
Hypoxia-Activated Prodrug Derivatives of Carbonic Anhydrase Inhibitors in Benzenesulfonamide Series: Synthesis and Biological Evaluation


 ,
,  ,
,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Results
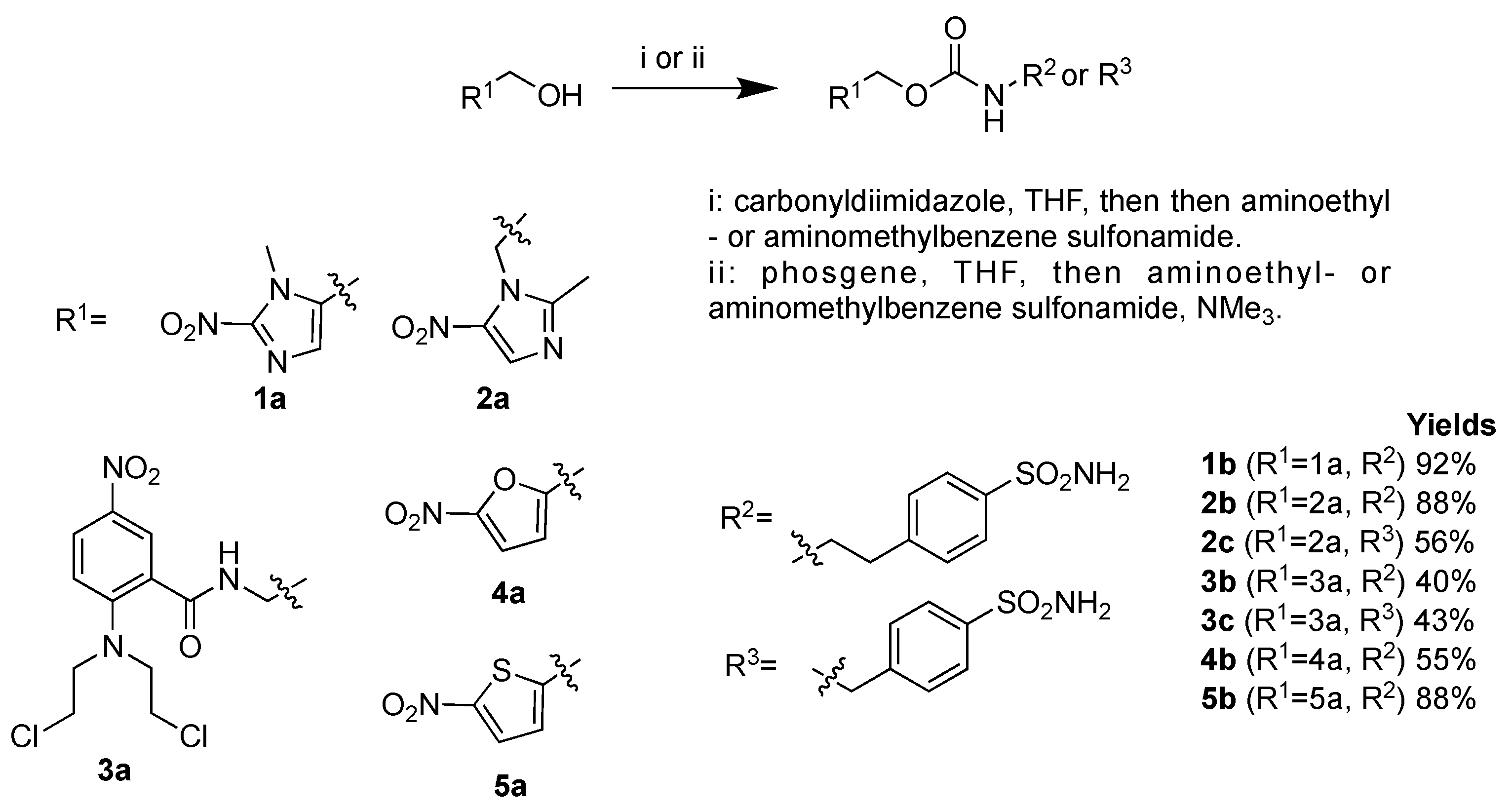
2.1. Chemistry
2.2. Carbonic Anhydrase Inhibition Assay
2.3. Stability of Carbamate Linker under Acidic Conditions
2.4. Biological Assays
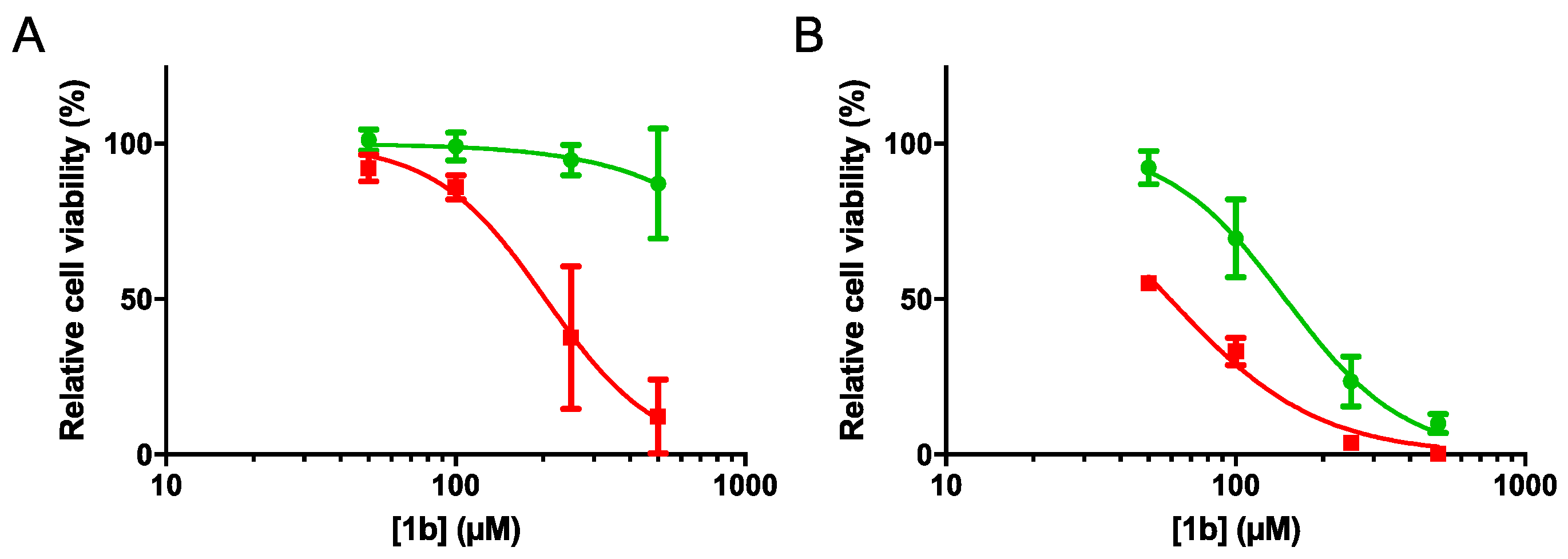
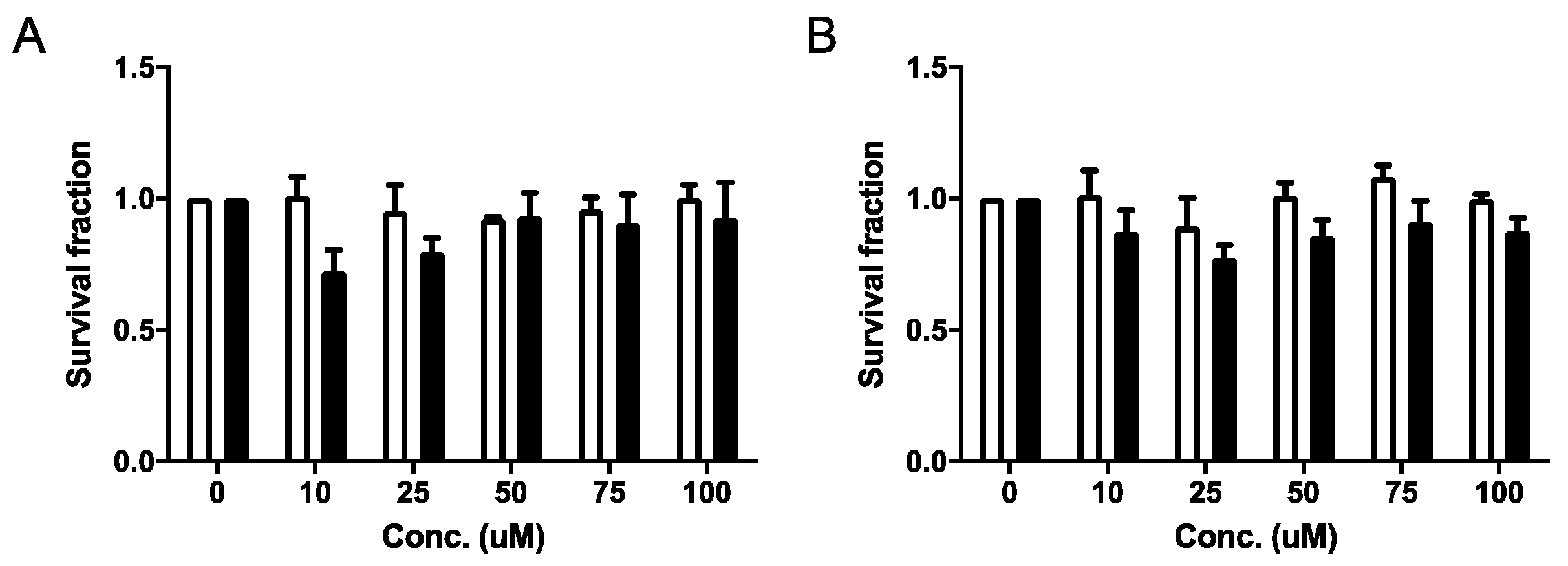
2.4.1. Cell Viability and Clonogenic Assays
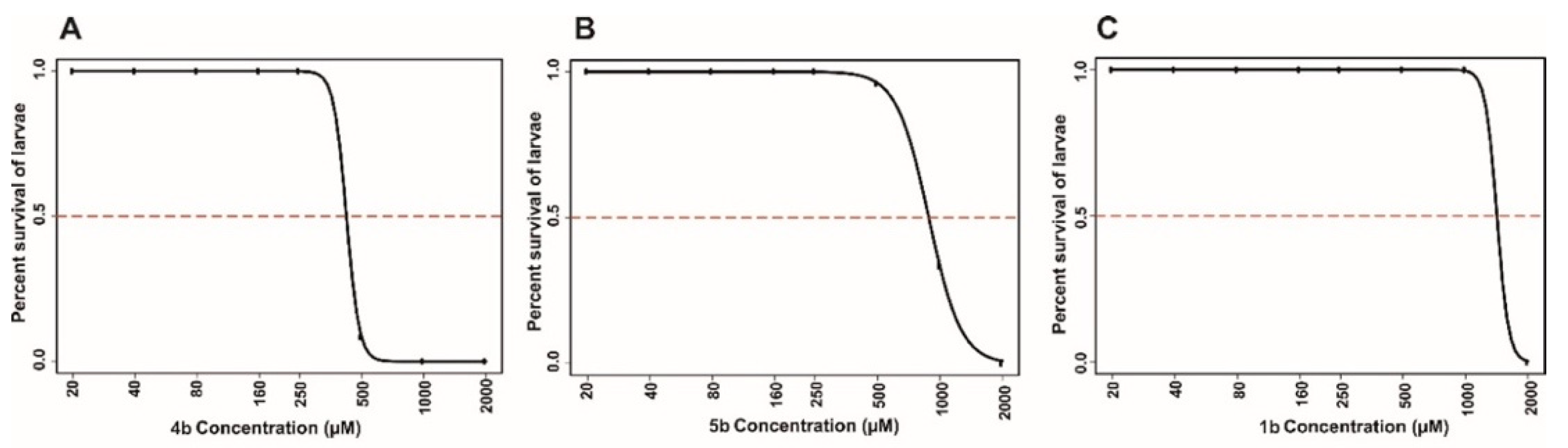
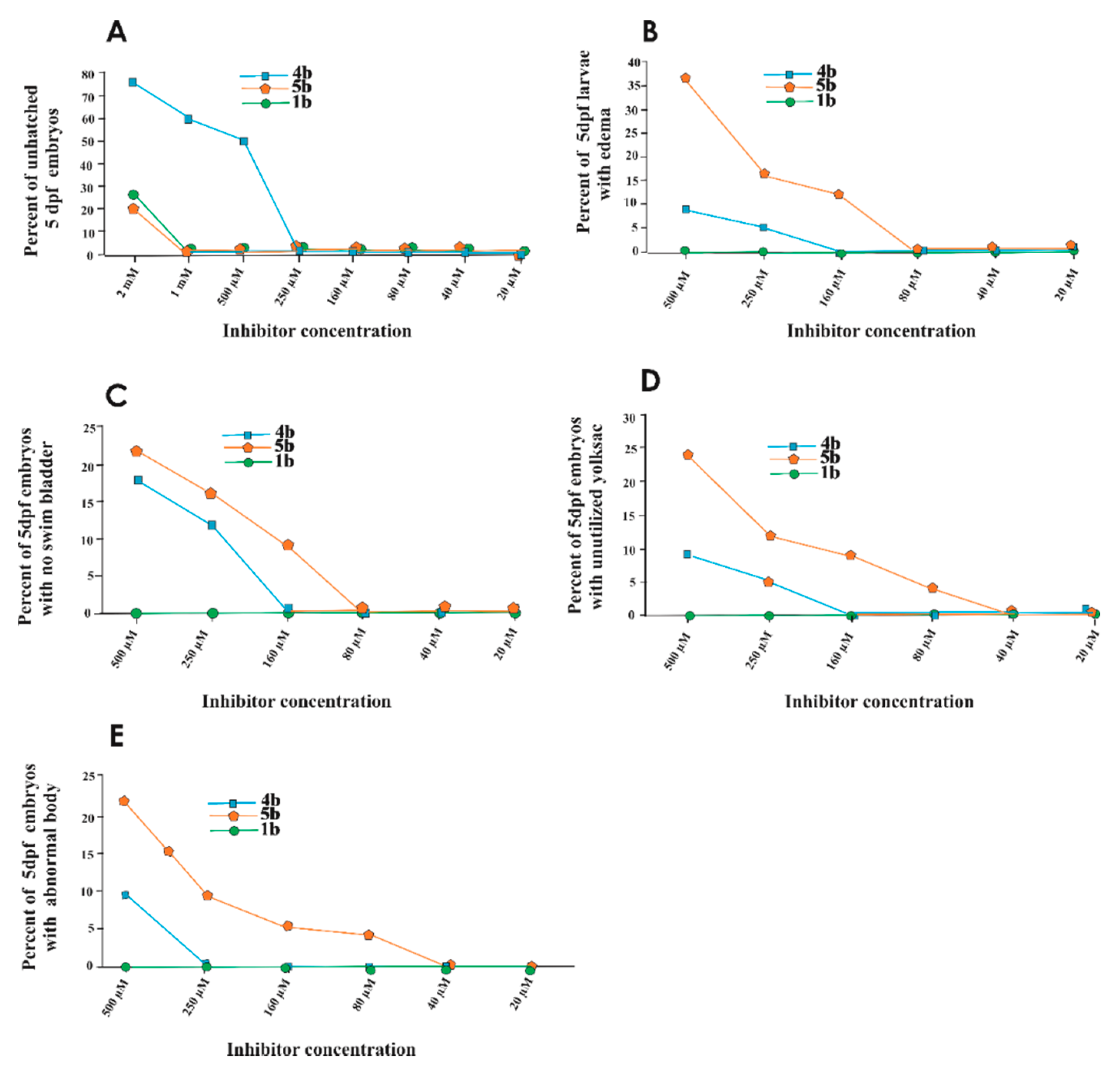
2.4.2. Toxicity Evaluation on Zebrafish
Determination of Half Maximal Lethal Concentration 50 (LC50)
Swim Pattern Analysis to Assess the Subtle Toxic Effects of the Prodrugs
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.2. Carbonic Anhydrase Inhibition Assays
4.3. In Vitro Chemical Stability and Analytical Method
4.4. Biological Assays
4.4.1. Cells
4.4.2. Cell Viability Assays
4.4.3. Clonogenic Assays
4.4.4. Statistical Analyses
4.5. Toxicity Assay
4.5.1. Preparation of Inhibitor Samples
4.5.2. Maintenance of the Zebrafish
4.5.3. Determination of half Maximal Lethal Concentration 50 (LC50)
4.5.4. Phenotypic Analysis of the Larvae
4.5.5. Swim Pattern Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Ramón, Y.; Cajal, S.; Sesé, M.; Capdevila, C.; Aasen, T.; De Mattos-Arruda, L.; Diaz-Cano, S.J.; Hernández-Losa, J.; Castellví, J. Clinical implications of intratumor heterogeneity: Challenges and opportunities. J. Mol. Med. 2020, 98, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; de Sauvage, F.J. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature 2013, 501, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Parks, S.K.; Cormerais, Y.; Pouysségur, J. Hypoxia and cellular metabolism in tumour pathophysiology. J. Physiol. 2017, 595, 2439–2450. [Google Scholar] [CrossRef] [PubMed]
