
Identification and Biological Evaluation of CK2 Allosteric Fragments through Structure-Based Virtual Screening


 , and
, and
Abstract
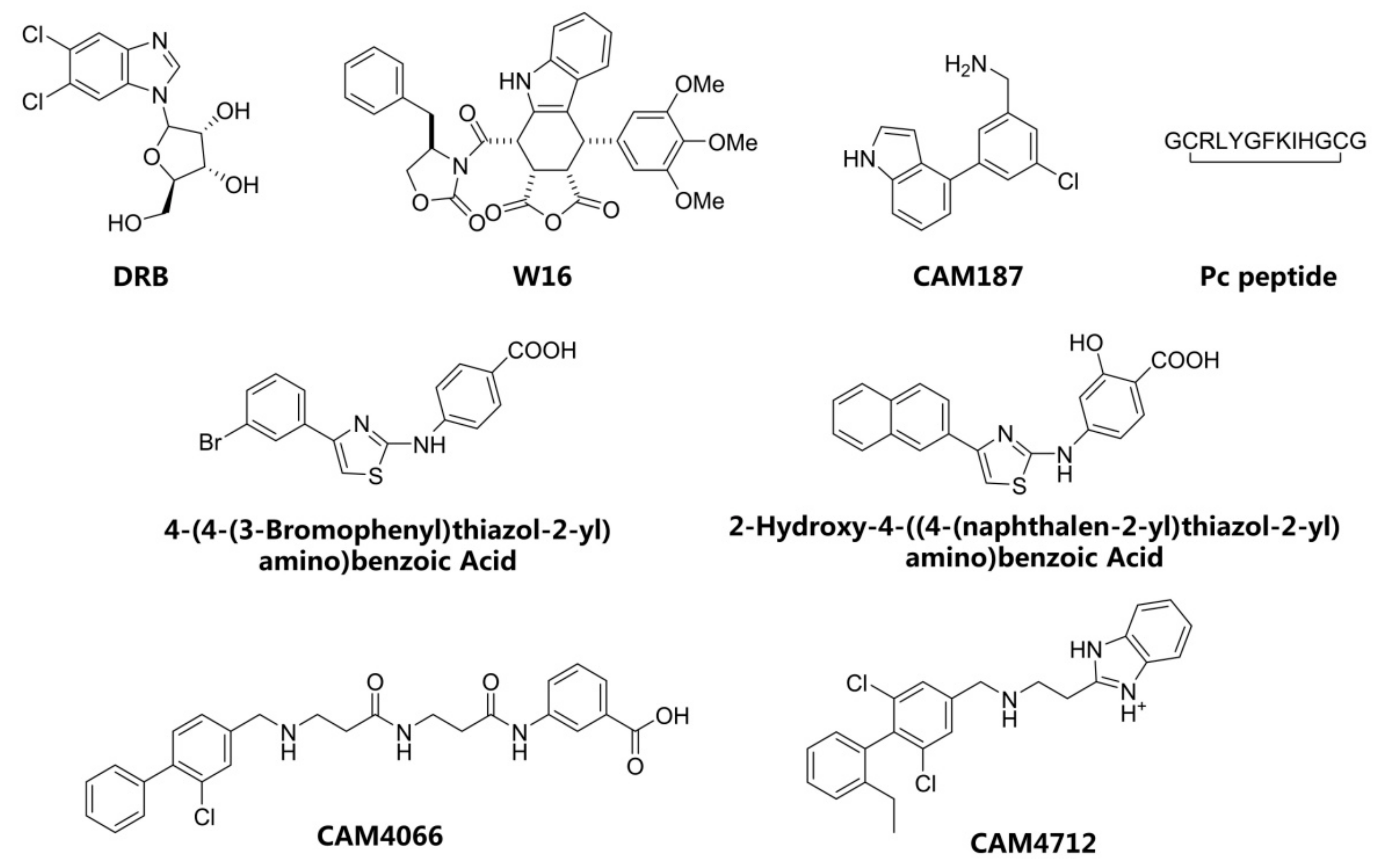
1. Introduction
2. Results and Discussion
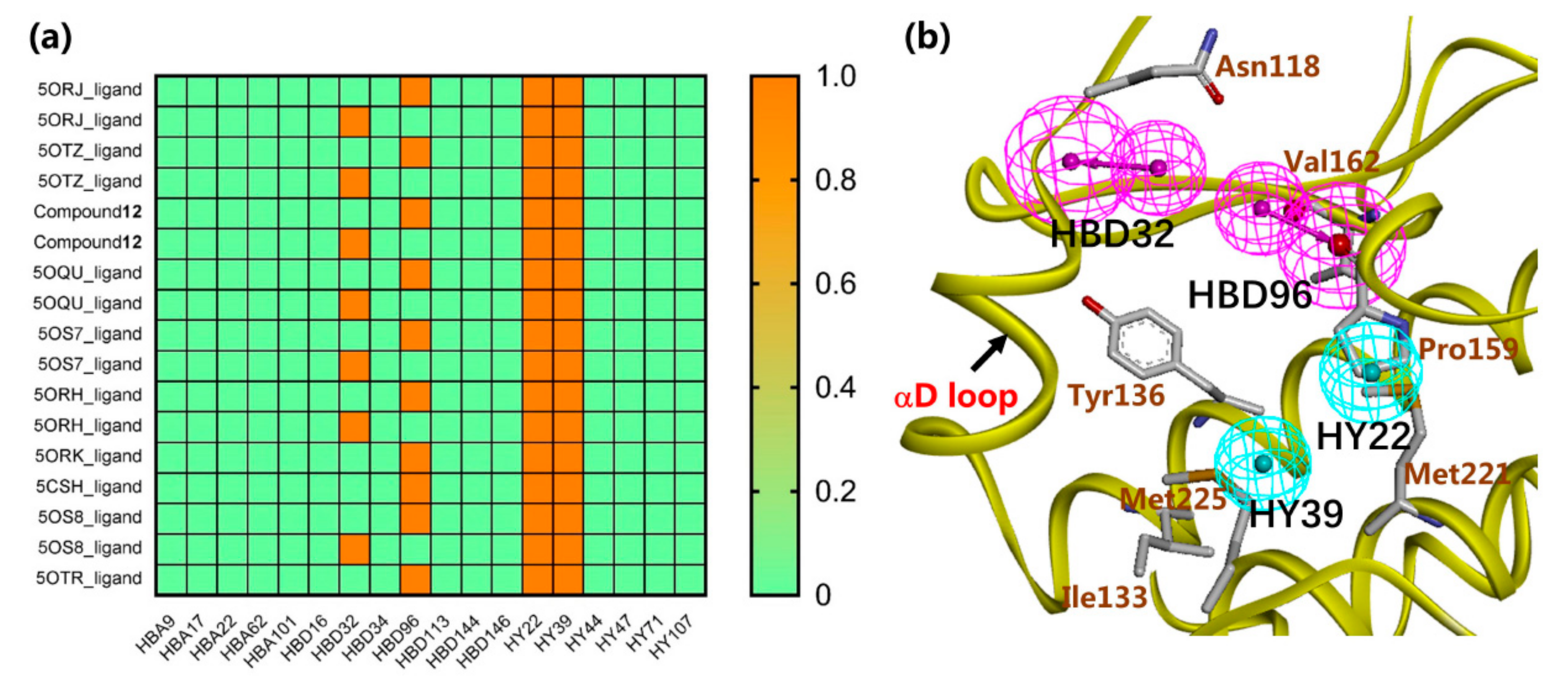
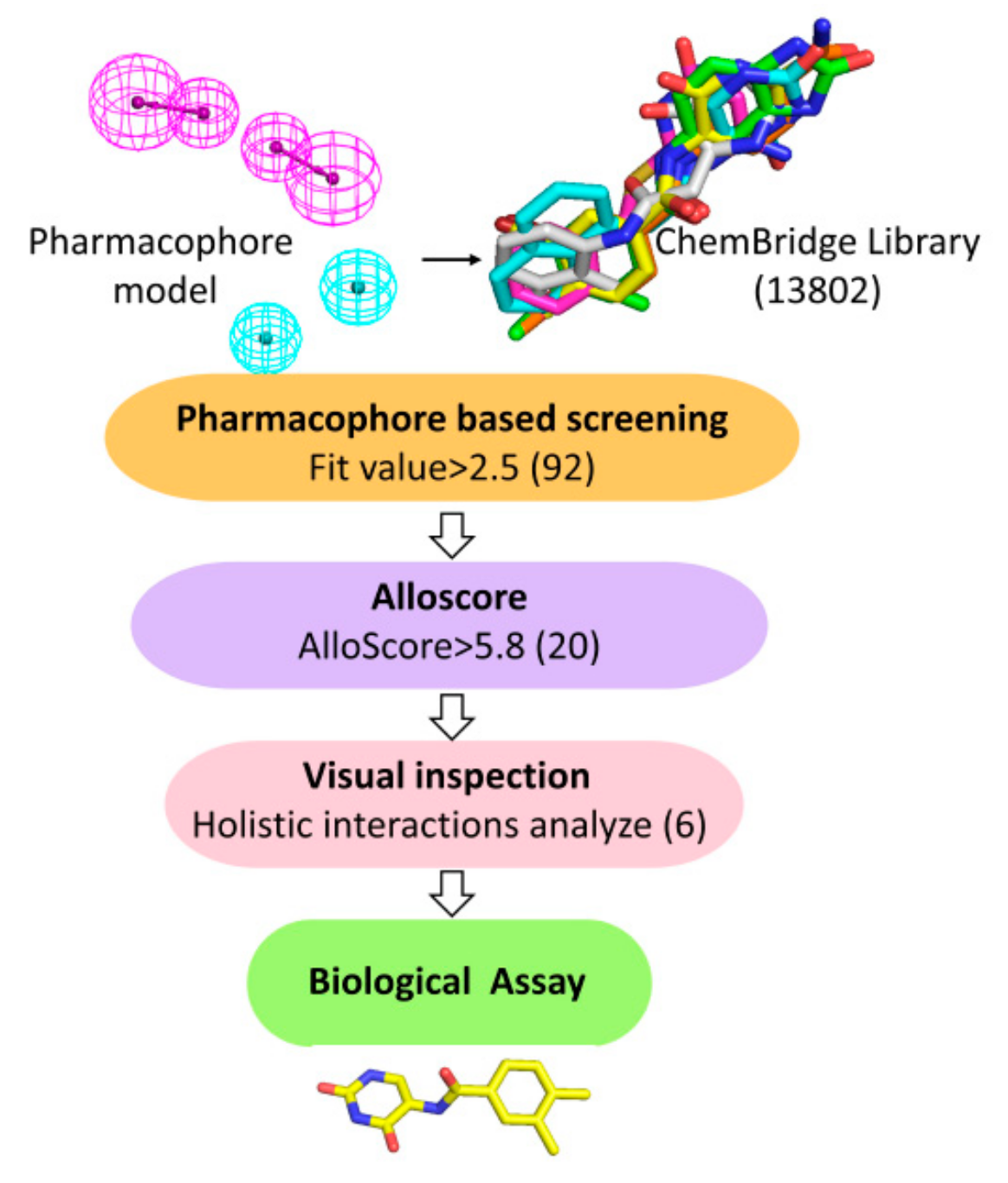
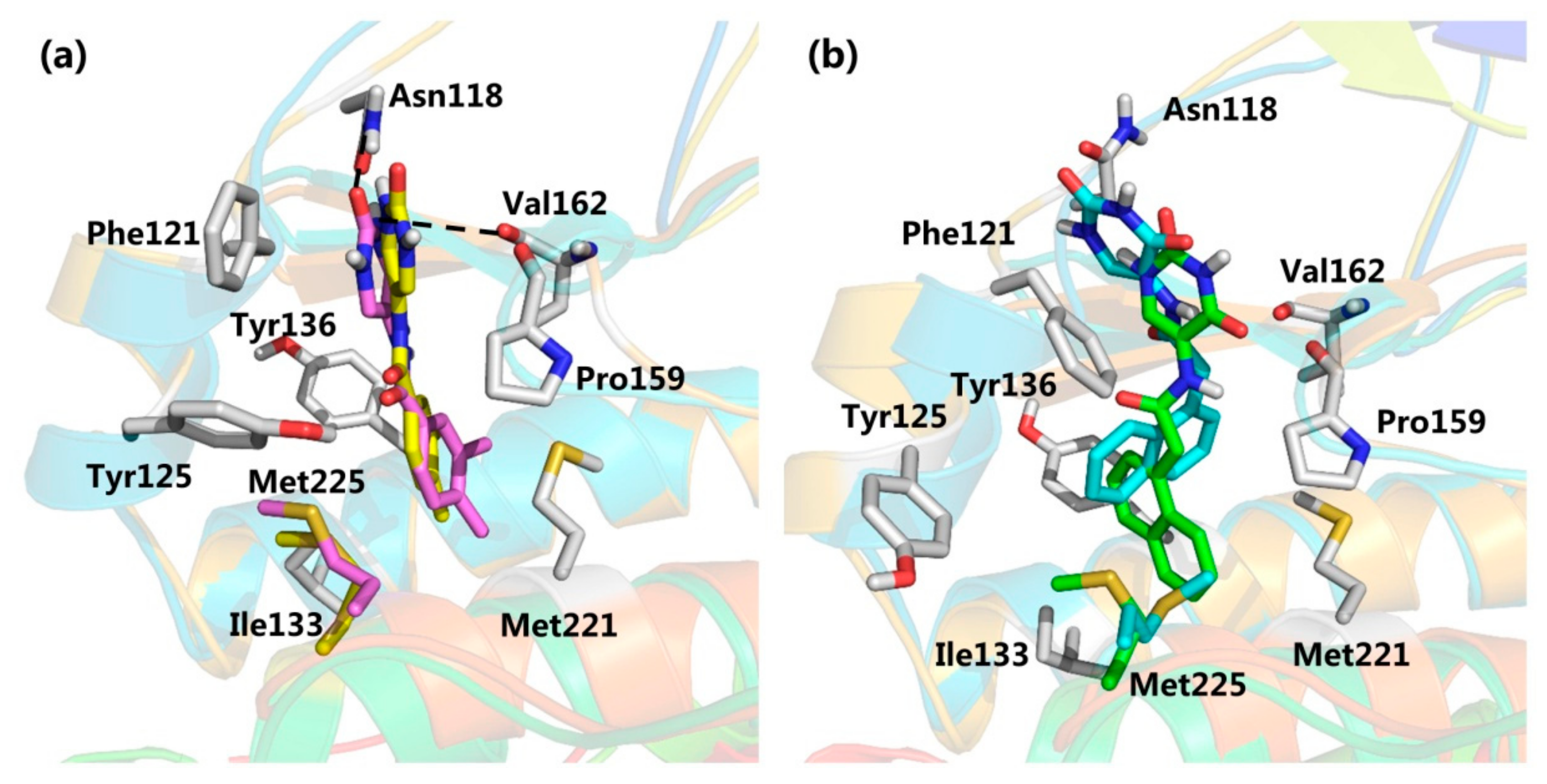
2.1. Structure-Based Pharmacophore Modeling
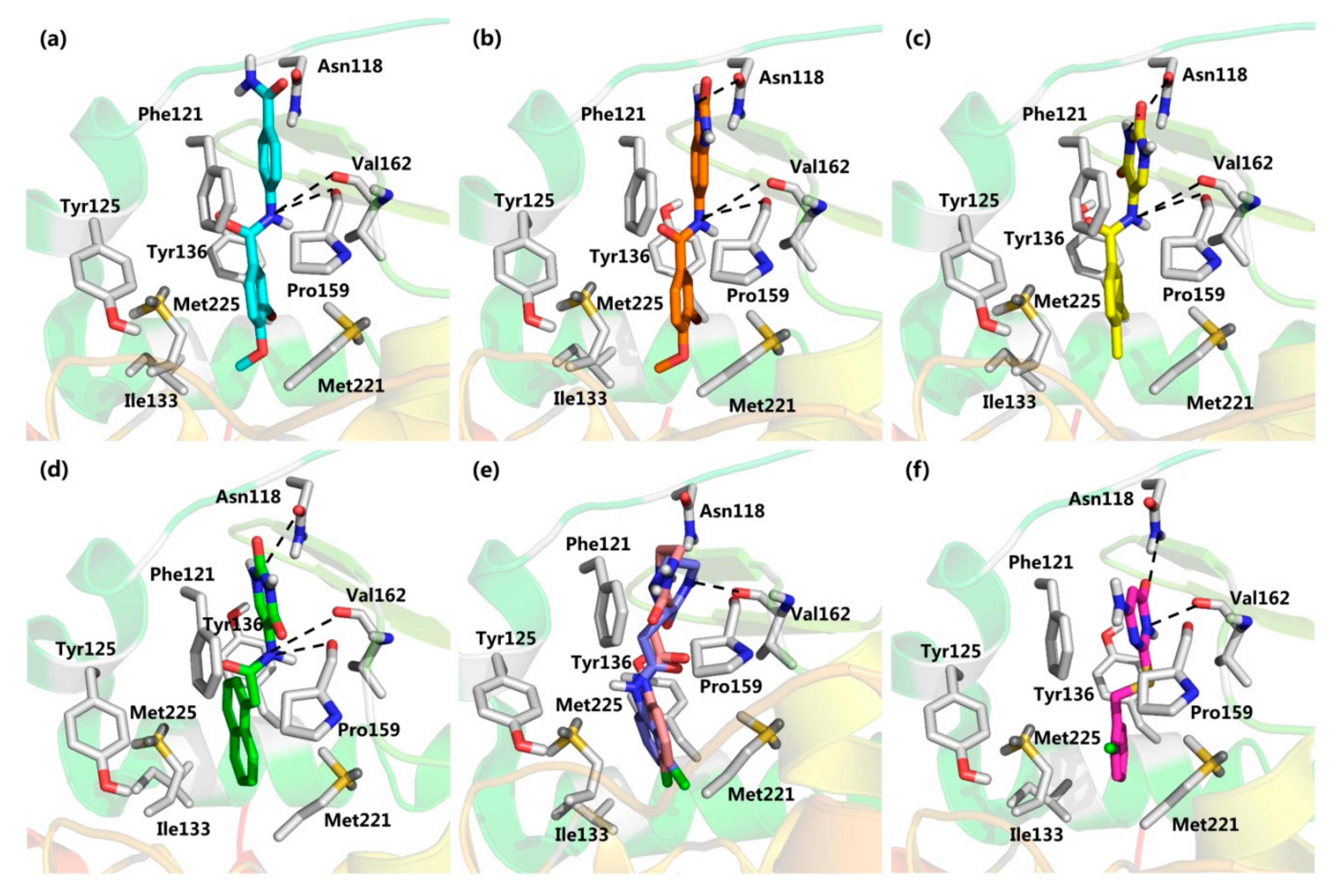
2.2. Virtual Screening
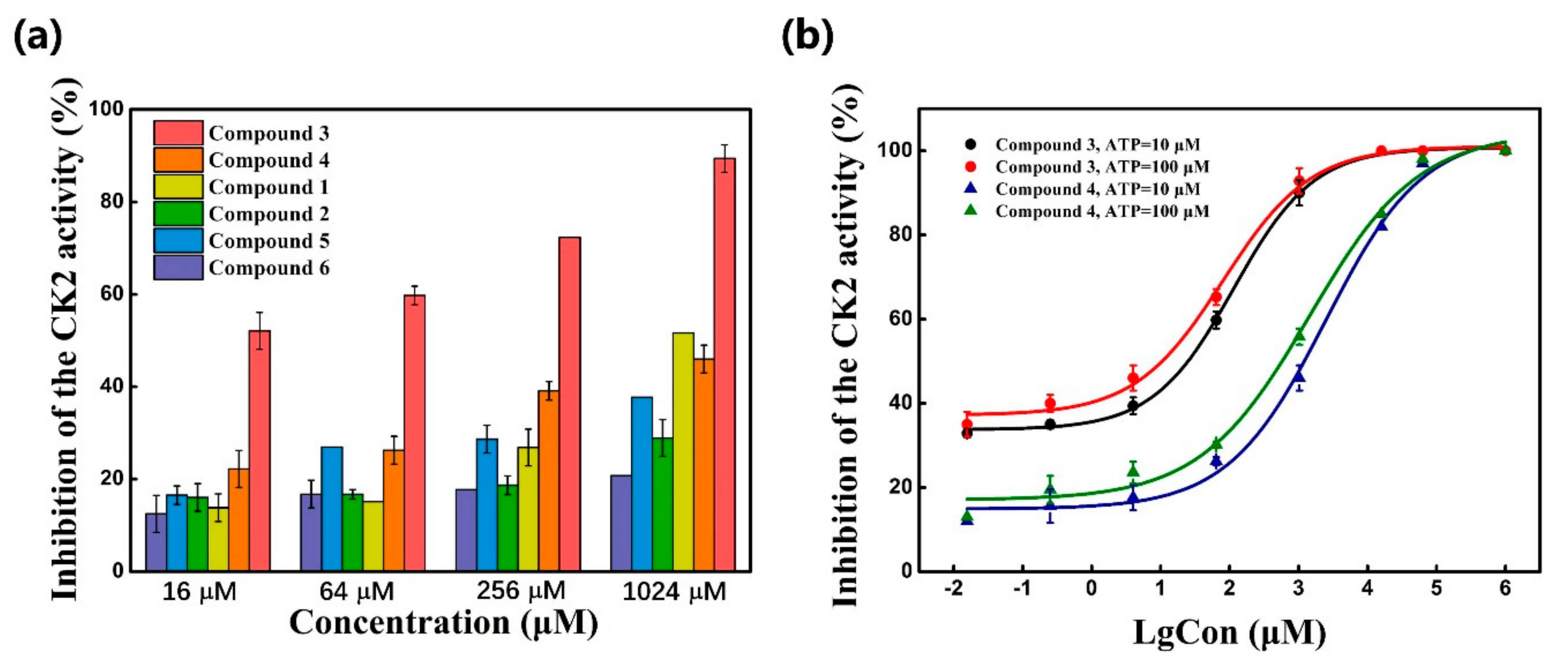
2.3. CK2 Kinase Assay
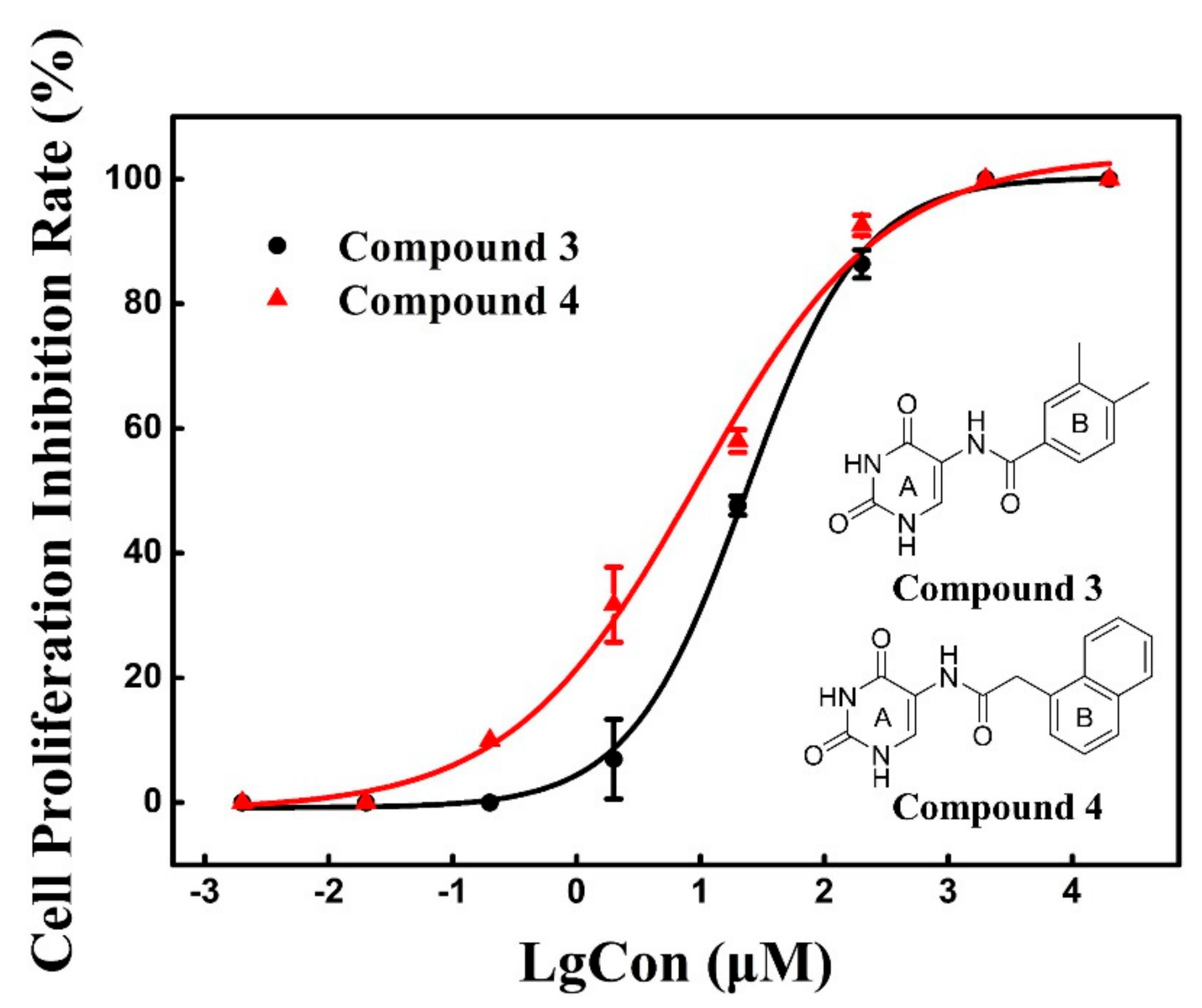
2.4. Cell Proliferation Assay
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cozza, G.; Pinna, L.A. Casein kinases as potential therapeutic targets. Expert Opin. Ther. Targets 2016, 20, 319–340. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Magid, A.F. Inhibition of CK2: An attractive therapeutic target for cancer treatment. ACS Med. Chem. Lett. 2013, 4, 1131–1132. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Pinna, L.A.; Moro, S. Kinase CK2 inhibition: An update. Curr. Med. Chem. 2013, 20, 671–693. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Pinna, L.A.; Moro, S. Protein kinase CK2 inhibitors: A patent review. Expert Opin. Ther. Pat. 2012, 22, 1081–1097. [Google Scholar] [CrossRef] [PubMed]
