
Mechanisms Regulating the UPS-ALS Crosstalk: The Role of Proteaphagy



Abstract
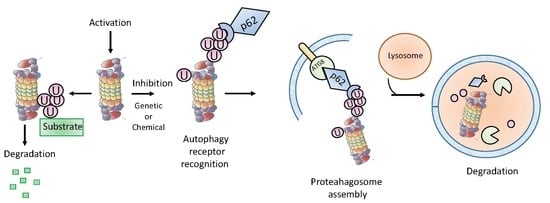
1. Introduction
2. The Ubiquitin Proteasome System (UPS)
2.1. The 20S Proteasome
2.2. The 19S Complex
2.3. Other CP Regulators
2.4. The Hybrid Proteasome
3. Autophagy Lysosome System (ALS)
3.1. Basal and Induced Autophagy
3.2. ATG8 Proteins Family, Characteristics and Functions
3.3. Autophagy Receptors
3.4. The Role of LIR Motifs
4. Crosstalk between ALS and UPS
4.1. Central Role of Ubiquitin in UPS/ALS Crosstalk
4.2. Compensatory UPS-ALS Mechanisms
4.3. Other Mechanisms Impacting the UPS-ALS Crosstalk
5. Role of Proteaphagy in the UPS-ALS Crosstalk
5.1. Proteaphagy upon Nutrient Starvation
5.2. Proteaphagy of Non-Functional Proteasomes
6. Proteaphagy in Pathology
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- van der Veen, A.G.; Ploegh, H.L. Ubiquitin-Like Proteins. Annu. Rev. Biochem. 2012, 81, 323–357. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Shabek, N. Ubiquitin Ligases: Structure, Function, and Regulation. Annu. Rev. Biochem. 2017, 86, 129–157. [Google Scholar] [CrossRef] [PubMed]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef] [PubMed]
- Seeler, J.-S.; Dejean, A. SUMO and the Robustness of Cancer. Nat. Rev. Cancer 2017, 17, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, H.M.; Shen, L.-N.; Botting, C.; Lewis, A.; Chen, J.; Ink, B.; Hay, R.T. NEDP1, a Highly Conserved Cysteine Protease That DeNEDDylates Cullins. J. Biol. Chem. 2003, 278, 25637–25643. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef] [PubMed]
- Hershko, A.; Ciechanover, A. The Ubiquitin System. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Komander, D.; Rape, M. The Ubiquitin Code. Annu. Rev. Biochem. 2012, 81, 203–229. [Google Scholar] [CrossRef]
- Zaffagnini, G.; Martens, S. Mechanisms of Selective Autophagy. J. Mol. Biol 2016, 428, 1714–1724. [Google Scholar] [CrossRef]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef]
- Komander, D. The Emerging Complexity of Protein Ubiquitination. Biochem. Soc. Trans. 2009, 37, 937–953. [Google Scholar] [CrossRef] [PubMed]
- Swatek, K.N.; Komander, D. Ubiquitin Modifications. Cell Res. 2016, 26, 399–422. [Google Scholar] [CrossRef] [PubMed]
- Livneh, I.; Cohen-Kaplan, V.; Cohen-Rosenzweig, C.; Avni, N.; Ciechanover, A. The Life Cycle of the 26S Proteasome: From Birth, through Regulation and Function, and onto Its Death. Cell Res. 2016, 26, 869–885. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K. The Proteasome: Overview of Structure and Functions. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2009, 85, 12–36. [Google Scholar] [CrossRef]
- Finley, D.; Chen, X.; Walters, K.J. Gates, Channels, and Switches: Elements of the Proteasome Machine. Trends Biochem. Sci. 2016, 41, 77–93. [Google Scholar] [CrossRef]
- Ben-Nissan, G.; Sharon, M. Regulating the 20S Proteasome Ubiquitin-Independent Degradation Pathway. Biomolecules 2014, 4, 862–884. [Google Scholar] [CrossRef]
- Ferrington, D.A.; Gregerson, D.S. Immunoproteasomes: Structure, Function, and Antigen Presentation. Prog. Mol. Biol. Transl. Sci. 2012, 109, 75–112. [Google Scholar] [CrossRef]
- Murata, S.; Udono, H.; Tanahashi, N.; Hamada, N.; Watanabe, K.; Adachi, K.; Yamano, T.; Yui, K.; Kobayashi, N.; Kasahara, M.; et al. Immunoproteasome Assembly and Antigen Presentation in Mice Lacking Both PA28α and PA28β. EMBO J. 2001, 20, 5898–5907. [Google Scholar] [CrossRef]
