Transfer Learning: Making Retrosynthetic Predictions Based on a Small Chemical Reaction Dataset Scale to a New Level



Abstract
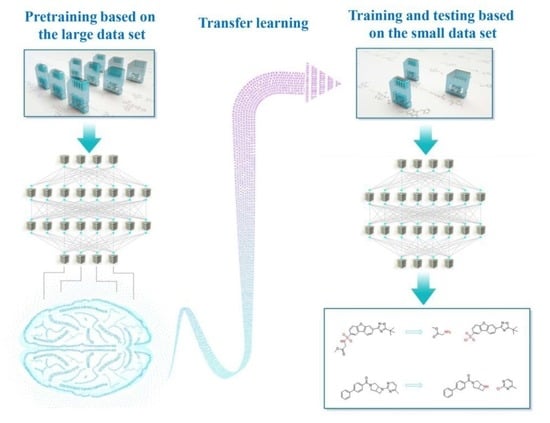
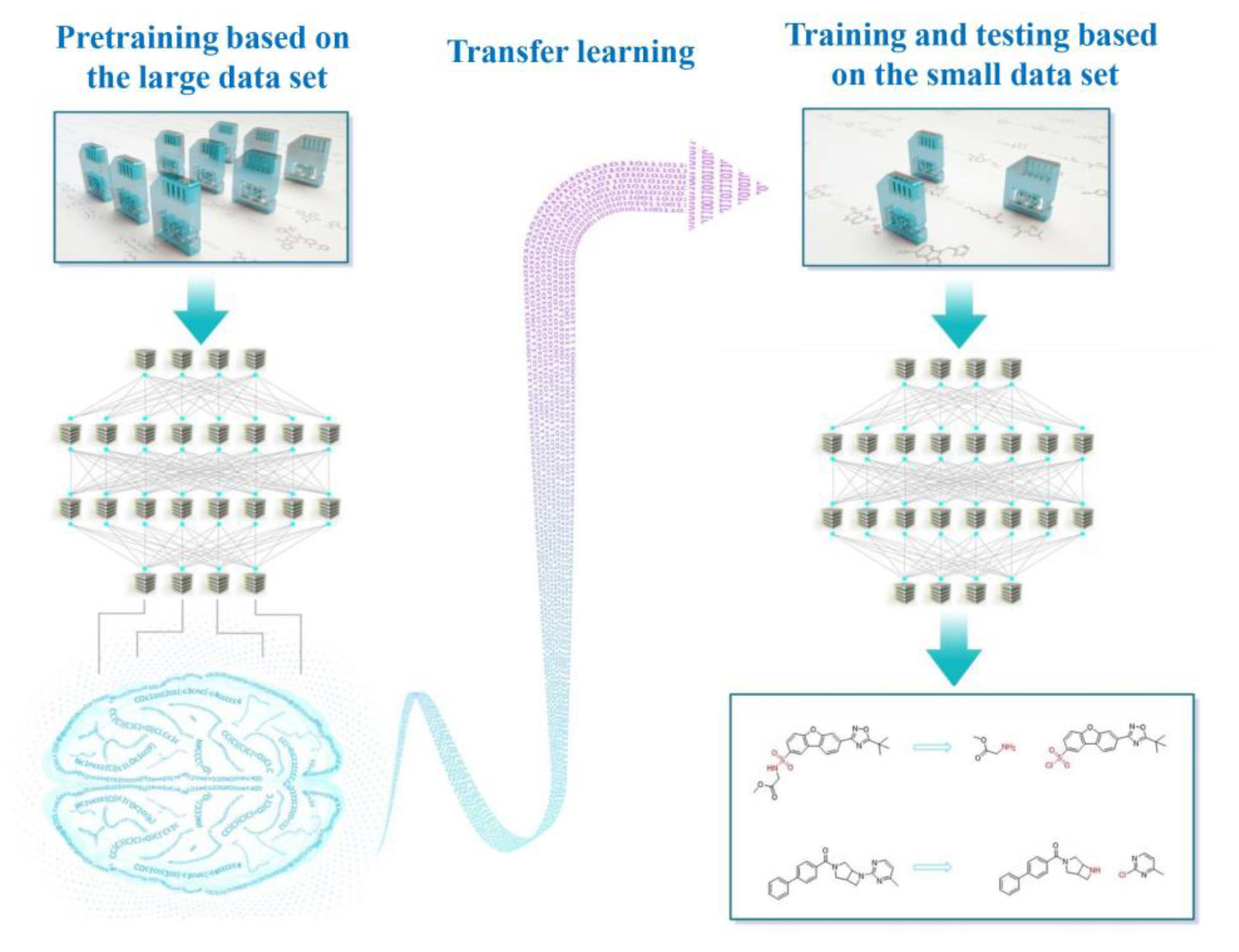
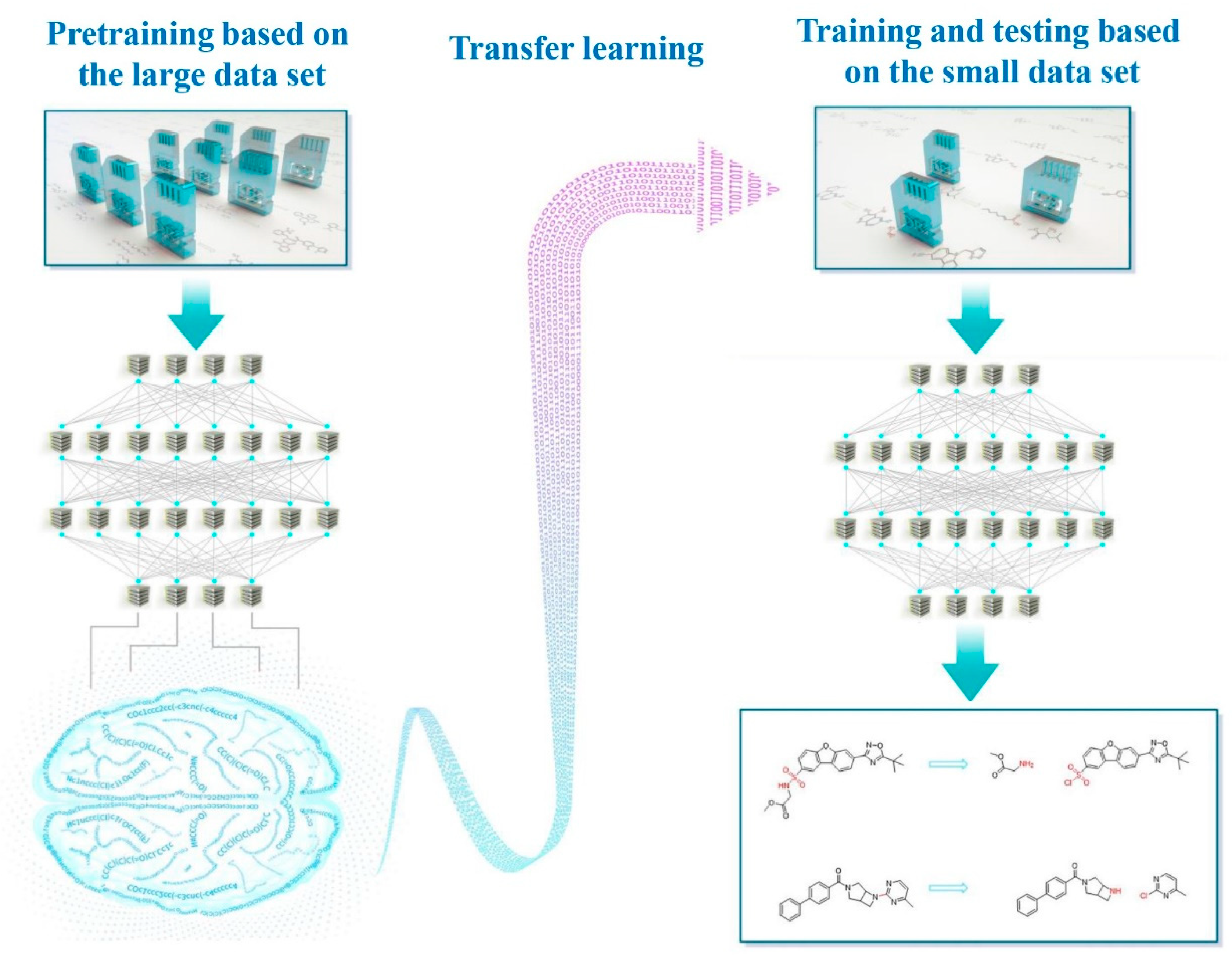
1. Introduction
2. Methods
2.1. Dataset Preparation
2.1.1. Pretraining Dataset Preparation: USPTO-380K
2.1.2. Small Dataset Preparation: USPTO-50K