- Cozza, G.; Bortolato, A.; Moro, S. How druggable is protein kinase CK2? Med. Res. Rev. 2010, 30, 419–462. [Google Scholar] [CrossRef]
- Kitagawa, D.; Yokota, K.; Gouda, M.; Narumi, Y.; Ohmoto, H.; Nishiwaki, E.; Akita, K.; Kirii, Y. Activity-based kinase profiling of approved tyrosine kinase inhibitors. Genes Cells 2013, 18, 110–122. [Google Scholar] [CrossRef]
- Battistutta, R.; Cozza, G.; Pierre, F.; Papinutto, E.; Lolli, G.; Sarno, S.; O’Brien, S.E.; Siddiqui-Jain, A.; Haddach, M.; Anderes, K.; et al. Unprecedented selectivity and structural determinants of a new class of protein kinase CK2 inhibitors in clinical trials for the treatment of cancer. Biochemistry 2011, 50, 8478–8488. [Google Scholar] [CrossRef]
- Guerra, B.; Hochscherf, J.; Jensen, N.B.; Issinger, O.-G. Identification of a novel potent, selective and cell permeable inhibitor of protein kinase CK2 from the NIH/NCI Diversity Set Library. Mol. Cell. Biochem. 2015, 406, 151–161. [Google Scholar] [CrossRef]
- Kim, H.; Lee, K.-S.; Kim, A.-K.; Choi, M.; Choi, K.; Kang, M.; Chi, S.-W.; Lee, M.-S.; Lee, J.-S.; Lee, S.-Y.; et al. A chemical with proven clinical safety rescues Down-syndrome-related phenotypes in through DYRK1A inhibition. Dis. Models Mech. 2016, 9, 839–848. [Google Scholar] [CrossRef]
