Evaluation of Transition Metal Catalysts in Electrochemically Induced Aromatic Phosphonation



Abstract
:1. Introduction
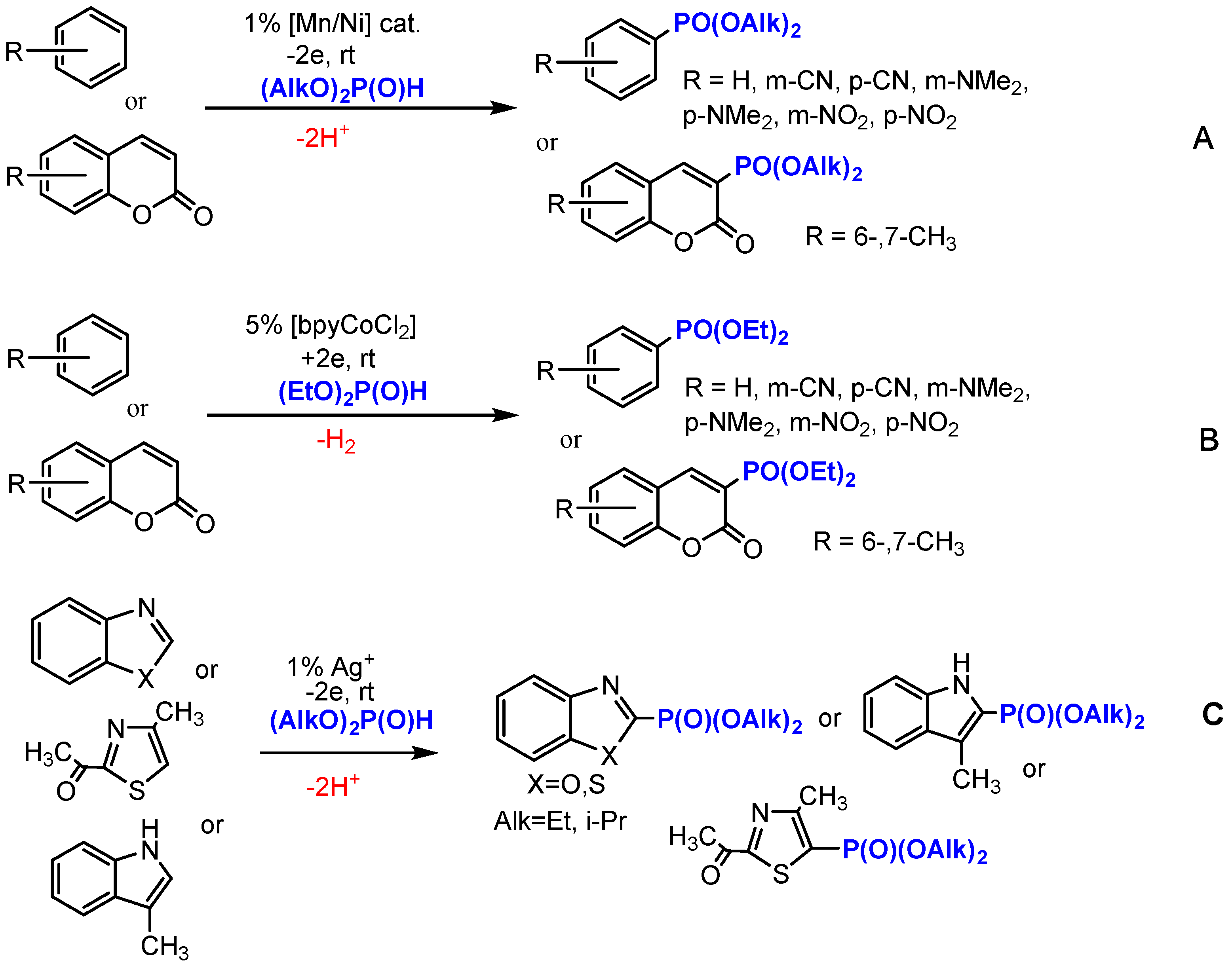
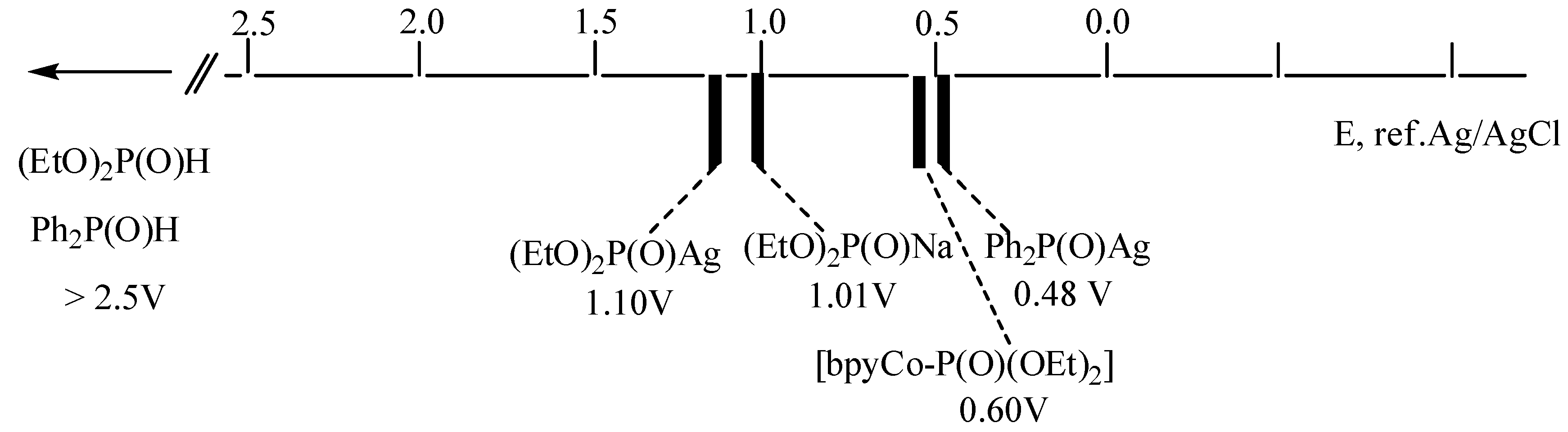
2. Results and Discussion
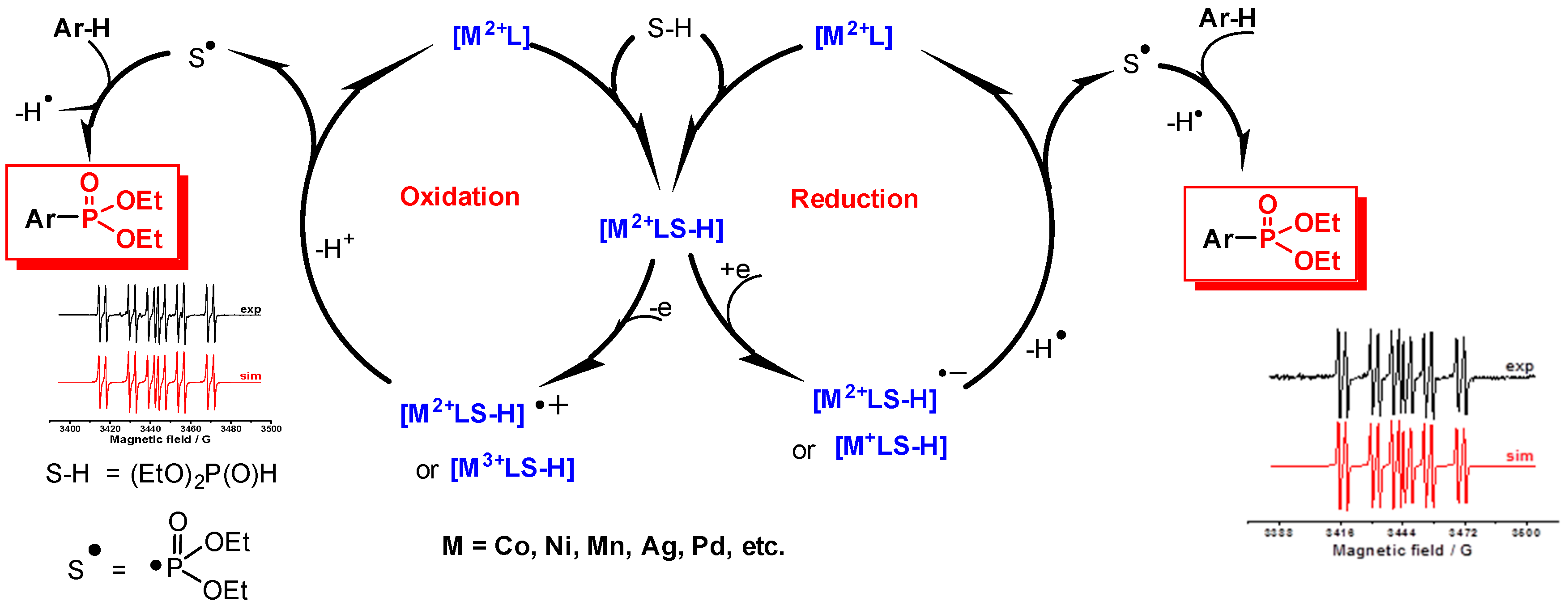
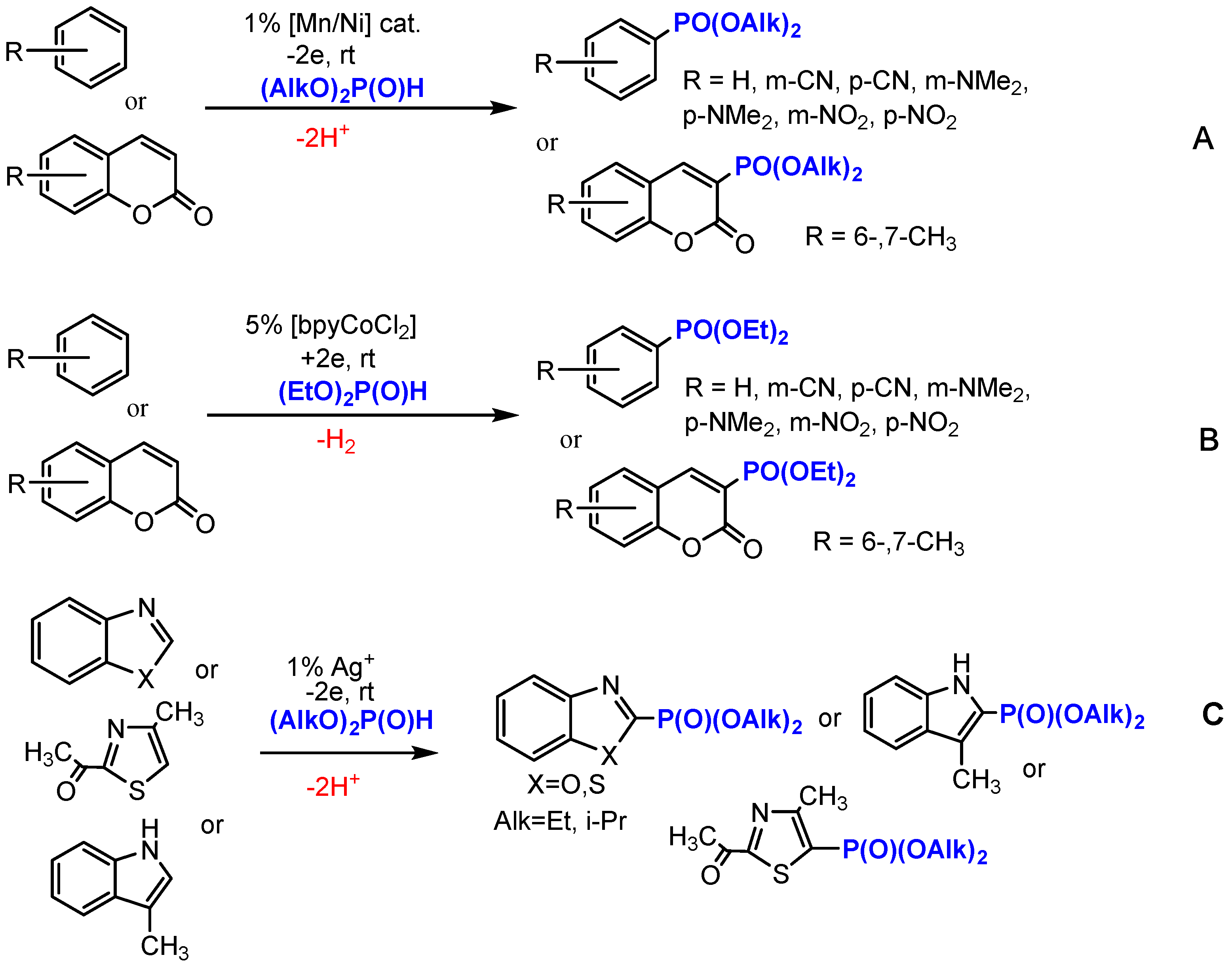
2.1. Oxidative Conditions
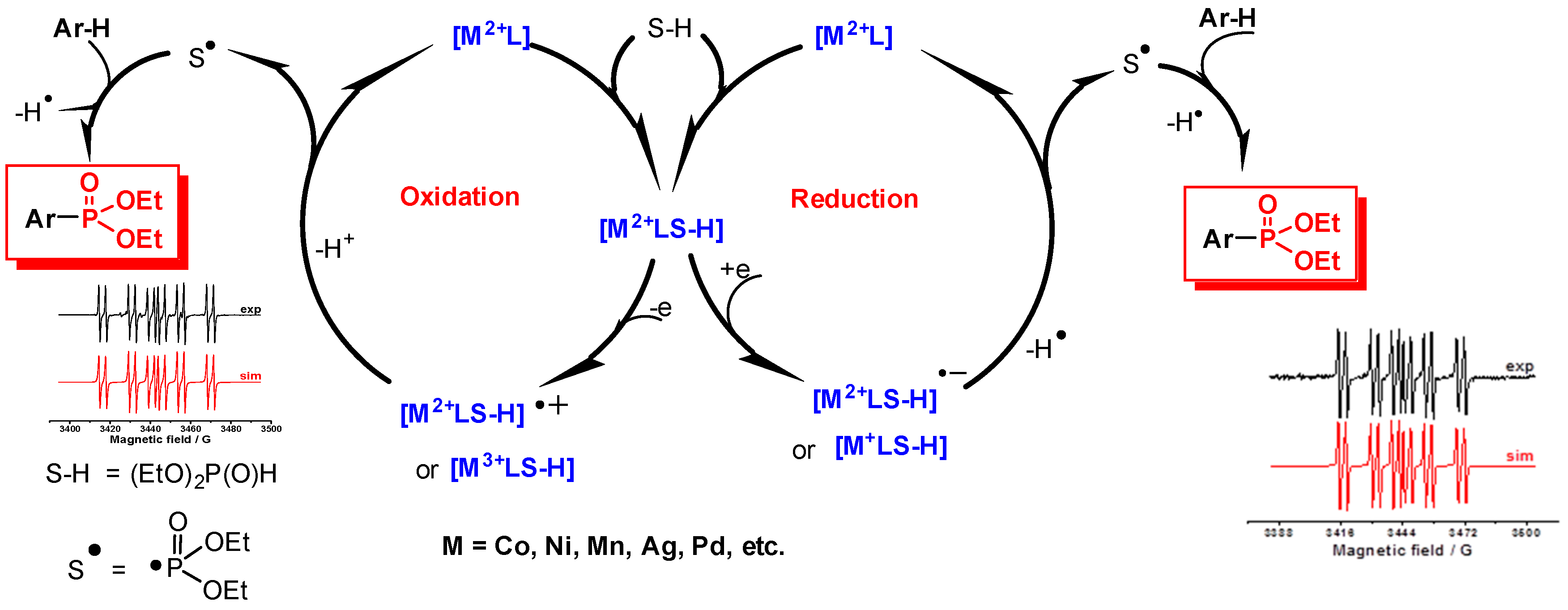
2.2. Electroreduction Conditions
3. Materials and Methods
3.1. Cyclic Voltammetry
3.2. Synthesis of the Metal Complexes