- Xing, Y.; Jameson, S.C.; Hogquist, K.A. Thymoproteasome Subunit-Β5T Generates Peptide-MHC Complexes Specialized for Positive Selection. Proc. Natl. Acad. Sci. USA 2013, 110, 6979–6984. [Google Scholar] [CrossRef]
- Dambacher, C.M.; Worden, E.J.; Herzik, M.A.; Martin, A.; Lander, G.C. Atomic Structure of the 26S Proteasome Lid Reveals the Mechanism of Deubiquitinase Inhibition. eLife 2016, 5, e3027. [Google Scholar] [CrossRef]
- Navon, A.; Goldberg, A.L. Proteins Are Unfolded on the Surface of the ATPase Ring before Transport into the Proteasome. Mol. Cell 2001, 8, 1339–1349. [Google Scholar] [CrossRef]
- Smith, D.M.; Chang, S.-C.; Park, S.; Finley, D.; Cheng, Y.; Goldberg, A. Docking of the Proteasomal ATPases’ C-Termini in the 20S Proteasomes Alpha Ring Opens the Gate for Substrate Entry. Mol. Cell 2007, 27, 731–744. [Google Scholar] [CrossRef] [PubMed]
- Snoberger, A.; Brettrager, E.J.; Smith, D.M. Conformational Switching in the Coiled-Coil Domains of a Proteasomal ATPase Regulates Substrate Processing. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rosenzweig, R.; Bronner, V.; Zhang, D.; Fushman, D.; Glickman, M.H. Rpn1 and Rpn2 Coordinate Ubiquitin Processing Factors at Proteasome. J. Biol. Chem. 2012, 287, 14659–14671. [Google Scholar] [CrossRef]
- Yu, Z.; Livnat-Levanon, N.; Kleifeld, O.; Mansour, W.; Nakasone, M.A.; Castaneda, C.A.; Dixon, E.K.; Fushman, D.; Reis, N.; Pick, E.; et al. Base-CP Proteasome Can Serve as a Platform for Stepwise Lid Formation. Biosci. Rep. 2015, 35, e00203. [Google Scholar] [CrossRef]
- Worden, E.J.; Padovani, C.; Martin, A. Structure of the Rpn11-Rpn8 Dimer Reveals Mechanisms of Substrate Deubiquitination during Proteasomal Degradation. Nat. Struct. Mol. Biol. 2014, 21, 220–227. [Google Scholar] [CrossRef]
- Worden, E.J.; Dong, K.C.; Martin, A. An AAA Motor-Driven Mechanical Switch in Rpn11 Controls Deubiquitination at the 26S Proteasome. Mol. Cell 2017, 67, 799–811.e8. [Google Scholar] [CrossRef]
- Lee, M.J.; Lee, B.-H.; Hanna, J.; King, R.W.; Finley, D. Trimming of Ubiquitin Chains by Proteasome-Associated Deubiquitinating Enzymes. Mol. Cell Proteom. 2011, 10, R110.003871. [Google Scholar] [CrossRef]
- Riedinger, C.; Boehringer, J.; Trempe, J.-F.; Lowe, E.D.; Brown, N.R.; Gehring, K.; Noble, M.E.M.; Gordon, C.; Endicott, J.A. Structure of Rpn10 and Its Interactions with Polyubiquitin Chains and the Proteasome Subunit Rpn12. J. Biol. Chem. 2010, 285, 33992–34003. [Google Scholar] [CrossRef]
- Isasa, M.; Katz, E.J.; Kim, W.; Yugo, V.; González, S.; Kirkpatrick, D.S.; Thomson, T.M.; Finley, D.; Gygi, S.P.; Crosas, B. Monoubiquitination of RPN10 Regulates Substrate Recruitment to the Proteasome. Mol. Cell 2010, 38, 733–745. [Google Scholar] [CrossRef]
- Keren-Kaplan, T.; Zeev Peters, L.; Levin-Kravets, O.; Attali, I.; Kleifeld, O.; Shohat, N.; Artzi, S.; Zucker, O.; Pilzer, I.; Reis, N.; et al. Structure of Ubiquitylated-Rpn10 Provides Insight into Its Autoregulation Mechanism. Nat. Commun. 2016, 7, 12960. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, P.; Chen, X.; Husnjak, K.; Randles, L.; Zhang, N.; Elsasser, S.; Finley, D.; Dikic, I.; Walters, K.J.; Groll, M. Ubiquitin Docking at the Proteasome through a Novel Pleckstrin-Homology Domain Interaction. Nature 2008, 453, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Chojnacki, M.; Mansour, W.; Hameed, D.S.; Singh, R.K.; El Oualid, F.; Rosenzweig, R.; Nakasone, M.A.; Yu, Z.; Glaser, F.; Kay, L.E.; et al. Polyubiquitin-Photoactivatable Crosslinking Reagents for Mapping Ubiquitin Interactome Identify Rpn1 as a Proteasome Ubiquitin-Associating Subunit. Cell Chem. Biol. 2017, 24, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.A.; Lawson, T.G.; Velayutham, M.; Zweier, J.L.; Pickart, C.M. A Proteasomal ATPase Subunit Recognizes the Polyubiquitin Degradation Signal. Nature 2002, 416, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Lin, Y.-L.; Fatimababy, A.S. Proteasomal Recognition of Ubiquitylated Substrates. Trends Plant. Sci. 2010, 15, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Paraskevopoulos, K.; Kriegenburg, F.; Tatham, M.H.; Rösner, H.I.; Medina, B.; Larsen, I.B.; Brandstrup, R.; Hardwick, K.G.; Hay, R.T.; Kragelund, B.B.; et al. Dss1 Is a 26S Proteasome Ubiquitin Receptor. Mol. Cell 2014, 56, 453–461. [Google Scholar] [CrossRef]
- Shi, Y.; Chen, X.; Elsasser, S.; Stocks, B.B.; Tian, G.; Lee, B.-H.; Shi, Y.; Zhang, N.; de Poot, S.A.H.; Tuebing, F.; et al. Rpn1 Provides Adjacent Receptor Sites for Substrate Binding and Deubiquitination by the Proteasome. Science 2016, 351, aad9421. [Google Scholar] [CrossRef]
- Olszewski, M.; Williams, C.; Dong, K.; Martin, A. The Cdc48 Unfoldase Prepares Well-Folded Protein Substrates for Degradation by the 26S Proteasome. Commun. Biol. 2019, 2, 1–8. [Google Scholar] [CrossRef]
- Fort, P.; Kajava, A.V.; Delsuc, F.; Coux, O. Evolution of Proteasome Regulators in Eukaryotes. Genome Biol Evol. 2015, 7, 1363–1379. [Google Scholar] [CrossRef]
- Murata, S.; Kawahara, H.; Tohma, S.; Yamamoto, K.; Kasahara, M.; Nabeshima, Y.; Tanaka, K.; Chiba, T. Growth Retardation in Mice Lacking the Proteasome Activator PA28γ. J. Biol. Chem. 1999, 274, 38211–38215. [Google Scholar] [CrossRef]
- Stohwasser, R. Proteasome Activator 28γ: Impact on Survival Signaling and Apoptosis. In Current Understanding Apoptosis-Program. Cell Death; Yusuf, T., Lutfi, T., Eds.; IntechOpen: London, UK, 2018. [Google Scholar]
- Ortega, J.; Heymann, J.B.; Kajava, A.V.; Ustrell, V.; Rechsteiner, M.; Steven, A.C. The Axial Channel of the 20S Proteasome Opens upon Binding of the PA200 Activator. J. Mol. Biol. 2005, 346, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Tanahashi, N.; Murakami, Y.; Minami, Y.; Shimbara, N.; Hendil, K.B.; Tanaka, K. Hybrid Proteasomes. Induction by Interferon-Gamma and Contribution to ATP-Dependent Proteolysis. J. Biol. Chem. 2000, 275, 14336–14345. [Google Scholar] [CrossRef] [PubMed]
- Cascio, P. PA28αβ: The Enigmatic Magic Ring of the Proteasome? Biomolecules 2014, 4, 566–584. [Google Scholar] [CrossRef] [PubMed]
- Olivier, C.; Barbara, A.Z.; Silke, M. The Proteasome System in Health and Disease. Adv. Exper. Med. Biol. 2020, 1233, 55–100. [Google Scholar]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in Yeast Demonstrated with Proteinase-Deficient Mutants and Conditions for Its Induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.J.; Simon, A.K. Autophagy in the Renewal, Differentiation and Homeostasis of Immune Cells. Nat. Rev. Immunol. 2019, 19, 170–183. [Google Scholar] [CrossRef]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo Recognition and Degradation by Selective Autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef]
- Khaminets, A.; Behl, C.; Dikic, I. Ubiquitin-Dependent And Independent Signals In Selective Autophagy. Trends Cell Biol. 2016, 26, 6–16. [Google Scholar] [CrossRef]