- Corbet, C.; Feron, O. Tumour acidosis: From the passenger to the driver’s seat. Nat. Rev. Cancer. 2017, 17, 577–593. [Google Scholar] [CrossRef]
- Pastorekova, S.; Gillies, R.J. The role of carbonic anhydrase IX in cancer development: Links to hypoxia, acidosis, and beyond. Cancer Metastasis Rev. 2019, 38, 65–77. [Google Scholar] [CrossRef]
- van Kuijk, S.J.; Yaromina, A.; Houben, R.; Niemans, R.; Lambin, P.; Dubois, L.J. Prognostic Significance of Carbonic Anhydrase IX Expression in Cancer Patients: A Meta-Analysis. Front. Oncol. 2016, 6, 69. [Google Scholar] [CrossRef]
- Dubois, L.J.; Niemans, R.; van Kuijk, S.J.; Panth, K.M.; Parvathaneni, N.K.; Peeters, S.G.; Zegers, C.M.; Rekers, N.H.; van Gisbergen, M.W.; Biemans, R.; et al. New ways to image and target tumour hypoxia and its molecular responses. Radiother. Oncol. 2015, 116, 352–357. [Google Scholar] [CrossRef]
- Eckert, A.W.; Horter, S.; Bethmann, D.; Kotrba, J.; Kaune, T.; Rot, S.; Bache, M.; Bilkenroth, U.; Reich, W.; Greither, T.; et al. Investigation of the Prognostic Role of Carbonic Anhydrase 9 (CAIX) of the Cellular mRNA/Protein Level or Soluble CAIX Protein in Patients with Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 375. [Google Scholar] [CrossRef]
- Yang, J.S.; Lin, C.W.; Chuang, C.Y.; Su, S.C.; Lin, S.H.; Yang, S.F. Carbonic anhydrase IX overexpression regulates the migration and progression in oral squamous cell carcinoma. Tumour Biol. 2015, 36, 9517–9524. [Google Scholar] [CrossRef] [PubMed]
