
Deboronation-Induced Ratiometric Emission Variations of Terphenyl-Based Closo-o-Carboranyl Compounds: Applications to Fluoride-Sensing

 , ,
, ,
Abstract
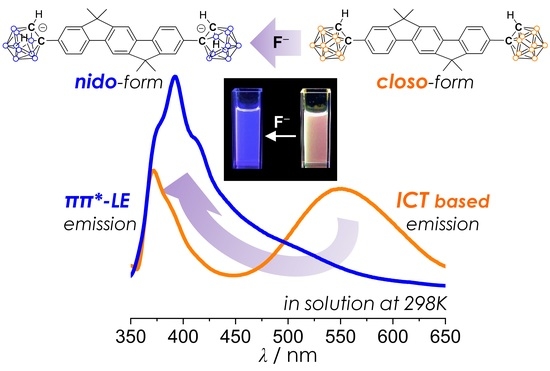
1. Introduction
2. Results and Discussion
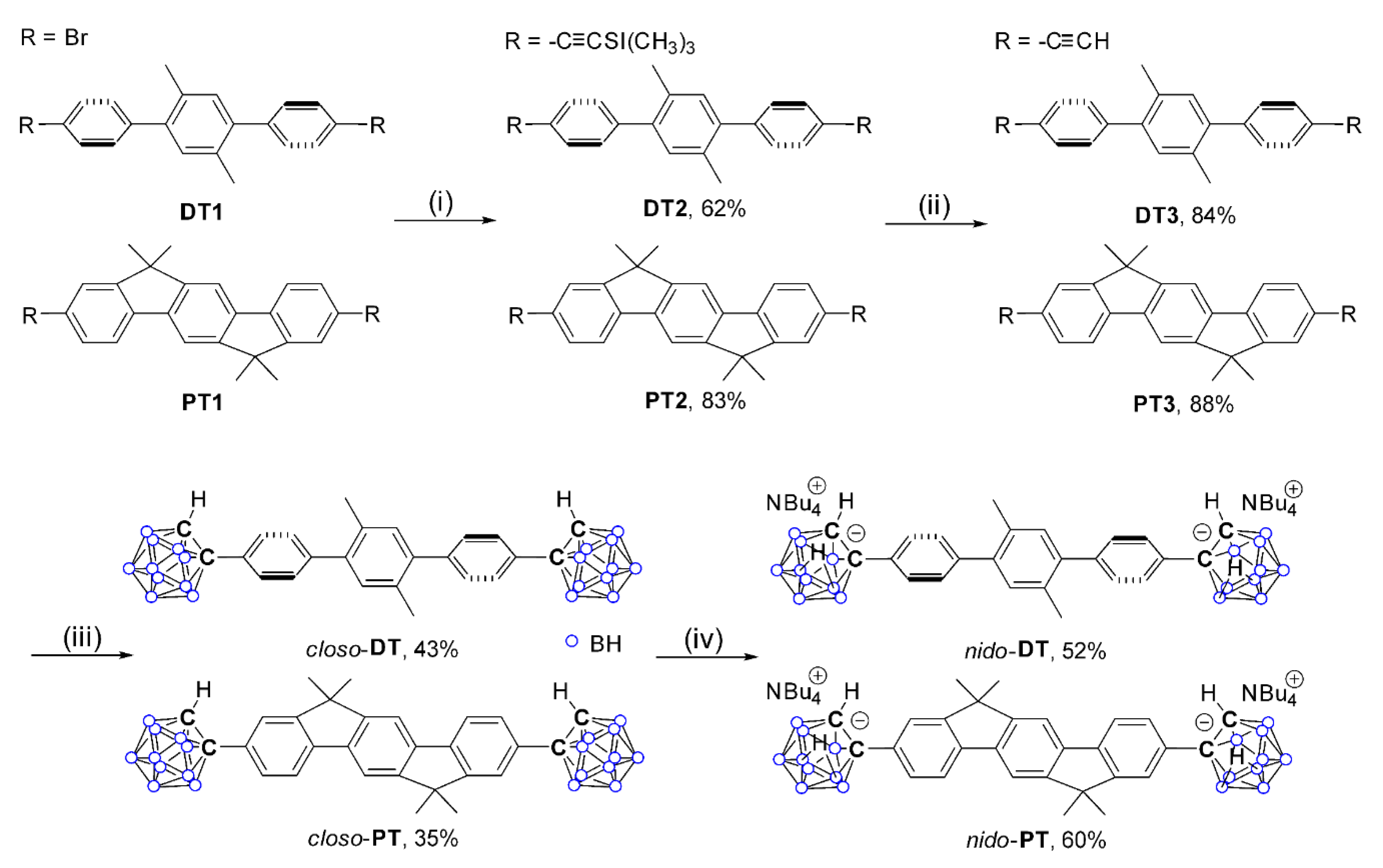
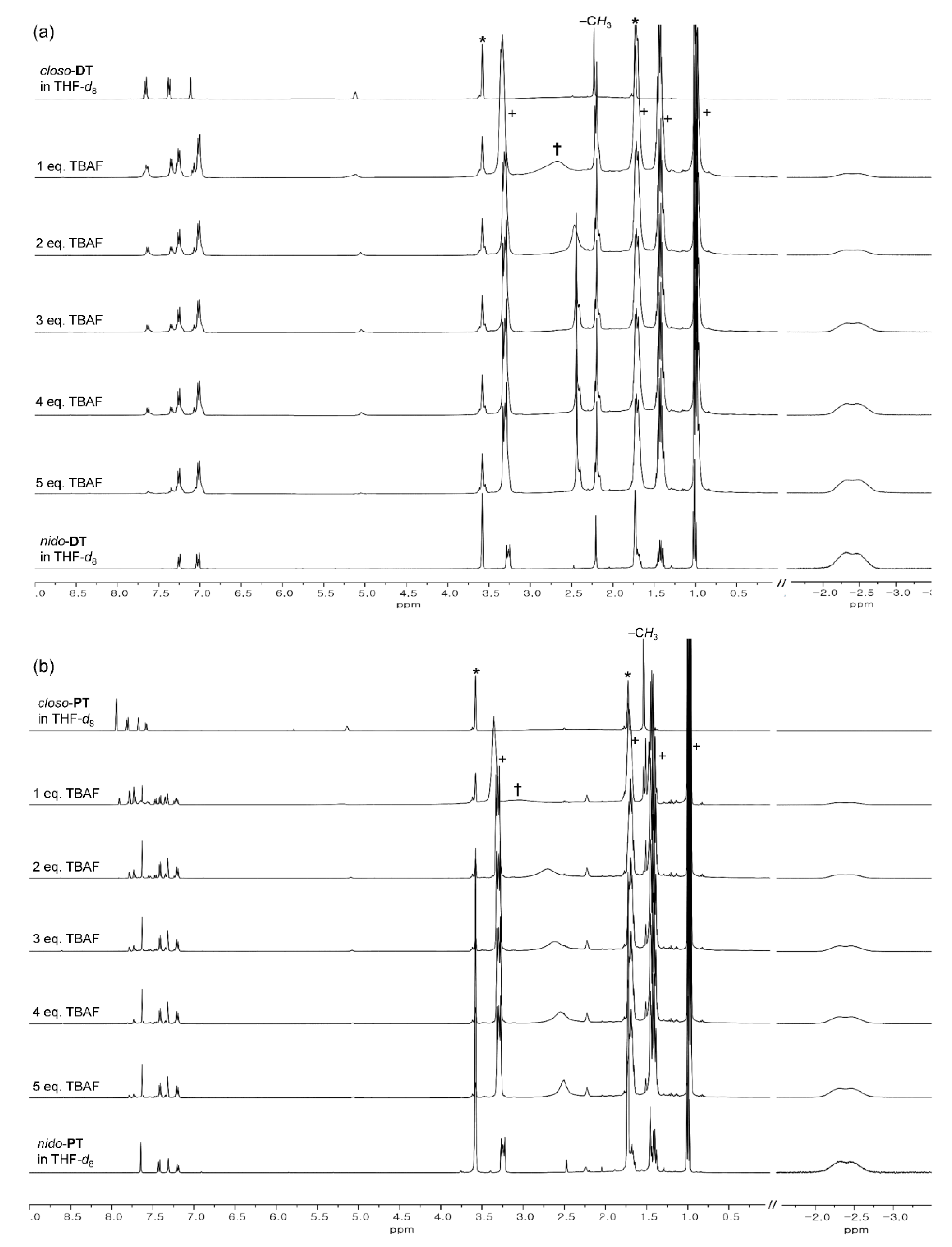
2.1. Synthesis and Characterization
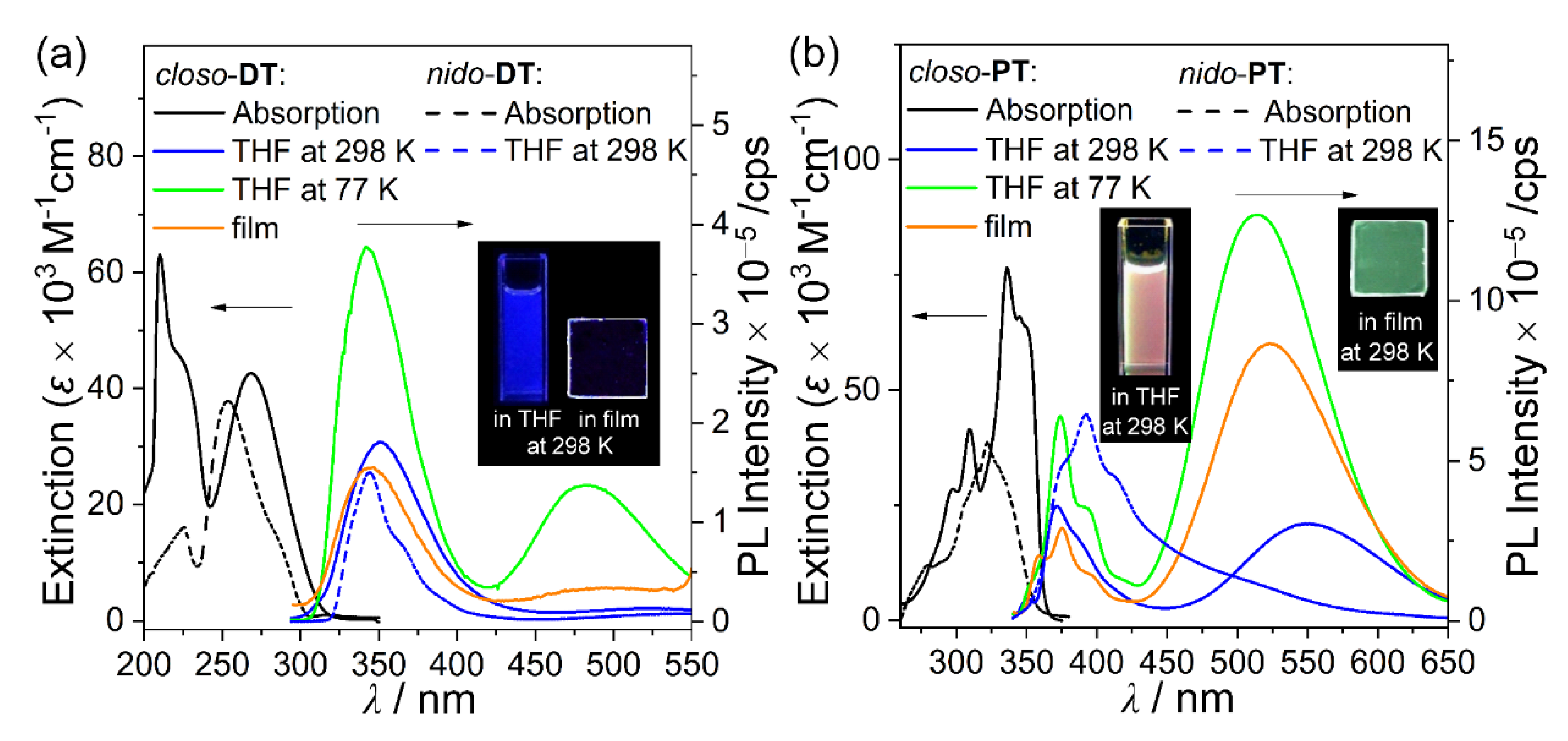
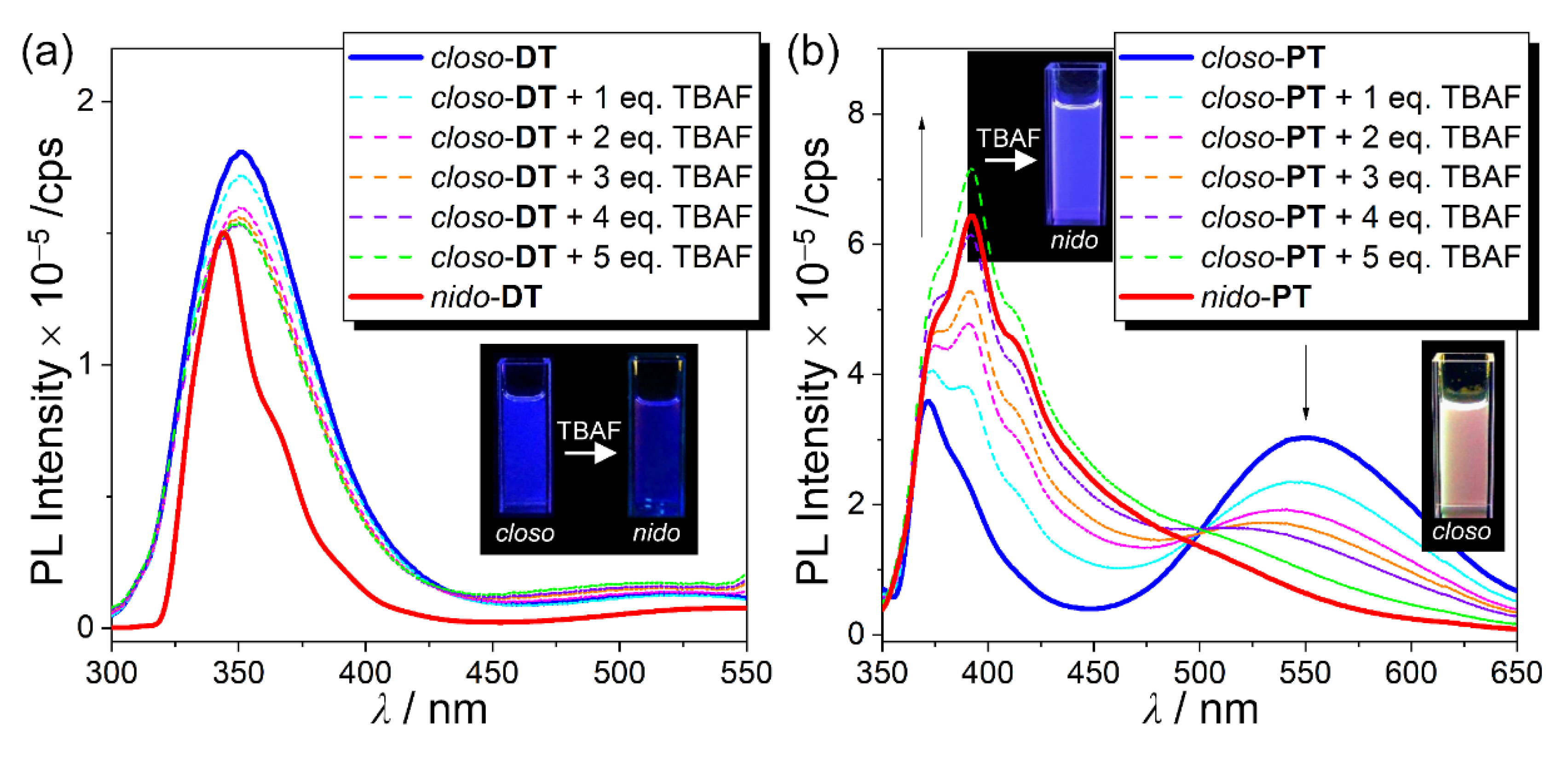
2.2. Photophysical Properties of the Closo- and Nido-o-Carboranyl Compounds
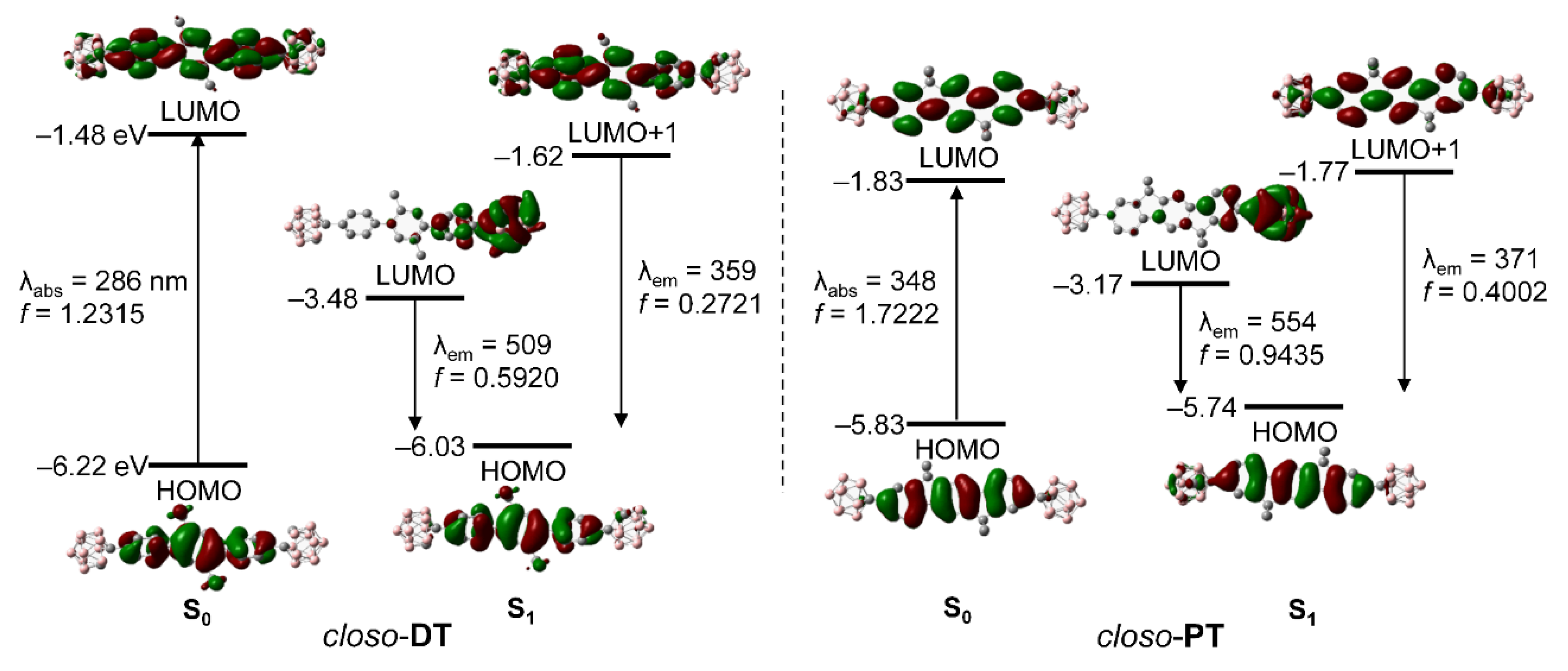
2.3. Computational Chemistry and Orbital Analyses for Closo-o-Carboranyl Compounds
2.4. Emission-Color Changes of Closo-o-Carboranyl Compounds Via Treatment of Fluoride Anion
3. Materials and Methods
3.1. General Considerations
3.2. Synthesis of DT2
3.3. Synthesis of PT2
3.4. Synthesis of DT3
3.5. Synthesis of PT3
3.6. Synthesis of closo-DT
3.7. Synthesis of closo-PT
3.8. Synthesis of nido-DT
3.9. Synthesis of nido-PT
3.10. UV/Vis Absorption and Photoluminescence (PL) Experiments
3.11. Computational Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bregadze, V.I. Dicarba-closo-dodecaboranes C2B10H12 and their derivatives. Chem. Rev. 1992, 92, 209–223. [Google Scholar] [CrossRef]
- González-Campo, A.; Juárez-Pérez, E.J.; Viñas, C.; Boury, B.; Sillanpää, R.; Kivekäs, R.; Núñez, R. Carboranyl Substituted Siloxanes and Octasilsesquioxanes: Synthesis, Characterization, and Reactivity. Macromolecules. 2008, 41, 8458–8466. [Google Scholar] [CrossRef]
- Issa, F.; Kassiou, M.; Rendina, L.M. Boron in Drug Discovery: Carboranes as Unique Pharmacophores in Biologically Active Compounds. Chem. Rev. 2011, 111, 5701–5722. [Google Scholar] [CrossRef]
- Wee, K.-R.; Cho, Y.-J.; Jeong, S.; Kwon, S.; Lee, J.-D.; Suh, I.-H.; Kang, S.O. Carborane-Based Optoelectronically Active Organic Molecules: Wide Band Gap Host Materials for Blue Phosphorescence. J. Am. Chem. Soc. 2012, 134, 17982–17990. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Ugalde, A.; Juárez-Pérez, E.J.; Teixidor, F.; Viñas, C.; Núñez, R. Synthesis, Characterization, and Thermal Behavior of Carboranyl–Styrene Decorated Octasilsesquioxanes: Influence of the Carborane Clusters on Photoluminescence. Chem. Eur. J. 2013, 19, 17021–17030. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Kim, H.; Lee, K.M.; Lee, Y.S.; Lee, M.H. Phosphorescence Color Tuning of Cyclometalated Iridium Complexes by o-Carborane Substitution. Inorg. Chem. 2013, 52, 160–168. [Google Scholar] [CrossRef]
- Bae, H.J.; Chung, J.; Kim, H.; Park, J.; Lee, K.M.; Koh, T.-W.; Lee, Y.S.; Yoo, S.; Do, Y.; Lee, M.H. Deep Red Phosphorescence of Cyclometalated Iridium Complexes by o-Carborane Substitution. Inorg. Chem. 2014, 53, 128–138. [Google Scholar] [CrossRef]
- Asay, M.J.; Fisher, S.P.; Lee, S.E.; Tham, F.S.; Borchardt, D.; Lavallo, V. Synthesis of unsymmetrical N-carboranyl NHCs: Directing effect of the carborane anion. Chem. Commun. 2015, 51, 5359–5362. [Google Scholar] [CrossRef]