- Xie, Y.; Kang, R.; Sun, X.; Zhong, M.; Huang, J.; Klionsky, D.J.; Tang, D. Posttranslational Modification of Autophagy-Related Proteins in Macroautophagy. Autophagy 2015, 11, 28–45. [Google Scholar] [CrossRef]
- Wei, Y.; Liu, M.; Li, X.; Liu, J.; Li, H. Origin of the Autophagosome Membrane in Mammals. Biomed. Res. Int. 2018, 2018, 1012789. [Google Scholar] [CrossRef]
- Tsuboyama, K.; Koyama-Honda, I.; Sakamaki, Y.; Koike, M.; Morishita, H.; Mizushima, N. The ATG Conjugation Systems Are Important for Degradation of the Inner Autophagosomal Membrane. Science 2016, 354, 1036–1041. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a Mammalian Homologue of Yeast Apg8p, Is Localized in Autophagosome Membranes after Processing. Embo J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Nair, U.; Klionsky, D.J. Atg8 Controls Phagophore Expansion during Autophagosome Formation. Mol. Biol. Cell 2008, 19, 3290–3298. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation Mechanisms and Signaling Pathways of Autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. LC3 Conjugation System in Mammalian Autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518. [Google Scholar] [CrossRef] [PubMed]
- Shpilka, T.; Weidberg, H.; Pietrokovski, S.; Elazar, Z. Atg8: An Autophagy-Related Ubiquitin-like Protein Family. Genome Biol. 2011, 12, 226. [Google Scholar] [CrossRef]
- Yu, Z.-Q.; Ni, T.; Hong, B.; Wang, H.-Y.; Jiang, F.-J.; Zou, S.; Chen, Y.; Zheng, X.-L.; Klionsky, D.J.; Liang, Y.; et al. Dual Roles of Atg8−PE Deconjugation by Atg4 in Autophagy. Autophagy 2012, 8, 883–892. [Google Scholar] [CrossRef]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. Ubiquitin-like System Mediates Protein Lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef]
- Rockenfeller, P.; Koska, M.; Pietrocola, F.; Minois, N.; Knittelfelder, O.; Sica, V.; Franz, J.; Carmona-Gutierrez, D.; Kroemer, G.; Madeo, F. Phosphatidylethanolamine Positively Regulates Autophagy and Longevity. Cell Death Differ. 2015, 22, 499–508. [Google Scholar] [CrossRef]
- Yang, A.; Li, Y.; Pantoom, S.; Triola, G.; Wu, Y.-W. Semisynthetic Lipidated LC3 Protein Mediates Membrane Fusion. Chembiochem 2013, 14, 1296–1300. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Padman, B.S.; Usher, J.; Oorschot, V.; Ramm, G.; Lazarou, M. Atg8 Family LC3/GABARAP Proteins Are Crucial for Autophagosome-Lysosome Fusion but Not Autophagosome Formation during PINK1/Parkin Mitophagy and Starvation. J. Cell Biol. 2016, 215, 857–874. [Google Scholar] [CrossRef] [PubMed]
- Grunwald, D.S.; Otto, N.M.; Park, J.-M.; Song, D.; Kim, D.-H. GABARAPs and LC3s Have Opposite Roles in Regulating ULK1 for Autophagy Induction. Autophagy 2020, 16, 600–614. [Google Scholar] [CrossRef] [PubMed]
- von Muhlinen, N.; Akutsu, M.; Ravenhill, B.J.; Foeglein, Á.; Bloor, S.; Rutherford, T.J.; Freund, S.M.V.; Komander, D.; Randow, F. LC3C, Bound Selectively by a Noncanonical LIR Motif in NDP52, Is Required for Antibacterial Autophagy. Mol. Cell 2012, 48, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Kirkin, V.; Rogov, V.V. A Diversity of Selective Autophagy Receptors Determines the Specificity of the Autophagy Pathway. Mol. Cell 2019, 76, 268–285. [Google Scholar] [CrossRef] [PubMed]