- Wenthur, C.J.; Gentry, P.R.; Mathews, T.P.; Lindsley, C.W. Drugs for allosteric sites on receptors. Annu. Rev. Pharmacool. Toxicol. 2014, 54, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; He, X.; Ni, D.; Zhang, J. Allosteric modulator discovery: From serendipity to structure-based design. J. Med. Chem. 2019, 62, 6405–6421. [Google Scholar] [CrossRef] [PubMed]
- Gurney, M.E.; Nugent, R.A.; Mo, X.; Sindac, J.A.; Hagen, T.J.; Fox, D.; O’Donnell, J.M.; Zhang, C.; Xu, Y.; Zhang, H.-T.; et al. Design and synthesis of selective phosphodiesterase 4D (PDE4D) allosteric inhibitors for the treatment of fragile X syndrome and other brain disorders. J. Med. Chem. 2019, 62, 4884–4901. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Jiang, L.; Liang, R.; Li, H.; Ruan, X.; Shan, C.; Ye, D.; Zhou, L. Synthesis and biological evaluation of anthraquinone derivatives as allosteric phosphoglycerate mutase 1 inhibitors for cancer treatment. Eur. J. Med. Chem. 2019, 168, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.-M.; Dong, J.-K.; Song, K.; Wang, T.-D.; Huang, W.-K.; Zhang, J.-M.; Yang, X.