- Torres López, M.; Pérez Sayáns, M.; Chamorro Petronacci, C.; Barros Angueira, F.; Gándara Vila, P.; Lorenzo Pouso, A.; García García, A. Determination and diagnostic value of CA9 mRNA in peripheral blood of patients with oral leukoplakia. J. Enzyme Inhib. Med. Chem. 2018, 33, 951–955. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Sayáns, M.; Suárez-Peñaranda, J.M.; Pilar, G.D.; Barros-Angueira, F.; Gándara-Rey, J.M.; García-García, A. Hypoxia-inducible factors in OSCC. Cancer Lett. 2011, 313, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ilies, M.; Winum, J.-Y. Carbonic Anhydrase Inhibitors for The Treatment of Tumors: Therapeutic, Immunologic, And Diagnostic Tools Targeting Isoforms IX And XII. In Carbonic Anhydrases. Biochemistry and Pharmacology of an Evergreen Pharmaceutical Target, 1st ed.; Supuran, C.T., Nocentini, A., Eds.; Academic Press: Amsterdam, The Netherlands, 2019; pp. 331–365. [Google Scholar]
- Supuran, C.T.; Alterio, V.; di Fiore, A.; d’ Ambrosio, K.; Carta, F.; Monti, S.M.; de Simone, G. Inhibition of carbonic anhydrase IX targets primary tumors, metastases, and cancer stem cells: Three for the price of one. Med. Res. Rev. 2018, 38, 1799–1836. [Google Scholar] [CrossRef] [PubMed]
- Nocentini, A.; Supuran, C.T. Carbonic anhydrase inhibitors as antitumor/antimetastatic agents: A patent review (2008–2018). Expert. Opin. Ther. Pat. 2018, 28, 729–740. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrase inhibitors as emerging agents for the treatment and imaging of hypoxic tumors. Expert Opin. Investig. Drugs 2018, 27, 963–970. [Google Scholar] [CrossRef]
- Sharma, A.; Arambula, J.F.; Koo, S.; Kumar, R.; Singh, H.; Sessler, J.L.; Kim, J.S. Hypoxia-targeted drug delivery. Chem. Soc. Rev. 2019, 48, 771–813. [Google Scholar] [CrossRef]