- Deosaran, E.; Larsen, K.B.; Hua, R.; Sargent, G.; Wang, Y.; Kim, S.; Lamark, T.; Jauregui, M.; Law, K.; Lippincott-Schwartz, J.; et al. NBR1 Acts as an Autophagy Receptor for Peroxisomes. J. Cell Sci. 2013, 126, 939–952. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-W.; Kwon, D.H.; Song, H.K. Structure Biology of Selective Autophagy Receptors. Bmb Rep. 2016, 49, 73–80. [Google Scholar] [CrossRef]
- Ciuffa, R.; Lamark, T.; Tarafder, A.K.; Guesdon, A.; Rybina, S.; Hagen, W.J.H.; Johansen, T.; Sachse, C. The Selective Autophagy Receptor P62 Forms a Flexible Filamentous Helical Scaffold. Cell Rep. 2015, 11, 748–758. [Google Scholar] [CrossRef]
- Padman, B.S.; Nguyen, T.N.; Uoselis, L.; Skulsuppaisarn, M.; Nguyen, L.K.; Lazarou, M. LC3/GABARAPs Drive Ubiquitin-Independent Recruitment of Optineurin and NDP52 to Amplify Mitophagy. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; et al. Phosphorylation of the Autophagy Receptor Optineurin Restricts Salmonella Growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef]
- Zhang, J.; Ney, P.A. Role of bnip3 and nix in cell death, autophagy, and mitophagy. Cell Death Differ 2009, 16, 939–946. [Google Scholar] [CrossRef]
- Clausen, T.H.; Lamark, T.; Isakson, P.; Finley, K.; Larsen, K.B.; Brech, A.; Øvervatn, A.; Stenmark, H.; Bjørkøy, G.; Simonsen, A.; et al. P62/SQSTM1 and ALFY Interact to Facilitate the Formation of P62 Bodies/ALIS and Their Degradation by Autophagy. Autophagy 2010, 6, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Isakson, P.; Holland, P.; Simonsen, A. The Role of ALFY in Selective Autophagy. Cell Death Differ. 2013, 20, 12–20. [Google Scholar] [CrossRef]
- Grumati, P.; Morozzi, G.; Hölper, S.; Mari, M.; Harwardt, M.-L.I.; Yan, R.; Müller, S.; Reggiori, F.; Heilemann, M.; Dikic, I. Full Length RTN3 Regulates Turnover of Tubular Endoplasmic Reticulum via Selective Autophagy. eLife 2017, 6, e25555. [Google Scholar] [CrossRef] [PubMed]
- Khaminets, A.; Heinrich, T.; Mari, M.; Grumati, P.; Huebner, A.K.; Akutsu, M.; Liebmann, L.; Stolz, A.; Nietzsche, S.; Koch, N.; et al. Regulation of Endoplasmic Reticulum Turnover by Selective Autophagy. Nature 2015, 522, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Wurzer, B.; Zaffagnini, G.; Fracchiolla, D.; Turco, E.; Abert, C.; Romanov, J.; Martens, S. Oligomerization of P62 Allows for Selection of Ubiquitinated Cargo and Isolation Membrane during Selective Autophagy. Elife 2015, 4, e0894. [Google Scholar] [CrossRef]
- Cha-Molstad, H.; Lee, S.H.; Kim, J.G.; Sung, K.W.; Hwang, J.; Shim, S.M.; Ganipisetti, S.; McGuire, T.; Mook-Jung, I.; Ciechanover, A.; et al. Regulation of Autophagic Proteolysis by the N-Recognin SQSTM1/P62 of the N-End Rule Pathway. Autophagy 2018, 14, 359–361. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The Ubiquitin Kinase PINK1 Recruits Autophagy Receptors to Induce Mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Sharma, V.; Verma, S.; Seranova, E.; Sarkar, S.; Kumar, D. Selective Autophagy and Xenophagy in Infection and Disease. Front. Cell Dev. Biol. 2018, 6, 147. [Google Scholar] [CrossRef]
- Birgisdottir, Å.B.; Lamark, T.; Johansen, T. The LIR Motif - Crucial for Selective Autophagy. J. Cell Sci. 2013, 126, 3237–3247. [Google Scholar] [CrossRef]
- Rogov, V.V.; Stolz, A.; Ravichandran, A.C.; Rios-Szwed, D.O.; Suzuki, H.; Kniss, A.; Löhr, F.; Wakatsuki, S.; Dötsch, V.; Dikic, I.; et al. Structural and Functional Analysis of the GABARAP Interaction Motif (GIM). EMBO Rep. 2017, 18, 1382–1396. [Google Scholar] [CrossRef]