-Y.; Shen, Y.; Zhang, J. A novel allosteric site in casein kinase 2α discovered using combining bioinformatics and biochemistry methods. Acta Pharmacol. Sin. 2017, 38, 1691–1698. [Google Scholar] [CrossRef]
- Brear, P.; North, A.; Iegre, J.; Hadje Georgiou, K.; Lubin, A.; Carro, L.; Green, W.; Sore, H.F.; Hyvönen, M.; Spring, D.R. Novel non-ATP competitive small molecules targeting the CK2 α/β interface. Biorg. Med. Chem. 2018, 26, 3016–3020. [Google Scholar] [CrossRef]
- Brear, P.; De Fusco, C.; Hadje Georgiou, K.; Francis-Newton, N.J.; Stubbs, C.J.; Sore, H.F.; Venkitaraman, A.R.; Abell, C.; Spring, D.R.; Hyvonen, M. Specific inhibition of CK2alpha from an anchor outside the active site. Chem. Sci. 2016, 7, 6839–6845. [Google Scholar] [CrossRef]
- De Fusco, C.; Brear, P.; Iegre, J.; Georgiou, K.H.; Sore, H.F.; Hyvonen, M.; Spring, D.R. A fragment-based approach leading to the discovery of a novel binding site and the selective CK2 inhibitor CAM4066. Bioorg. Med. Chem. 2017, 25, 3471–3482. [Google Scholar] [CrossRef]
- Iegre, J.; Brear, P.; De Fusco, C.; Yoshida, M.; Mitchell, S.L.; Rossmann, M.; Carro, L.; Sore, H.F.; Hyvonen, M.; Spring, D.R. Second-generation CK2alpha inhibitors targeting the alphaD pocket. Chem. Sci. 2018, 9, 3041–3049. [Google Scholar] [CrossRef]