2.2. Models
2.2.1. Seq2seq Model
2.2.2. Transformer Model
2.3. Performance Evaluation
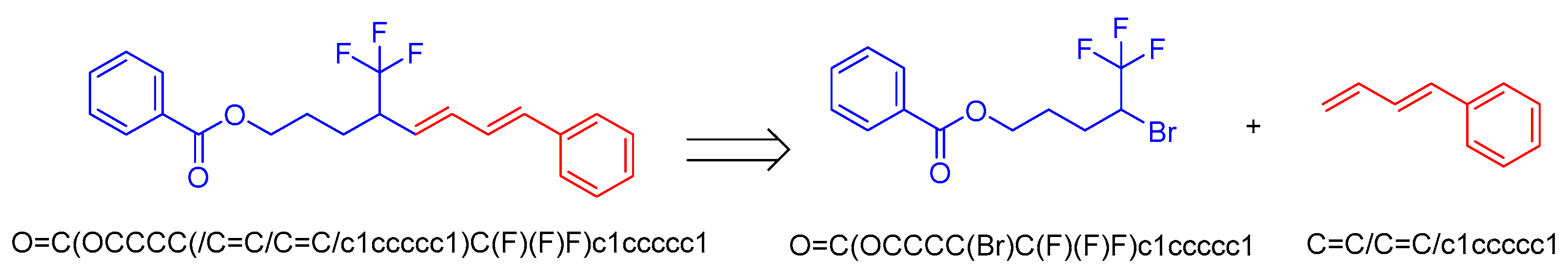
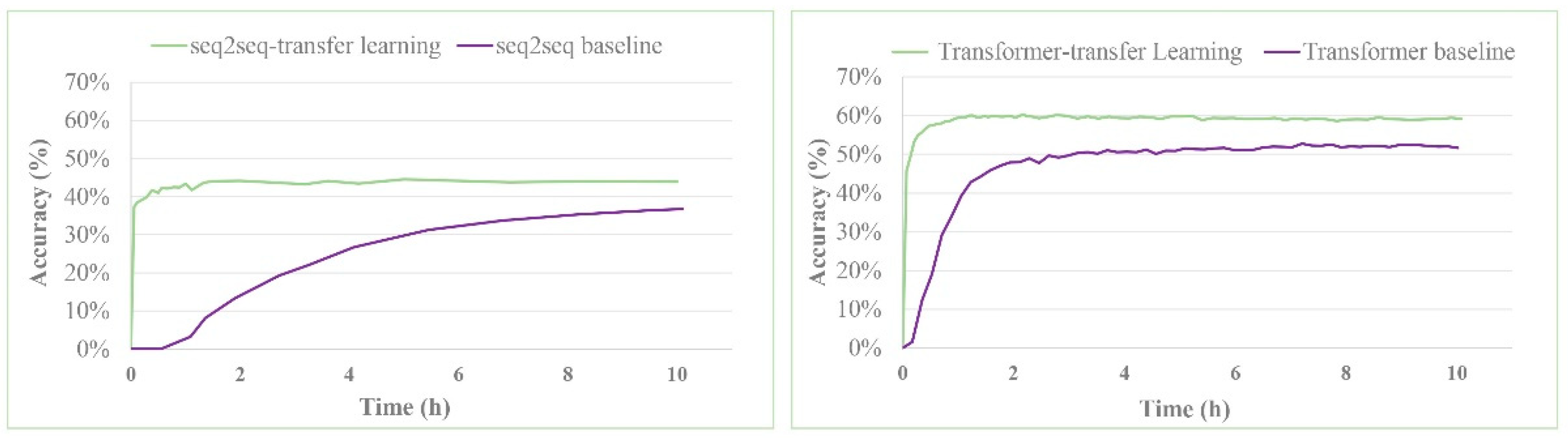
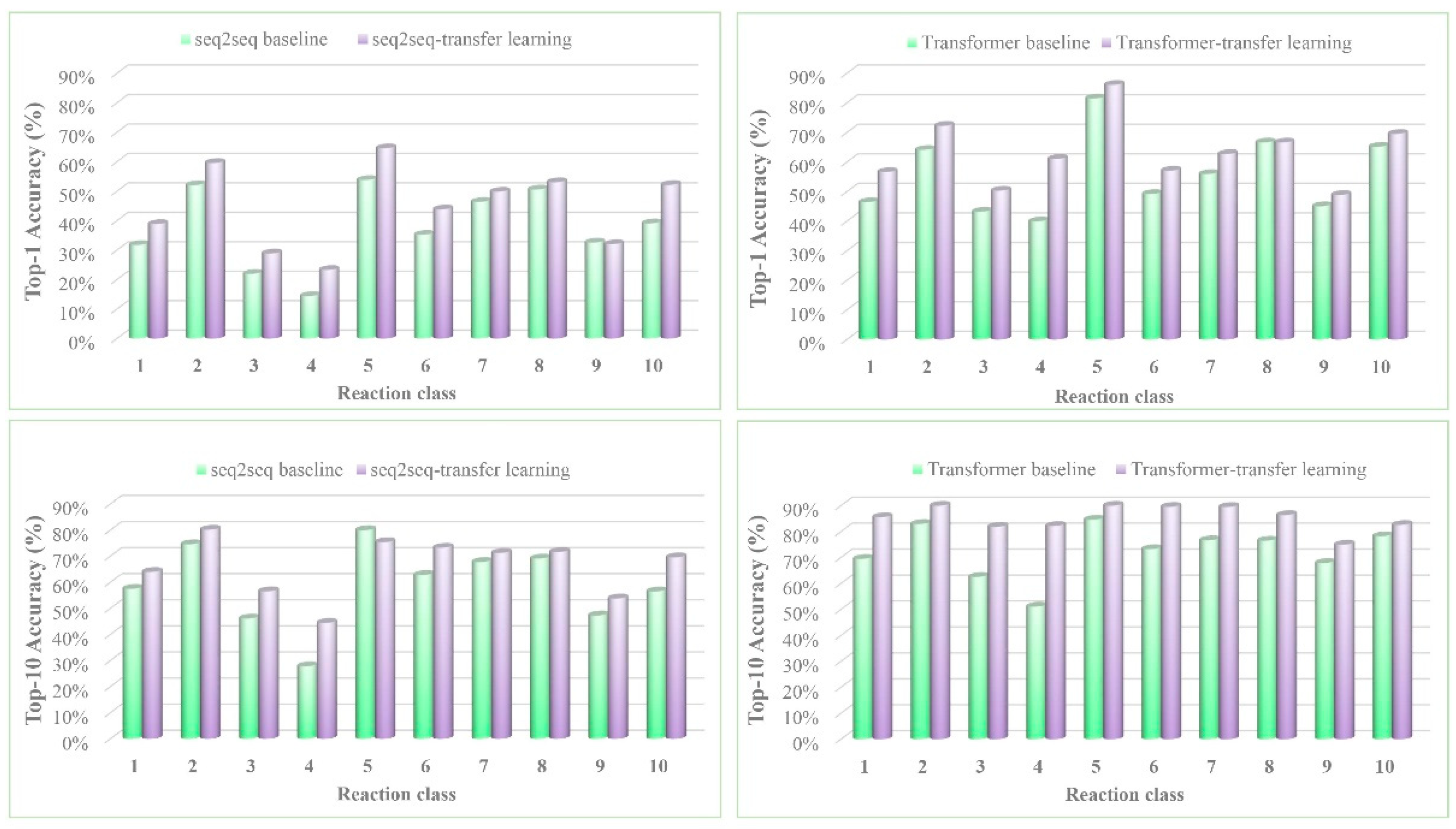
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Almeida, A.F.; Moreira, R.; Rodrigues, T. Synthetic organic chemistry driven by artificial intelligence. Nat. Rev. Chem. 2019, 3, 589–604. [Google Scholar] [CrossRef]
- Judson, P. Knowledge-Based Expert Systems in Chemistry; Royal Society of Chemistry (RSC): Cambridge, UK, 2009. [Google Scholar]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Model. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Nam, J.; Kim, J. Linking the Neural Machine Translation and the Prediction of Organic Chemistry Reactions. 2016. Available online: https://arxiv.org/abs/1612.09529 (accessed on 29 December 2016).
- Vaswani, A. Attention is All You Need. 2017. Available online: https://arxiv.org/pdf/1706.03762 (accessed on 6 December 2017).
- Lowe, D.M. Extraction of Chemical Structures and Reactions from the Literature. 2012. Available online: https://doi.org/10.17863/CAM.16293 (accessed on 9 October 2012).