- Su, M.X.; Zhang, L.L.; Huang, Z.J.; Shi, J.J.; Lu, J.J. Investigational Hypoxia-Activated Prodrugs: Making Sense of Future Development. Curr. Drug Targets 2019, 20, 668–678. [Google Scholar] [CrossRef]
- Baran, N.; Konopleva, M. Molecular Pathways: Hypoxia-Activated Prodrugs in Cancer Therapy. Clin. Cancer Res. 2017, 23, 2382–2391. [Google Scholar] [CrossRef]
- Dickson, B.D.; Wong, W.W.; Wilson, W.R.; Hay, M.P. Studies Towards Hypoxia-Activated Prodrugs of PARP Inhibitors. Molecules 2019, 24, 1559. [Google Scholar] [CrossRef]
- Liew, L.P.; Singleton, D.C.; Wong, W.W.; Cheng, G.J.; Jamieson, S.M.F.; Hay, M.P. Hypoxia-Activated Prodrugs of PERK Inhibitors. Chem. Asian J. 2019, 14, 1238–1248. [Google Scholar] [CrossRef] [PubMed]
- Bielec, B.; Schueffl, H.; Terenzi, A.; Berger, W.; Heffeter, P.; Keppler, B.; Kowol, C.R. Development and biological investigations of hypoxia-sensitive prodrugs of the tyrosine kinase inhibitor crizotinib. Bioorg. Chem. 2020, 99, 103778. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Xie, Y.; Xu, S.; Xin, J.; Wang, J.; Han, T.; Ting, R.; Zhang, J.; An, F. Hypoxia-activated nanomedicines for effective cancer therapy. Eur. J. Med. Chem. 2020, 195, 112274. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Ma, J.; Zhan, Y.; Xu, X.; Zeng, Q.; Liang, J.; Chen, X. Hypoxia-activated prodrugs and redox-responsive nanocarriers. Int. J. Nanomed. 2018, 13, 6551–6574. [Google Scholar] [CrossRef]
- de Simone, G.; Vitale, R.M.; di Fiore, A.; Pedone, C.; Scozzafava, A.; Montero, J.-L.; Winum, J.-Y.; Supuran, C.T. Carbonic anhydrase inhibitors: Hypoxia-activatable sulfonamides incorporating disulfide bonds that target the tumor-associated isoform IX. J. Med. Chem. 2006, 49, 5544–5551. [Google Scholar] [CrossRef]