- Bestgen, B.; Krimm, I.; Kufareva, I.; Kamal, A.A.M.; Seetoh, W.G.; Abell, C.; Hartmann, R.W.; Abagyan, R.; Cochet, C.; Le Borgne, M.; et al. 2-Aminothiazole derivatives as selective allosteric modulators of the protein kinase CK2. 1. identification of an allosteric binding site. J. Med. Chem. 2019, 62, 1803–1816. [Google Scholar] [CrossRef]
- Bestgen, B.; Kufareva, I.; Seetoh, W.; Abell, C.; Hartmann, R.W.; Abagyan, R.; Le Borgne, M.; Filhol, O.; Cochet, C.; Lomberget, T.; et al. 2-Aminothiazole derivatives as selective allosteric modulators of the protein kinase CK2. 2. structure-based optimization and investigation of effects specific to the allosteric mode of action. J. Med. Chem. 2019, 62, 1817–1836. [Google Scholar] [CrossRef]
- Hardy, J.A.; Wells, J.A. Searching for new allosteric sites in enzymes. Curr. Opin. Struct. Biol. 2004, 14, 706–715. [Google Scholar] [CrossRef]
- Wagner, J.R.; Lee, C.T.; Durrant, J.D.; Malmstrom, R.D.; Feher, V.A.; Amaro, R.E. Emerging computational methods for the rational discovery of allosteric drugs. Chem. Rev. 2016, 116, 6370–6390. [Google Scholar] [CrossRef]