- Lee, Y.H.; Park, J.; Jo, S.-J.; Kim, M.; Lee, J.; Lee, S.U.; Lee, M.H. Manipulation of Phosphorescence Efficiency of Cyclometalated Iridium Complexes by Substituted o-Carboranes. Chem. Eur. J. 2015, 21, 2052–2061. [Google Scholar] [CrossRef]
- Núñez, R.; Tarrés, M.; Ferrer-Ugalde, A.; Fabrizi de Biani, F.; Teixidor, F. Electrochemistry and Photoluminescence of Icosahedral Carboranes, Boranes, Metallacarboranes, and Their Derivatives. Chem. Rev. 2016, 116, 14307–14378. [Google Scholar] [CrossRef]
- Mukherjee, S.; Thilagar, P. Boron clusters in luminescent materials. Chem. Commun. 2016, 52, 1070–1093. [Google Scholar] [CrossRef]
- Dziedzic, R.M.; Saleh, L.M.A.; Axtell, J.C.; Martin, J.L.; Stevens, S.L.; Royappa, A.T.; Rheingold, A.L.; Spokoyny, A.M. B–N, B–O, and B–CN Bond Formation via Palladium-Catalyzed Cross-Coupling of B-Bromo-Carboranes. J. Am. Chem. Soc. 2016, 138, 9081–9084. [Google Scholar] [CrossRef]
- Kirlikovali, K.O.; Axtell, J.C.; Gonzalez, A.; Phung, A.C.; Khan, S.I.; Spokoyny, A.M. Luminescent metal complexes featuring photophysically innocent boron cluster ligands. Chem. Sci. 2016, 7, 5132–5138. [Google Scholar] [CrossRef]
- Saleh, L.M.A.; Dziedzic, R.M.; Khan, S.I.; Spokoyny, A.M. Forging Unsupported Metal–Boryl Bonds with Icosahedral Carboranes. Chem. Eur. J. 2016, 22, 8466–8470. [Google Scholar] [CrossRef]
- Eleazer, B.J.; Smith, M.D.; Popov, A.A.; Peryshkov, D.V. (BB)-Carboryne Complex of Ruthenium: Synthesis by Double B–H Activation at a Single Metal Center. J. Am. Chem. Soc. 2016, 138, 10531–10538. [Google Scholar] [CrossRef]
- Wong, Y.O.; Smith, M.D.; Peryshkov, D.V. Synthesis of the First Example of the 12-Vertex-closo/12-Vertex-nido Biscarborane Cluster by a Metal-Free B−H Activation at a Phosphorus(III) Center. Chem. Eur. J. 2016, 22, 6764–6767. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.L.; Estrada, J.; Kefalidis, C.E.; Lavallo, V. Changing the Charge: Electrostatic Effects in Pd-Catalyzed Cross-Coupling. Organometallics 2016, 35, 3257–3260. [Google Scholar] [CrossRef]
- Fisher, S.P.; El-Hellani, A.; Tham, F.S.; Lavallo, V. Anionic and zwitterionic carboranyl N-heterocyclic carbene Au(I) complexes. Dalton Trans. 2016, 45, 9762–9765. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Park, S.; Lee, Y.H.; Jung, J.; Yoo, S.; Lee, M.H. Homoleptic Tris-Cyclometalated Iridium Complexes with Substituted o-Carboranes: Green Phosphorescent Emitters for Highly Efficient Solution-Processed Organic Light-Emitting Diodes. Inorg. Chem. 2016, 55, 909–917. [Google Scholar] [CrossRef]
- Tu, D.; Leong, P.; Guo, S.; Yan, H.; Lu, C.; Zhao, Q. Highly Emissive Organic Single-Molecule White Emitters by Engineering o-Carborane-Based Luminophores. Angew. Chem. Int. Ed. 2017, 56, 11370–11374. [Google Scholar] [CrossRef]
- Kirlikovali, K.O.; Axtell, J.C.; Anderson, K.; Djurovich, P.I.; Rheingold, A.L.; Spokoyny, A.M. Fine-Tuning Electronic Properties of Luminescent Pt(II) Complexes via Vertex-Differentiated Coordination of Sterically Invariant Carborane-Based Ligands. Organometallics 2018, 37, 3122–3131. [Google Scholar] [CrossRef]