- Schwaller, P.; Gaudin, T.; Lanyi, D.; Bekas, C.; Laino, T. “Found in Translation”: Predicting outcomes of complex organic chemistry reactions using neural sequence-to-sequence models. Chem. Sci. 2018, 9, 6091–6098. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Ramsundar, B.; Kawthekar, P.; Shi, J.; Gomes, J.; Nguyen, Q.L.; Ho, S.; Sloane, J.; Wender, P.A.; Pande, V.S. Retrosynthetic Reaction Prediction Using Neural Sequence-to-Sequence Models. ACS Central Sci. 2017, 3, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Schwaller, P.; Laino, T.; Gaudin, T.; Bolgar, P.; Hunter, C.A.; Bekas, C.; Lee, A.A. Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction. ACS Central Sci. 2019, 5, 1572–1583. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.A.; Yang, Q.; Sresht, V.; Bolgar, P.; Hou, X.; Klug-McLeod, J.L.; Butler, C.R. Molecular Transformer unifies reaction prediction and retrosynthesis across pharma chemical space. Chem. Commun. 2019, 55, 12152–12155. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.; Khoshgoftaar, T.M.; Wang, D. A survey of transfer learning. J. Big Data 2016, 3, 1817. [Google Scholar] [CrossRef]
- Pan, S.J.; Yang, Q. A Survey on Transfer Learning Sinno Jialin Pan and Qiang Yang Fellow. IEEE T. Knowl. Data Eng. 2009, 22, 1345–1359. [Google Scholar] [CrossRef]
- Olivas, E.S.; Guerrero, J.D.M.; Sober, M.M.; Benedito, J.R.M.; Lopez, A.J.S. Handbook of Research on Machine Learning Applications and Trends: Algorithms, Methods and Techniques-2 Volumes; Information Science Reference: Hershey, NY, USA, 2009. [Google Scholar]
- Caruana, R. Multitask learning. Mach. Learn. 1997, 28, 41–75. [Google Scholar] [CrossRef]
- Luo, Y.; Liu, T.; Tao, D.; Xu, C. Decomposition-Based Transfer Distance Metric Learning for Image Classification. IEEE Trans. Image Process. 2014, 23, 3789–3801. [Google Scholar] [CrossRef]
- Wang, C.; Mahadevan, S. Heterogeneous domain adaptation using manifold alignment. In Proceedings of the Twenty-Second International Joint Conference on Artificial Intelligence, Barcelona, Catalonia, Spain, 16–22 July 2011. [Google Scholar]
- Prettenhofer, P.; Stein, B. Cross-language text classification using structural correspondence learning. In Proceedings of the 48th Annual Meeting of the Association for Computational Linguistics, Uppsala, Sweden, 11–16 July 2010. [Google Scholar]
- Schneider, N.; Stiefl, N.; Landrum, G.A. What’s What: The (Nearly) Definitive Guide to Reaction Role Assignment. J. Chem. Inf. Model. 2016, 56, 2336–2346. [Google Scholar] [CrossRef] [PubMed]
- Seq2seq Model. Available online: https://github.com/pandegroup/reaction_prediction_seq2seq.git (accessed on 30 November 2017).
- Hochreiter, S.; Schmidhuber, J. Long Short-Term Memory. Neural Comput. 1997, 9, 1735–1780. [Google Scholar] [CrossRef] [PubMed]
- Graves, A.; Schmidhuber, J. Framewise phoneme classification with bidirectional LSTM and other neural network architectures. Neural Netw. 2005, 18, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Bahdanau, D.; Cho, K.; Bengio, Y. Neural Machine Translation by Jointly Learning to Align and Translate. 2014. Available online: https://arxiv.org/abs/1409.0473 (accessed on 19 May 2016).
- Duan, H.; Wang, L.; Zhang, C.; Li, J. Retrosynthesis with Attention-Based NMT Model and Chemical Analysis of the “Wrong” Predictions. RSC Adv. 2020, 10, 1371–1378. [Google Scholar] [CrossRef]
- Transformer Model. Available online: https://github.com/hongliangduan/RetroSynthesisT2T.git (accessed on 22 August 2019).