- Perna, A.M.; Reisen, F.; Schmidt, T.P.; Geppert, T.; Pillong, M.; Weisel, M.; Hoy, B.; Simister, P.C.; Feller, S.M.; Wessler, S.; et al. Inhibiting Helicobacter pylori HtrA protease by addressing a computationally predicted allosteric ligand binding site. Chem. Sci. 2014, 5, 3583–3590. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhao, J.; Deng, W.; Chen, Y.; Shang, J.; Song, K.; Zhang, L.; Wang, C.; Lu, S.; Yang, X.; et al. Identification of a cellularly active SIRT6 allosteric activator. Nat. Chem. Biol. 2018, 14, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Apetoh, L. Enhancing the anticancer effects of 5-fluorouracil: Current challenges and future perspectives. Biomed. J. 2015, 38, 111–116. [Google Scholar] [PubMed]
- Li, S.; Shen, Q.; Su, M.; Liu, X.; Lu, S.; Chen, Z.; Wang, R.; Zhang, J. Alloscore: A method for predicting allosteric ligand–protein interactions. Bioinformatics 2016, 32, 1574–1576. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking11Edited by F. E. Cohen. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. Amber 14; University of California: San Francisco, CA, USA, 2014. [Google Scholar]
- Zegzouti, H.; Zdanovskaia, M.; Hsiao, K.; Goueli, S.A. ADP-Glo: A Bioluminescent and homogeneous ADP monitoring assay for kinases. Assay Drug Dev. Technol. 2009, 7, 560–572. [Google Scholar] [CrossRef]