- Nar, I.; Atsay, A.; Altındal, A.; Hamuryudan, E. o-Carborane, Ferrocene, and Phthalocyanine Triad for High-Mobility Organic Field-Effect Transistors. Inorg. Chem. 2018, 57, 2199–2208. [Google Scholar] [CrossRef] [PubMed]
- Grimes, R.N. Carboranes, 2nd ed.; Academic Press: London, UK, 2011. [Google Scholar]
- Spokoyny, A.M. New ligand platforms featuring boron-rich clusters as organomimetic substituents. Pure Appl. Chem. 2013, 85, 903–919. [Google Scholar] [CrossRef]
- Poater, J.; Solà, M.; Viñas, C.; Teixidor, F. π Aromaticity and Three-Dimensional Aromaticity: Two sides of the Same Coin? Angew. Chem. Int. Ed. 2014, 53, 12191–12195. [Google Scholar] [CrossRef]
- Poater, J.; Solà, M.; Viñas, C.; Teixidor, F. Hückel's Rule of Aromaticity Categorizes Aromatic closo Boron Hydride Clusters. Chem. Eur. J. 2016, 22, 7437–7443. [Google Scholar] [CrossRef] [PubMed]
- Núñez, R.; Romero, I.; Teixidor, F.; Viñas, C. Icosahedral boron clusters: A perfect tool for the enhancement of polymer features. Chem. Soc. Rev. 2016, 45, 5147–5173. [Google Scholar] [CrossRef]
- Cabrera-González, J.; Sánchez-Arderiu, V.; Viñas, C.; Parella, T.; Teixidor, F.; Náñez, R. Redox-Active Metallacarborane-Decorated Octasilsesquioxanes. Electrochemical and Thermal Properties. Inorg. Chem. 2016, 55, 11630–11634. [Google Scholar] [CrossRef]
- Kokado, K.; Chujo, Y. Multicolor Tuning of Aggregation-Induced Emission through Substituent Variation of Diphenyl-o-carborane. J. Org. Chem. 2011, 76, 316–319. [Google Scholar] [CrossRef]
- Dash, B.P.; Satapathy, R.; Gaillard, E.R.; Norton, K.M.; Maguire, J.A.; Chug, N.; Hosmane, N.S. Enhanced π-Conjugation and Emission via Icosahedral Carboranes: Synthetic and Spectroscopic Investigation. Inorg. Chem. 2011, 50, 5485–5493. [Google Scholar] [CrossRef]
- Wee, K.-R.; Han, W.-S.; Cho, D.W.; Kwon, S.; Pac, C.; Kang, S.O. Carborane photochemistry triggered by aryl substitution: Carborane-based dyads with phenyl carbazoles. Angew. Chem. Int. Ed. 2012, 51, 2677–2680. [Google Scholar] [CrossRef]
- Weber, L.; Kahlert, J.; Brockhinke, R.; Böhling, L.; Brockhinke, A.; Stammler, H.-G.; Neumann, B.; Harder, R.A.; Fox, M.A. Luminescence Properties of C-Diazaborolyl-ortho-Carboranes as Donor–Acceptor Systems. Chem. Eur. J. 2012, 18, 8347–8357. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.J.; Kim, H.; Lee, K.M.; Kim, T.; Eo, M.; Lee, Y.S.; Do, Y.; Lee, M.H. Heteroleptic tris-cyclometalated iridium(III) complexes supported by an o-carboranyl-pyridine ligand. Dalton Trans. 2013, 42, 8549–8552. [Google Scholar] [CrossRef] [PubMed]
- Weber, L.; Kahlert, J.; Brockhinke, R.; Böhling, L.; Halama, J.; Brockhinke, A.; Stammler, H.-G.; Neumann, B.; Nervi, C.; Harder, R.A.; et al. C,C′-Bis(benzodiazaborolyl)dicarba-closo-dodecaboranes: Synthesis, structures, photophysics and electrochemistry. Dalton Trans. 2013, 42, 10982–10996. [Google Scholar] [CrossRef] [PubMed]
