Heteropolyacids in the Test Reaction of CH3OH Oxidation. J. Catal. 1993, 139, 455–467. [Google Scholar] [CrossRef]
- Chung, J.S.; Miranda, R.; Bennett, C.O. Mechanism of partial oxidation of methanol over MoO3. J. Catal. 1988, 114, 398–410. [Google Scholar] [CrossRef]
- Van Oeffelen, D.; Van Hooff, J.; Schuit, G. In situ measurements of the electrical conductivity of bismuth molybdate catalysts in operation for oxidative dehydrogenation of butene. J. Catal. 1985, 95, 84–100. [Google Scholar] [CrossRef]
- Raun, K.V.; Lundegaard, L.F.; Beato, P.; Appel, C.C.; Nielsen, K.; Thorhauge, M.; Schumann, M.; Jensen, A.D.; Grunwaldt, J.-D.; Høj, M. Stability of Iron-Molybdate Catalysts for Selective Oxidation of Methanol to Formaldehyde: Influence of Preparation Method. Catal. Lett. 2019, 150, 1434–1444. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds Cat-SS and Cat-FS are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mo/Fe Mole Ratio-EDS | Mo/Fe Mole Ratio | Surface Area/m2·g−1 | |||
|---|---|---|---|---|---|
| Plate | Particles | ICP | XPS | ||
| Cat-SS | 5.5 | 1.6 | 2.50/2.50 | 2.49/2.51 | 15.6 |
| Cat-FS | 4.8 | 1.8 | 2.50/2.51 | 2.54/2.53 | 18.8 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Han, M. Effect of Mo Dispersion on the Catalytic Properties and Stability of Mo–Fe Catalysts for the Partial Oxidation of Methanol. Molecules 2020, 25, 2410. https://doi.org/10.3390/molecules25102410
Zhang S, Han M. Effect of Mo Dispersion on the Catalytic Properties and Stability of Mo–Fe Catalysts for the Partial Oxidation of Methanol. Molecules. 2020; 25(10):2410. https://doi.org/10.3390/molecules25102410
Chicago/Turabian StyleZhang, Shuai, and Minghan Han. 2020. "Effect of Mo Dispersion on the Catalytic Properties and Stability of Mo–Fe Catalysts for the Partial Oxidation of Methanol" Molecules 25, no. 10: 2410. https://doi.org/10.3390/molecules25102410
APA StyleZhang, S., & Han, M. (2020). Effect of Mo Dispersion on the Catalytic Properties and Stability of Mo–Fe Catalysts for the Partial Oxidation of Methanol. Molecules, 25(10), 2410. https://doi.org/10.3390/molecules25102410