- Nocentini, A.; Trallori, E.; Singh, S.; Lomelino, C.L.; Bartolucci, G.; di Cesare Mannelli, L.; Ghelardini, C.; McKenna, R.; Gratteri, P.; Supuran, C.T. 4-Hydroxy-3-nitro-5-ureido-benzenesulfonamides Selectively Target the Tumor-Associated Carbonic Anhydrase Isoforms IX and XII Showing Hypoxia-Enhanced Antiproliferative Profiles. J. Med. Chem. 2018, 61, 10860–10874. [Google Scholar] [CrossRef]
- Aspatwar, A.; Parvathaneni, N.K.; Barker, H.; Anduran, E.; Supuran, C.T.; Dubois, L.; Lambin, P.; Parkkila, S.; Winum, J.Y. Design, synthesis, in vitro inhibition and toxicological evaluation of human carbonic anhydrases I, II and IX inhibitors in 5-nitroimidazole series. J. Enzyme Inhib. Med. Chem. 2020, 35, 109–117. [Google Scholar] [CrossRef]
- van Kuijk, S.J.A.; Parvathaneni, N.K.; Niemans, R.; van Gisbergen, M.W.; Carta, F.; Vullo, D.; Pastorekova, S.; Yaromina, A.; Supuran, C.T.; Dubois, L.J.; et al. New approach of delivering cytotoxic drugs towards CAIX expressing cells: A concept of dual-target drugs. Eur. J. Med. Chem. 2017, 127, 691–702. [Google Scholar] [CrossRef]
- Aspatwar, A.; Becker, H.M.; Parvathaneni, N.K.; Hammaren, M.; Svorjova, A.; Barker, H.; Supuran, C.T.; Dubois, L.; Lambin, P.; Parikka, M.; et al. Nitroimidazole-based inhibitors DTP338 and DTP348 are safe for zebrafish embryos and efficiently inhibit the activity of human CA IX in Xenopus oocytes. J. Enzyme Inhib. Med. Chem. 2018, 33, 1064–1073. [Google Scholar] [CrossRef]
- Musser, J.H.; Chakrabortv, U.; Bailey, K.; Sciortino, S.; Whyzmuzis, C.; Amin, D.; Sutherland, C.A. Synthesis and Antilipolytic Activities of Quinolyl Carbanilates and Related Analogues. J. Med. Chem. 1987, 30, 62–67. [Google Scholar] [CrossRef]
- Khalifah, R.G. The Carbon Dioxide Hydration Activity of Carbonic Anhydrase. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar] [PubMed]