- Weber, L.; Kahlert, J.; Böhling, L.; Brockhinke, A.; Stammler, H.-G.; Neumann, B.; Harder, R.A.; Low, P.J.; Fox, M.A. Electrochemical and spectroelectrochemical studies of C-benzodiazaborolyl-ortho-carboranes. Dalton Trans. 2013, 42, 2266–2281. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.; Wee, K.-R.; Cho, Y.-J.; Kang, S.O. Carborane Dyads for Photoinduced Electron Transfer: Photophysical Studies on Carbazole and Phenyl-o-carborane Molecular Assemblies. Chem. Eur. J. 2014, 20, 5953–5960. [Google Scholar] [CrossRef]
- Ferrer-Ugalde, A.; González-Campo, A.; Viñas, C.; Rodríguez-Romero, J.; Santillan, R.; Farfán, N.; Sillanpää, R.; Sousa-Pedrares, A.; Núñez, R.; Teixidor, F. Fluorescence of New o-Carborane Compounds with Different Fluorophores: Can it be Tuned? Chem. Eur. J. 2014, 20, 9940–9951. [Google Scholar] [CrossRef]
- Bae, H.J.; Kim, H.; Lee, K.M.; Kim, T.; Lee, Y.S.; Do, Y.; Lee, M.H. Through-space charge transfer and emission color tuning of di-o-carborane substituted benzene. Dalton Trans. 2014, 43, 4978–4985. [Google Scholar] [CrossRef]
- Lee, Y.H.; Park, J.; Lee, J.; Lee, S.U.; Lee, M.H. Iridium Cyclometalates with Tethered o-Carboranes: Impact of Restricted Rotation of o-Carborane on Phosphorescence Efficiency. J. Am. Chem. Soc. 2015, 137, 8018–8021. [Google Scholar] [CrossRef]
- Naito, H.; Morisaki, Y.; Chujo, Y. o-Carborane-Based Anthracene: A Variety of Emission Behaviors. Angew. Chem. Int. Ed. 2015, 54, 5084–5087. [Google Scholar] [CrossRef]
- Kim, T.; Lee, J.; Lee, S.U.; Lee, M.H. o-Carboranyl–Phosphine as a New Class of Strong-Field Ancillary Ligand in Cyclometalated Iridium(III) Complexes: Toward Blue Phosphorescence. Organometallics 2015, 34, 3455–3458. [Google Scholar] [CrossRef]
- Choi, B.H.; Lee, J.H.; Hwang, H.; Lee, K.M.; Park, M.H. Novel Dimeric o-Carboranyl Triarylborane: Intriguing Ratiometric Color-Tunable Sensor via Aggregation-Induced Emission by Fluoride Anions. Organometallics 2016, 35, 1771–1777. [Google Scholar] [CrossRef]
- Wee, K.-R.; Cho, Y.-J.; Song, J.K.; Kang, S.O. Multiple photoluminescence from 1,2-dinaphthyl-ortho-carborane. Angew. Chem. Int. Ed. 2013, 52, 9682–9685. [Google Scholar] [CrossRef] [PubMed]
- Naito, H.; Nishino, K.; Morisaki, Y.; Tanaka, K.; Chujo, Y. Solid-State Emission of the Anthracene-o-Carborane Dyad from the Twisted-Intramolecular Charge Transfer in the Crystalline State. Angew. Chem. Int. Ed. 2017, 56, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Guo, J.; Cao, Y.; Zhao, J.; Jia, W.; Chen, Y.; Jia, D. Mechanically triggered reversible stepwise tricolor switching and thermochromism of anthracene-o-carborane dyad. Chem. Sci. 2018, 9, 5270–5277. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yang, C.; Peng, X.; Chen, Y.; Qi, Q.; Luo, X.; Lai, W.-Y.; Huang, W. Stimuli-responsive solid-state emission from o-carborane–tetraphenylethene dyads induced by twisted intramolecular charge transfer in the crystalline state. J. Mater. Chem. C. 2018, 6, 19–28. [Google Scholar] [CrossRef]