- Liu, S.; Hsieh, D.; Yang, Y.L.; Xu, Z.D.; Peto, C.; Jablons, D.M.; You, L. Coumestrol from the national cancer Institute’s natural product library is a novel inhibitor of protein kinase CK2. BMC Pharmacol. Toxicol. 2013, 14, 9. [Google Scholar] [CrossRef]
- Davis, M.I.; Auld, D.S.; Inglese, J. Bioluminescence methods for assaying kinases in quantitative high-throughput screening (qHTS) format applied to yes1 tyrosine kinase, glucokinase, and PI5P4Kalpha lipid kinase. Methods Mol. Biol. 2016, 1360, 47–58. [Google Scholar] [PubMed]
- Daya-Makin, M.; Sanghera, J.S.; Mogentale, T.L.; Lipp, M.; Parchomchuk, J.; Hogg, J.C.; Pelech, S.L. Activation of a tumor-associated protein kinase (p40TAK) and casein kinase 2 in human squamous cell carcinomas and adenocarcinomas of the lung. Cancer Res. 1994, 54, 2262–2268. [Google Scholar] [PubMed]
- Ortega, C.E.; Seidner, Y.; Dominguez, I. Mining CK2 in cancer. PLoS ONE 2014, 9, e115609. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | 2D Structure | AlloScore | %Inhibition at 16 μM | %Inhibition at 256 μM |
|---|---|---|---|---|
| 1 |  | 6.38 | 14 | 27 |
| 2 |  | 6.38 | 16 | 19 |