- Tarzia, G.; Duranti, A.; Gatti, G.; Piersanti, G.; Tontini, A.; Rivara, S.; Lodola, A.; Plazzi, P.V.; Mor, M.; Kathuria, S.; et al. Synthesis and Structure-Activity Relationships of FAAH Inhibitors: Cyclohexylcarbamic Acid Biphenyl Esters with Chemical Modulation at the Proximal Phenyl Ring. ChemMedChem 2006, 1, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Evans, J.W.; Bhupathi, D.; Banica, M.; Lan, L.; Lorente, G.; Duan, J.X.; Cai, X.; Mowday, A.M.; Guise, C.P.; et al. Molecular and Cellular Pharmacology of the Hypoxia-Activated Prodrug TH-302. Mol. Cancer Ther. 2012, 11, 740–751. [Google Scholar] [CrossRef] [PubMed]
- Aspatwar, A.; Hammaren, M.M.; Parikka, M.; Parkkila, S. Rapid Evaluation of Toxicity of Chemical Compounds Using Zebrafish Embryos. J. Vis. Exp. 2019, 2019, 1–7. [Google Scholar] [CrossRef]
- Aspatwar, A.; Hammarén, M.; Koskinen, S.; Luukinen, B.; Barker, H.; Carta, F.; Supuran, C.T.; Parikka, M.; Parkkila, S. β-CA-Specific Inhibitor Dithiocarbamate Fc14–584B: A Novel Antimycobacterial Agent with Potential to Treat Drug-Resistant Tuberculosis. J. Enzyme Inhib. Med. Chem. 2017, 32, 832–840. [Google Scholar] [CrossRef]
- Kazokaitė, J.; Aspatwar, A.; Kairys, V.; Parkkila, S.; Matulis, D. Fluorinated benzenesulfonamide anticancer inhibitors of carbonic anhydrase IX exhibit lower toxic effects on zebrafish embryonic development than ethoxzolamide. Drug Chem. Toxicol. 2017, 40, 309–319. [Google Scholar] [CrossRef]
- Sharma, A.; Tiwari, M.; Supuran, C.T. Novel coumarins and benzocoumarins acting as isoform-selective inhibitors against the tumor-associated carbonic anhydrase IX. J. Enzyme Inhib. Med. Chem. 2014, 29, 292–296. [Google Scholar] [CrossRef]