- Nishino, K.; Yamamoto, H.; Tanaka, K.; Chujo, Y. Development of Solid-State Emissive Materials Based on Multifunctional o-Carborane–Pyrene Dyads. Org. Lett. 2016, 18, 4064–4067. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Guo, J.; Zhao, J.; Che, Y.; Jia, D.; Chen, Y. Multifunctional luminescent molecules of o-carborane-pyrene dyad/triad: Flexible synthesis and study of the photophysical properties. Dyes and Pigm. 2018, 154, 44–51. [Google Scholar] [CrossRef]
- Marsh, A.V.; Cheetham, N.J.; Little, M.; Dyson, M.; White, A.J.P.; Beavis, P.; Warriner, C.N.; Swain, A.C.; Stavrinou, P.N.; Heeney, M. Carborane-Induced Excimer Emission of Severely Twisted Bis-o-Carboranyl Chrysene. Angew. Chem. Int. Ed. 2018, 57, 10640–10645. [Google Scholar] [CrossRef]
- Kim, S.-Y.; Cho, Y.-J.; Jin, G.F.; Han, W.-S.; Son, H.-J.; Cho, D.W.; Kang, S.O. Intriguing emission properties of triphenylamine–carborane systems. Phys. Chem. Chem. Phys. 2015, 17, 15679–15682. [Google Scholar] [CrossRef]
- Wan, Y.; Li, J.; Peng, X.; Huang, C.; Qi, Q.; Lai, W.-Y.; Huang, W. Intramolecular charge transfer induced emission from triphenylamine-o-carborane dyads. RSC Adv. 2017, 7, 35543–35548. [Google Scholar] [CrossRef]
- Nishino, K.; Uemura, K.; Gon, M.; Tanaka, K.; Chujo, Y. Enhancement of Aggregation-Induced Emission by Introducing Multiple o-Carborane Substitutions into Triphenylamine. Molecules. 2017, 22, 2009. [Google Scholar] [CrossRef] [PubMed]
- Nishino, K.; Uemura, K.; Tanaka, K.; Morisaki, Y.; Chujo, Y. Modulation of the cis- and trans-Conformations in Bis-ocarborane Substituted Benzodithiophenes and Emission Enhancement Effect on Luminescent Efficiency by Solidification. Eur. J. Org. Chem. 2018, 12, 1507–1512. [Google Scholar] [CrossRef]
- Ferrer-Ugalde, A.; Juárez-Pérez, E.J.; Teixidor, F.; Viñas, C.; Sillanpää, R.; Pérez-Inestrosa, E.; Núñez, R. Synthesis and Characterization of New Fluorescent Styrene-Containing Carborane Derivatives: The Singular Quenching Role of a Phenyl Substituent. Chem. Eur. J. 2012, 18, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Tu, D.; Leong, P.; Li, Z.; Hu, R.; Shi, C.; Zhang, K.Y.; Yan, H.; Zhao, Q. A carborane-triggered metastable charge transfer state leading to spontaneous recovery of mechanochromic luminescence. Chem. Commun. 2016, 52, 12494–12497. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Ugalde, A.; Cabrera-González, J.; Juárez-Pérez, E.J.; Teixidor, F.; Pérez-Inestrosa, E.; Montenegro, J.M.; Sillanpää, R.; Haukka, M.; Núñez, R. Carborane–stilbene dyads: The influence of substituents and cluster isomers on photoluminescence properties. Dalton Trans. 2017, 46, 2091–2104. [Google Scholar] [CrossRef]
- Li, X.; Yin, Y.; Yan, H.; Lu, C. Aggregation-Induced Emission Characteristics of o-Carborane-Functionalized Tetraphenylethylene Luminogens: The Influence of Carborane Cages on Photoluminescence. Chem. Asian J. 2017, 12, 2207–2210. [Google Scholar] [CrossRef]