| 3 |  | 6.07 | 52 | 72 |
| 4 |  | 6.00 | 22 | 40 |
| 5 |  | 5.89 (S) 5.80 (R) | 16 | 29 |
| 6 |  | 5.81 | 12 | 18 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Zhang, X.; Zhang, N.; Zhou, Y.; Sun, G.; Zhao, L.; Zhong, R. Identification and Biological Evaluation of CK2 Allosteric Fragments through Structure-Based Virtual Screening. Molecules 2020, 25, 237. https://doi.org/10.3390/molecules25010237
Li C, Zhang X, Zhang N, Zhou Y, Sun G, Zhao L, Zhong R. Identification and Biological Evaluation of CK2 Allosteric Fragments through Structure-Based Virtual Screening. Molecules. 2020; 25(1):237. https://doi.org/10.3390/molecules25010237
Chicago/Turabian StyleLi, Chunqiong, Xuewen Zhang, Na Zhang, Yue Zhou, Guohui Sun, Lijiao Zhao, and Rugang Zhong. 2020. "Identification and Biological Evaluation of CK2 Allosteric Fragments through Structure-Based Virtual Screening" Molecules 25, no. 1: 237. https://doi.org/10.3390/molecules25010237
APA StyleLi, C., Zhang, X., Zhang, N., Zhou, Y., Sun, G., Zhao, L., & Zhong, R. (2020). Identification and Biological Evaluation of CK2 Allosteric Fragments through Structure-Based Virtual Screening. Molecules, 25(1), 237. https://doi.org/10.3390/molecules25010237