- Durdagi, S.; Scozzafava, G.; Vullo, D.; Sahin, H.; Kolayli, S.; Supuran, C.T. Inhibition of mammalian carbonic anhydrases I-XIV with grayanotoxin III: Solution and in silico studies. J. Enzyme Inhib. Med. Chem. 2014, 29, 469–475. [Google Scholar] [CrossRef]
- Alterio, V.; Hilvo, M.; Di Fiore, A.; Supuran, C.T.; Pan, P.; Parkkila, S.; Scaloni, A.; Pastorek, J.; Pastorekova, S.; Pedone, C.; et al. Crystal structure of the catalytic domain of the tumor-associated human carbonic anhydrase IX. Proc. Natl. Acad. Sci. USA 2009, 106, 16233–16238. [Google Scholar] [CrossRef]
- Ditte, P.; Dequiedt, F.; Svastova, E.; Hulikova, A.; Ohradanova-Repic, A.; Zatovicova, M.; Csaderova, L.; Kopacek, J.; Supuran, C.T.; Pastorekova, S.; et al. Phosphorylation of carbonic anhydrase IX controls its ability to mediate extracellular acidification in hypoxic tumors. Cancer Res. 2011, 71, 7558–7567. [Google Scholar] [CrossRef]
- Gourmelon, A.; Delrue, N. Validation in Support of Internationally Harmonised OECD Test Guidelines for Assessing the Safety of Chemicals. Adv. Exp. Med. Biol. 2016, 856, 9–32. [Google Scholar] [PubMed]
- Ritz, C.; Baty, F.; Streibig, J.C.; Gerhard, D. Dose-Response Analysis Using R. PLoS ONE 2015, 10, e0146021. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | KI (nM) * | Selectivity Ratio | ||
|---|---|---|---|---|
| hCA I | hCA II | hCA IX | KI hCA II/KI hCA IX | |
| 1b | 166.7 | 30.6 | 7.6 | 1.19 |
| 2b | 3.7 | 4.3 | 12.1 | 0.35 |
| 2c | 2.3 | 4.0 | 14.1 | 0.28 |
| 3b | 83.0 | 3.1 | 32.3 | 0.09 |
| 3c | 2179.9 | 83.7 | 88.7 | 0.94 |
| 4b | 83.8 | 15.5 | 5.7 | 2.72 |
| 5b | 64.7 | 19.8 | 15.1 | 1.31 |
| AAZ | 250 | 12.0 | 25 | 0.48 |
| Compounds | HT29 | HCT116 | ||||
|---|---|---|---|---|---|---|
| IC50N | IC50A | HCR (IC50N/IC50A) | IC50N | IC50A | HCR (IC50N/IC50A) | |