- Noda, N.N.; Kumeta, H.; Nakatogawa, H.; Satoo, K.; Adachi, W.; Ishii, J.; Fujioka, Y.; Ohsumi, Y.; Inagaki, F. Structural Basis of Target Recognition by Atg8/LC3 during Selective Autophagy. Genes Cells 2008, 13, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Ji, C.H.; Kwon, Y.T. Crosstalk and Interplay between the Ubiquitin-Proteasome System and Autophagy. Mol. Cells 2017, 40, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Liebl, M.P.; Hoppe, T. It’s All about Talking: Two-Way Communication between Proteasomal and Lysosomal Degradation Pathways via Ubiquitin. Am. J. Physiol. Cell Physiol. 2016, 311, C166–C178. [Google Scholar] [CrossRef] [PubMed]
- Nam, T.; Han, J.H.; Devkota, S.; Lee, H.-W. Emerging Paradigm of Crosstalk between Autophagy and the Ubiquitin-Proteasome System. Mol. Cells 2017, 40, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Locke, M.; Toth, J.I.; Petroski, M.D. K11- and K48-Linked Ubiquitin Chains Interact with P97 during Endoplasmic Reticulum-Associated Degradation. BiochemJ 2014, 459, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.M.M.; Wong, E.S.P.; Kirkpatrick, D.S.; Pletnikova, O.; Ko, H.S.; Tay, S.-P.; Ho, M.W.L.; Troncoso, J.; Gygi, S.P.; Lee, M.K.; et al. Lysine 63-Linked Ubiquitination Promotes the Formation and Autophagic Clearance of Protein Inclusions Associated with Neurodegenerative Diseases. Hum. Mol. Genet. 2008, 17, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Mattern, M.; Sutherland, J.; Kadimisetty, K.; Barrio, R.; Rodriguez, M.S. Using Ubiquitin Binders to Decipher the Ubiquitin Code. Trends Biochem. Sci. 2019, 44, 599–615. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, S. TRIM Family Proteins: Roles in Autophagy, Immunity, and Carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef]
- Boccitto, M.; Kalb, R.G. Regulation of Foxo-Dependent Transcription by Post-Translational Modifications. Curr. Drug Targets 2011, 12, 1303–1310. [Google Scholar] [CrossRef]
- Larrue, C.; Saland, E.; Boutzen, H.; Vergez, F.; David, M.; Joffre, C.; Hospital, M.-A.; Tamburini, J.; Delabesse, E.; Manenti, S.; et al. Proteasome Inhibitors Induce FLT3-ITD Degradation through Autophagy in AML Cells. Blood 2016, 127, 882–892. [Google Scholar] [CrossRef]
- Zhu, K.; Dunner, K.; McConkey, D.J. Proteasome Inhibitors Activate Autophagy as a Cytoprotective Response in Human Prostate Cancer Cells. Oncogene 2010, 29, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Cha-Molstad, H.; Sung, K.S.; Hwang, J.; Kim, K.A.; Yu, J.E.; Yoo, Y.D.; Jang, J.M.; Han, D.H.; Molstad, M.; Kim, J.G.; et al. Amino-Terminal Arginylation Targets Endoplasmic Reticulum Chaperone BiP for Autophagy through P62 Binding. Nat. Cell Biol. 2015, 17, 917–929. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis Control by the Unfolded Protein Response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef] [PubMed]
- White, E. Autophagy and P53. Cold Spring Harb. Perspect. Med. 2016, 6, a026120. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Yao, Z.; Klionsky, D.J. How to Control Self-Digestion: Transcriptional, Post-Transcriptional, and Post-Translational Regulation of Autophagy. Trends Cell Biol 2015, 25, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol 2018, 6, 128. [Google Scholar] [CrossRef]