- Kaiser, R.P.; Mosinger, J.; Císařová, I.; Kotora, M. Synthesis of selectively 4-substituted 9,9′-spirobifluorenes and modulation of their photophysical properties. Org. Biomol. Chem. 2017, 15, 6913–6920. [Google Scholar] [CrossRef]
- Santos, W.G.; Budkina, D.S.; Deflon, V.M.; Tarnovsky, A.N.; Cardoso, D.R.; Forbes, M.D.E. Photoinduced Charge Shifts and Electron Transfer in Viologen–Tetraphenylborate Complexes: Push–Pull Character of the Exciplex. J. Am. Chem. Soc. 2017, 139, 7681–7684. [Google Scholar] [CrossRef]
- Naito, H.; Nishino, K.; Morisaki, Y.; Tanaka, K.; Chujo, Y. Luminescence Color Tuning from Blue to Near Infrared of Stable Luminescent Solid Materials Based on Bis-o-Carborane-Substituted Oligoacenes. Chem. Asian J. 2017, 12, 2134–2138. [Google Scholar] [CrossRef]
- Naito, H.; Nishino, K.; Morisaki, Y.; Tanaka, K.; Chujo, Y. Highly-efficient solid-state emissions of anthracene–o-carborane dyads with various substituents and their thermochromic luminescence properties. J. Mater. Chem. C. 2017, 5, 10047–10054. [Google Scholar] [CrossRef]
- Wu, X.; Guo, J.; Quan, Y.; Jia, W.; Jia, D.; Chen, Y.; Xie, Z. Cage carbon-substitute does matter for aggregation-induced emission features of o-carborane-functionalized anthracene triads. J. Mater. Chem. C. 2018, 6, 4140–4149. [Google Scholar] [CrossRef]
- Mori, H.; Nishino, K.; Wada, K.; Morisaki, Y.; Tanaka, K.; Chujo, Y. Modulation of luminescence chromic behaviors and environment-responsive intensity changes by substituents in bis-o-carborane-substituted conjugated molecules. Mater. Chem. Front. 2018, 2, 573–579. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, J.; Wu, X.; Jia, D.; Tong, F. Color-tuning aggregation-induced emission of o-Carborane-bis(1,3,5-triaryl-2-pyrazoline) triads: Preparation and investigation of the photophysics. Dyes Pigm. 2018, 148, 180–188. [Google Scholar] [CrossRef]
- Kim, S.-Y.; Lee, J.-D.; Cho, Y.-J.; Son, M.R.; Son, H.-J.; Cho, D.W.; Kang, S.O. Excitation spectroscopic and synchronous fluorescence spectroscopic analysis of the origin of aggregation-induced emission in N,N-diphenyl-1-naphthylamine-o-carborane derivatives. Phys. Chem. Chem. Phys. 2018, 20, 17458–17463. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.; Yu, S.; Lee, J.H.; Hwang, H.; Lee, K.M. Biphenyl- and Fluorene-Based o-Carboranyl Compounds: Alteration of Photophysical Properties by Distortion of Biphenyl Rings. Organometallics 2017, 36, 1522–1529. [Google Scholar] [CrossRef]
- Jin, H.; Bae, H.J.; Kim, S.; Lee, J.H.; Hwang, H.; Park, M.H.; Lee, K.M. 2-Phenylpyridine- and 2-(benzo[b]thiophen-2-yl)pyridine-based o-carboranyl compounds: Impact of the structural formation of aromatic rings on photophysical properties. Dalton Trans. 2019, 48, 1467–1476. [Google Scholar] [CrossRef]
- Jin, H.; Kim, S.; Bae, H.J.; Lee, J.H.; Hwang, H.; Park, M.H.; Lee, K.M. Effect of Planarity of Aromatic Rings Appended to o-Carborane on Photophysical Properties: A Series of o-Carboranyl Compounds Based on 2-Phenylpyridine- and 2-(Benzo[b]thiophen-2-yl)pyridine. Molecules 2019, 24, 201. [Google Scholar] [CrossRef]