| 1b | >500 | 204.5 | >2.44 | 148.6 | 59.36 | 2.50 |
| 2b | >500 | >500 | - | >500 | >500 | - |
| 2c | >500 | >500 | - | >500 | >500 | - |
| 3b | >500 | >500 | - | 267.1 | 97.27 | 2.74 |
| 3c | >500 | >500 | - | >500 | >500 | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anduran, E.; Aspatwar, A.; Parvathaneni, N.-K.; Suylen, D.; Bua, S.; Nocentini, A.; Parkkila, S.; Supuran, C.T.; Dubois, L.; Lambin, P.; et al. Hypoxia-Activated Prodrug Derivatives of Carbonic Anhydrase Inhibitors in Benzenesulfonamide Series: Synthesis and Biological Evaluation. Molecules 2020, 25, 2347. https://doi.org/10.3390/molecules25102347
Anduran E, Aspatwar A, Parvathaneni N-K, Suylen D, Bua S, Nocentini A, Parkkila S, Supuran CT, Dubois L, Lambin P, et al. Hypoxia-Activated Prodrug Derivatives of Carbonic Anhydrase Inhibitors in Benzenesulfonamide Series: Synthesis and Biological Evaluation. Molecules. 2020; 25(10):2347. https://doi.org/10.3390/molecules25102347
Chicago/Turabian StyleAnduran, Emilie, Ashok Aspatwar, Nanda-Kumar Parvathaneni, Dennis Suylen, Silvia Bua, Alessio Nocentini, Seppo Parkkila, Claudiu T. Supuran, Ludwig Dubois, Philippe Lambin, and et al. 2020. "Hypoxia-Activated Prodrug Derivatives of Carbonic Anhydrase Inhibitors in Benzenesulfonamide Series: Synthesis and Biological Evaluation" Molecules 25, no. 10: 2347. https://doi.org/10.3390/molecules25102347
APA StyleAnduran, E., Aspatwar, A., Parvathaneni, N.-K., Suylen, D., Bua, S., Nocentini, A., Parkkila, S., Supuran, C. T., Dubois, L., Lambin, P., & Winum, J.-Y. (2020). Hypoxia-Activated Prodrug Derivatives of Carbonic Anhydrase Inhibitors in Benzenesulfonamide Series: Synthesis and Biological Evaluation. Molecules, 25(10), 2347. https://doi.org/10.3390/molecules25102347