- Wang, X.J.; Yu, J.; Wong, S.H.; Cheng, A.S.L.; Chan, F.K.L.; Ng, S.S.M.; Cho, C.H.; Sung, J.J.Y.; Wu, W.K.K. A Novel Crosstalk between Two Major Protein Degradation Systems: Regulation of Proteasomal Activity by Autophagy. Autophagy 2013, 9, 1500–1508. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Mansilla, A.; Menzies, F.M.; Rubinsztein, D.C. Autophagy Inhibition Compromises Degradation of Ubiquitin-Proteasome Pathway Substrates. Mol. Cell 2009, 33, 517–527. [Google Scholar] [CrossRef]
- McDonough, H.; Patterson, C. CHIP: A Link between the Chaperone and Proteasome Systems. Cell Stress Chaperones 2003, 8, 303–308. [Google Scholar] [CrossRef]
- Zhao, B.; Qiang, L.; Joseph, J.; Kalyanaraman, B.; Viollet, B.; He, Y.-Y. Mitochondrial Dysfunction Activates the AMPK Signaling and Autophagy to Promote Cell Survival. Genes Dis 2016, 3, 82–87. [Google Scholar] [CrossRef]
- Kawamata, T.; Horie, T.; Matsunami, M.; Sasaki, M.; Ohsumi, Y. Zinc Starvation Induces Autophagy in Yeast. J. Biol. Chem. 2017, 292, 8520–8530. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.R.; Doelling, J.H.; Suttangkakul, A.; Vierstra, R.D. Autophagic Nutrient Recycling in Arabidopsis Directed by the ATG8 and ATG12 Conjugation Pathways. Plant. Physiol. 2005, 138, 2097–2110. [Google Scholar] [CrossRef] [PubMed]
- Wójcik, C.; DeMartino, G.N. Intracellular Localization of Proteasomes. Int. J. Biochem. Cell Biol. 2003, 35, 579–589. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Palmer, A.; Rivett, A.J.; Knecht, E. Degradation of Proteasomes by Lysosomes in Rat Liver. Eur. J. Biochem. 1995, 227, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Dengjel, J.; Høyer-Hansen, M.; Nielsen, M.O.; Eisenberg, T.; Harder, L.M.; Schandorff, S.; Farkas, T.; Kirkegaard, T.; Becker, A.C.; Schroeder, S.; et al. Identification of Autophagosome-Associated Proteins and Regulators by Quantitative Proteomic Analysis and Genetic Screens. Mol. Cell Proteom. 2012, 11, M111.014035. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Shen, S.; Qu, J.; Ghaemmaghami, S. Global Analysis of Cellular Protein Flux Quantifies the Selectivity of Basal Autophagy. Cell Rep. 2016, 14, 2426–2439. [Google Scholar] [CrossRef] [PubMed]
- Marshall, R.S.; Li, F.; Gemperline, D.C.; Book, A.J.; Vierstra, R.D. Autophagic Degradation of the 26S Proteasome Is Mediated by the Dual ATG8/Ubiquitin Receptor RPN10 in Arabidopsis. Mol. Cell 2015, 58, 1053–1066. [Google Scholar] [CrossRef]
- Marshall, R.S.; McLoughlin, F.; Vierstra, R.D. Autophagic Turnover of Inactive 26S Proteasomes in Yeast Is Directed by the Ubiquitin Receptor Cue5 and the Hsp42 Chaperone. Cell Rep. 2016, 16, 1717–1732. [Google Scholar] [CrossRef]
- Waite, K.A.; De-La Mota-Peynado, A.; Vontz, G.; Roelofs, J. Starvation Induces Proteasome Autophagy with Different Pathways for Core and Regulatory Particles. J. Biol. Chem. 2016, 291, 3239–3253. [Google Scholar] [CrossRef]
- Nemec, A.A.; Howell, L.A.; Peterson, A.K.; Murray, M.A.; Tomko, R.J. Autophagic Clearance of Proteasomes in Yeast Requires the Conserved Sorting Nexin Snx4. J. Biol. Chem. 2017, 292, 21466–21480. [Google Scholar] [CrossRef]
- Mochida, K.; Oikawa, Y.; Kimura, Y.; Kirisako, H.; Hirano, H.; Ohsumi, Y.; Nakatogawa, H. Receptor-Mediated Selective Autophagy Degrades the Endoplasmic Reticulum and the Nucleus. Nature 2015, 522, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Kaplan, V.; Livneh, I.; Avni, N.; Fabre, B.; Ziv, T.; Kwon, Y.T.; Ciechanover, A. P62- and Ubiquitin-Dependent Stress-Induced Autophagy of the Mammalian 26S Proteasome. Proc. Natl. Acad. Sci. USA 2016, 113, E7490–E7499. [Google Scholar] [CrossRef] [PubMed]