- So, H.; Kim, J.H.; Lee, J.H.; Hwang, H.; An, D.K.; Lee, K.M. Planarity of terphenyl rings possessing o-carborane cages: Turning on intramolecular-charge-transfer-based emission. Chem. Commun. 2019, 55, 14518. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J.H.; So, H.; Ryu, J.; Lee, J.; Hwang, H.; Kim, Y.; Park, M.H.; Lee, K.M. Spirobifluorene-Based o-Carboranyl Compounds: Insights into the Rotational Effect of Carborane Cages on Photoluminescence. Chem. Eur. J. 2020, 26, 548. [Google Scholar] [CrossRef]
- Nishino, K.; Morisaki, Y.; Tanaka, K.; Chujo, Y. Electron-donating abilities and luminescence properties of tolane-substituted nido-carboranes. New J. Chem. 2017, 41, 10550. [Google Scholar] [CrossRef]
- Fox, M.A.; Gill, W.R.; Herbertson, P.L.; MacBride, J.A.H.; Wade, K.; Colquhoun, H.M. Deboronation of C-substituted ortho- and meta-closo-carboranes using “wet” fluoride ion solutions. Polyhedron 1996, 15, 565. [Google Scholar] [CrossRef]
- Yoo, J.; Hwang, J.-W.; Do, Y. Facile and Mild Deboronation of o-Carboranes Using Cesium Fluoride. Inorg. Chem. 2001, 40, 568. [Google Scholar] [CrossRef]
- Peterson, J.J.; Werre, M.; Simon, Y.C.; Coughlin, E.B.; Carter, K.R. Carborane-Containing Polyfluorene: O-Carborane in the Main Chain. Macromolecules 2009, 42, 8594. [Google Scholar] [CrossRef]
- Lerouge, F.; Ferrer-Ugalde, A.; Viñas, C.; Teixidor, F.; Sillanpää, R.; Abreu, A.; Xochitiotzi, E.; Farfán, N.; Santillan, R.; Núñez, R. Synthesis and fluorescence emission of neutral and anionic di- and tetra-carboranyl compounds. Dalton Trans. 2011, 40, 7541. [Google Scholar] [CrossRef]
- Park, M.H.; Lee, K.M.; Kim, T.; Do, Y.; Lee, M.H. Ortho-Carborane-Functionalized Luminescent Polyethylene: Potential Chemodosimeter for the Sensing of Nucleophilic Anions. Chem. Asian J. 2011, 6, 1362. [Google Scholar] [CrossRef]
- You, D.K.; Lee, J.H.; Hwang, H.; Kwon, H.; Park, M.H.; Lee, K.M. Deboronation-induced ratiometric emission sensing of fluoride by 1,3,5-tris-(o-carboranyl-methyl)benzene. Tetrahedron Lett. 2017, 58, 3246. [Google Scholar] [CrossRef]
- Song, K.C.; Kim, H.; Lee, K.M.; Lee, Y.S.; Do, Y.; Lee, M.H. Dual sensing of fluoride ions by the o-carborane–triarylborane dyad. Dalton Trans. 2013, 42, 2351. [Google Scholar] [CrossRef]
- Hawthorne, M.F.; Berry, T.E.; Wegner, P.A. The Electronic Properties of the 1,2- and 1,7-Dicarbaclovododecaborane(12) Groups Bonded at Carbon. J. Am. Chem. Soc. 1965, 87, 4746–4750. [Google Scholar] [CrossRef]
- Paxson, T.E.; Callahan, K.P.; Hawthorne, M.F. Improved synthesis of biscarborane and its precursor ethynylcarborane. Inorg. Chem. 1973, 12, 708–709. [Google Scholar] [CrossRef]
- Jiang, W.; Knobler, C.B.; Hawthorne, M.F. Synthesis and Structural Characterization of Bis- and Tris(closo-1,2-C2B10H11-1-yl)-Substituted Biphenyl and Benzene. Inorg. Chem. 1996, 35, 3056–3058. [Google Scholar] [CrossRef]