- Lučić, B.; Batista, J.; Bojović, V.; Lovrić, M.; Sović-Kržić, A.; Beslo, D.; Nadramija, D.; Vikić-Topić, D. Estimation of Random Accuracy and its Use in Validation of Predictive Quality of Classification Models within Predictive Challenges. Croat. Chem. Acta 2019, 92, 379–391. [Google Scholar] [CrossRef]
- Batista, J.; Vikić-Topić, D.; Lućič, B. The Difference between the Accuracy of Real and the Corresponding Random Model is a Useful Parameter for Validation of Two-State Classification Model Quality. Croat. Chem. Acta 2016, 89, 527–534. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Description | No. of Examples | Percentage of Dataset (%) |
|---|---|---|---|
| 1 | heteroatom alkylation and arylation | 15122 | 30.3 |
| 2 | acylation and related processes | 11913 | 23.8 |
| 3 | C−C bond formation | 5639 | 11.3 |
| 4 | heterocycle formation | 900 | 1.8 |
| 5 | protection | 650 | 1.3 |
| 6 | deprotection | 8353 | 16.5 |
| 7 | reduction | 4585 | 9.2 |
| 8 | oxidation | 814 | 1.6 |
| 9 | functional group interconversion (FGI) | 1834 | 3.7 |
| 10 | functional group addition (FGA) | 227 | 0.5 |
| Model | Top-N Accuracy (%) | |||||
|---|---|---|---|---|---|---|
| Top-1 | Top-2 | Top-3 | Top-5 | Top-10 | Top-20 | |
| seq2seq baseline b | 37.4% | -- | 52.4% | 57.0% | 61.7% | 65.9% |
| seq2seq-transfer learning | 44.6% | 54.8% | 59.4% | 64.1% | 68.8% | 72.1% |
| Model | Top-N Accuracy (%) | |||||
|---|---|---|---|---|---|---|
| Top-1 | Top-2 | Top-3 | Top-5 | Top-10 | Top-20 | |
| Transformer baseline | 52.4% | 63.3% | 67.1% | 70.8% | 73.2% | 74.3% |
| Transformer-transfer learning | 60.7% | 74.0% | 79.4% | 83.5% | 87.6% | 88.9% |
| Positive (exp.) | Negative (exp.) | |
|---|---|---|
| Positive (pred.) | 2021 | 776 |
| Negative (pred.) | 1191 | 1016 |
| Total | 5004 | |
| Target Compound | Retrosynthetic Analysis | ||
|---|---|---|---|
| Transformer Model (Incorrect Prediction) | Transformer-Transfer-Learning Model (Correct Prediction) | ||
| 1 |  |  |  |
| 2 |  |  |  |
| 3 |  |  |  |
| 4 |  |  |  |
| 5 |  |  |  |
| Target Compound | Retrosynthetic Analysis | ||
|---|---|---|---|
| Transformer Model (Incorrect Prediction) | Transformer-Transfer-Learning Model (Correct Prediction) | ||
| 1 |  |  |  |
| 2 |  |  |  |
| 3 |  |  |  |
| 4 |  |  |  |
| 5 |  |  |  |
| 6 |  |  |  |
| 7 |  |  |  |
| Target Compound | Retrosynthetic Analysis | ||
|---|---|---|---|
| Transformer Model (Incorrect Prediction) | Transformer-Transfer-Learning Model (Correct Prediction) | ||
| 1 |  |  |  |
| 2 |  |  |  |
| 3 |  |  |  |
| 4 |  |  |  |
| 5 |  |  |  |
| 6 |  |  |  |
| 7 |  |  |  |
| Target Compound | Retrosynthetic Analysis | ||
|---|---|---|---|
| Transformer Model (Incorrect prediction) | Transformer-Transfer-Learning Model (Correct Prediction) | ||
| 1 |  | SMILES code error |  |
| 2 |  | SMILES code error |  |
| 3 |  |  |  |
| 4 |  |  |  |
| 5 |  |  |  |
| 6 |  |  |  |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, R.; Zhang, C.; Wang, L.; Yao, C.; Ge, J.; Duan, H. Transfer Learning: Making Retrosynthetic Predictions Based on a Small Chemical Reaction Dataset Scale to a New Level. Molecules 2020, 25, 2357. https://doi.org/10.3390/molecules25102357
Bai R, Zhang C, Wang L, Yao C, Ge J, Duan H. Transfer Learning: Making Retrosynthetic Predictions Based on a Small Chemical Reaction Dataset Scale to a New Level. Molecules. 2020; 25(10):2357. https://doi.org/10.3390/molecules25102357
Chicago/Turabian StyleBai, Renren, Chengyun Zhang, Ling Wang, Chuansheng Yao, Jiamin Ge, and Hongliang Duan. 2020. "Transfer Learning: Making Retrosynthetic Predictions Based on a Small Chemical Reaction Dataset Scale to a New Level" Molecules 25, no. 10: 2357. https://doi.org/10.3390/molecules25102357
APA StyleBai, R., Zhang, C., Wang, L., Yao, C., Ge, J., & Duan, H. (2020). Transfer Learning: Making Retrosynthetic Predictions Based on a Small Chemical Reaction Dataset Scale to a New Level. Molecules, 25(10), 2357. https://doi.org/10.3390/molecules25102357