- Marshall, R.S.; Vierstra, R.D. Proteasome Storage Granules Protect Proteasomes from Autophagic Degradation upon Carbon Starvation. eLife 2018, 7, e34532. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.; Fischer, S.; Karow, M.; Müller, R.; Meßling, S.; Eichinger, L. ATG16 Mediates the Autophagic Degradation of the 19S Proteasomal Subunits PSMD1 and PSMD2. Eur. J. Cell Biol. 2018, 97, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Üstün, S.; Sheikh, A.; Gimenez-Ibanez, S.; Jones, A.; Ntoukakis, V.; Börnke, F. The Proteasome Acts as a Hub for Plant Immunity and Is Targeted by Pseudomonas Type III Effectors. Plant. Physiol. 2016, 172, 1941–1958. [Google Scholar] [CrossRef] [PubMed]
- Üstün, S.; Hafrén, A.; Liu, Q.; Marshall, R.S.; Minina, E.A.; Bozhkov, P.V.; Vierstra, R.D.; Hofius, D. Bacteria Exploit Autophagy for Proteasome Degradation and Enhanced Virulence in Plants. Plant. Cell 2018, 30, 668–685. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Youle, R.J. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef]
- Irvine, G.B.; El-Agnaf, O.M.; Shankar, G.M.; Walsh, D.M. Protein Aggregation in the Brain: The Molecular Basis for Alzheimer’s and Parkinson’s Diseases. Mol. Med. 2008, 14, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Santamarta; Quinet, G.; Reyes-Garau, D.; Sola, B.; Roué, G.; Manuel, S.R. Resistance to the Proteasome Inhibitors: Lessons from Multiple Myeloma and Mantle Cell Lymphoma. In Proteostasis and Disease From Basic Mechanisms to Clinics; Springer International Publishing: Cham, Switzerland, 2020. [Google Scholar]
- Lopez, R. New insights into Proteasome Inhibition-Induced Proteaphagy in Acute Myeloid Leukaemia. 2020; unpublished work. [Google Scholar]
- Quinet, G. Targeting p62/Sequestosome-1Impairs Constitutively Active Proteaphagy and Enhances Apoptosis of BTZ-Resistant MCL. 2020; unpublished work. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Autophagy Receptor | Structure | Selective Autophagy | Collaboration | References |
| p62/SQSTM1 |  | Aggrephagy; Mitophagy; Xenophagy; Lysophagy; Pexophagy; Proteaphagy | NBR1 (aggregaphagy, pexophagy) NDP52 + OPTN (xenophagy) | [66,67,68] |
| NBR1 |  | Pexophagy; Aggrephagy | p62 (aggregaphagy, pexophagy); | [64,66,67] |
| NDP52 |  | Mitophagy | p62 + OPTN (xenophagy) | [67,69] |
| OPTN |  | Mitophagy; Xenophagy | NDP52 + p62 (xenophagy) | [67,69,70] |
| BNIP3/NIX |  | Mitophagy | [71] | |
| ALFY |  | Aggrephagy | p62 (aggrephagy) | [72,73] |
| RTN3 |  | ER-phagy | [74] | |
| FAM134B |  | ER-phagy; Aggrephagy | [75] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quinet, G.; Gonzalez-Santamarta, M.; Louche, C.; Rodriguez, M.S. Mechanisms Regulating the UPS-ALS Crosstalk: The Role of Proteaphagy. Molecules 2020, 25, 2352. https://doi.org/10.3390/molecules25102352
Quinet G, Gonzalez-Santamarta M, Louche C, Rodriguez MS. Mechanisms Regulating the UPS-ALS Crosstalk: The Role of Proteaphagy. Molecules. 2020; 25(10):2352. https://doi.org/10.3390/molecules25102352
Chicago/Turabian StyleQuinet, Grégoire, Maria Gonzalez-Santamarta, Clara Louche, and Manuel S. Rodriguez. 2020. "Mechanisms Regulating the UPS-ALS Crosstalk: The Role of Proteaphagy" Molecules 25, no. 10: 2352. https://doi.org/10.3390/molecules25102352
APA StyleQuinet, G., Gonzalez-Santamarta, M., Louche, C., & Rodriguez, M. S. (2020). Mechanisms Regulating the UPS-ALS Crosstalk: The Role of Proteaphagy. Molecules, 25(10), 2352. https://doi.org/10.3390/molecules25102352