- Lee, J.H.; Hwang, H.; Lee, K.M. p-Terphenyl-based di-o-carboranyl compounds: Alteration of electronic transition state by terminal phenyl groups. J. Organomet. Chem. 2016, 825–826, 69–74. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A. 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Binkley, J.S.; Pople, J.A.; Hehre, W.J. Self-consistent molecular orbital methods. 21. Small split-valence basis sets for first-row elements. J. Am. Chem. Soc. 1980, 102, 939–947. [Google Scholar] [CrossRef]
- Runge, E.; Gross, E.K.U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997. [Google Scholar] [CrossRef]
- Gaussian, version 09 B.01; Gaussian. Inc.: Wallingford, CT, USA, 2016.
- O'Boyle, N.M.; Tenderholt, A.L.; Langner, K.M. cclib: A library for package-independent computational chemistry algorithms. J. Comp. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef]
- GaussView, version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2016.
Sample Availability: Samples of the o-carboranyl compounds are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | λabs1/nm (ε × 10−3 M−1 cm−1) | λex/nm | λem/nm | ||||
|---|---|---|---|---|---|---|---|
| Tol 2 | THF 2 | DCM 2 | 77 K 1 | Film 3 | |||
| closo-DT | 268 (42.6) | 292 | 349 | 350 | 349 | 343, 482 | 345, 492(sh) |
| nido-DT | 254 (37.8) | 292 | - | 343 | - | - | - |
| closo-PT | 336 (84.8) | 345 | 376, 521 | 374, 549 | 375, 560 | 374, 514 | 375, 524 |
| nido-PT | 323 (38.2) | 345 | - | 390 | - | - | - |
| State | λcalc/nm | fcalc | Assignment | |
|---|---|---|---|---|
| closo-DT | S0 | 285.7 | 1.2315 | HOMO → LUMO (98.0%) |
| S1 | 509.37 | 0.592 | HOMO → LUMO (99.6%) | |
| 359.14 | 0.2721 | HOMO → LUMO+1 (87.7%) | ||
| closo-PT | S0 | 348.17 | 1.7222 | HOMO → LUMO (98.8%) |
| S1 | 554.44 | 0.9435 | HOMO → LUMO (99.7%) | |
| 371.34 | 0.4002 | HOMO → LUMO+1 (78.9%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
So, H.; Mun, M.S.; Kim, M.; Kim, J.H.; Lee, J.H.; Hwang, H.; An, D.K.; Lee, K.M. Deboronation-Induced Ratiometric Emission Variations of Terphenyl-Based Closo-o-Carboranyl Compounds: Applications to Fluoride-Sensing. Molecules 2020, 25, 2413. https://doi.org/10.3390/molecules25102413
So H, Mun MS, Kim M, Kim JH, Lee JH, Hwang H, An DK, Lee KM. Deboronation-Induced Ratiometric Emission Variations of Terphenyl-Based Closo-o-Carboranyl Compounds: Applications to Fluoride-Sensing. Molecules. 2020; 25(10):2413. https://doi.org/10.3390/molecules25102413
Chicago/Turabian StyleSo, Hyunhee, Min Sik Mun, Mingi Kim, Jea Ho Kim, Ji Hye Lee, Hyonseok Hwang, Duk Keun An, and Kang Mun Lee. 2020. "Deboronation-Induced Ratiometric Emission Variations of Terphenyl-Based Closo-o-Carboranyl Compounds: Applications to Fluoride-Sensing" Molecules 25, no. 10: 2413. https://doi.org/10.3390/molecules25102413
APA StyleSo, H., Mun, M. S., Kim, M., Kim, J. H., Lee, J. H., Hwang, H., An, D. K., & Lee, K. M. (2020). Deboronation-Induced Ratiometric Emission Variations of Terphenyl-Based Closo-o-Carboranyl Compounds: Applications to Fluoride-Sensing. Molecules, 25(10), 2413. https://doi.org/10.3390/molecules25102413