3.3. Data Processing of Voltammetric Measurements of Complexes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Demmer, C.S.; Krogsgaard-Larsen, N.; Bunch, L. Review on Modern Advances of Chemical Methods for the Introduction of a Phosphonic Acid Group. Chem. Rev. 2011, 111, 7981–8006. [Google Scholar] [CrossRef] [PubMed]
- Budnikova, Y.H. Opportunities and challenges for combining electro-and organometallic catalysis in C(sp2)-H phosphonation. Pure Appl. Chem. 2019, 91, 17–31. [Google Scholar] [CrossRef]
- Budnikova, Y.H.; Sinyashin, O.G. Phosphorylation of C–H bonds of aromatic compounds using metals and metal complexes. Russ. Chem. Rev. 2015, 84, 917–951. [Google Scholar] [CrossRef]
- Budnikova, Y.H. Transition metal-promoted reactions of diarylphosphine oxides as a synthetic method for organophosphorus heterocyclic compounds. Chem. Heterocycl. Com. 2018, 54, 269–279. [Google Scholar] [CrossRef]
- Abakumov, G.A.; Piskunov, A.V.; Cherkasov, V.K.; Fedushkin, I.L.; Ananikov, V.P.; Eremin, D.B.; Gordeev, E.G.; Beletskaya, I.P.; Averin, A.D.; Bochkarev, M.N.; et al. Organoelement chemistry: Promising growth areas and challenges. Russ. Chem. Rev. 2018, 87, 393–507. [Google Scholar] [CrossRef]
- Gao, Y.; Tang, G.; Zhao, Y. Recent progress toward organophosphorus compounds based on phosphorus-centered radical difunctionalizations. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 589–596. [Google Scholar] [CrossRef]
- Montchamp, J.-L. Phosphinate Chemistry in the 21st Century. Acc. Chem. Res. 2014, 47, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Quin, L.D. A Guide to Organophosphorus Chemistry; Wiley Interscience: New York, NY, USA, 2000. [Google Scholar]
- Corbridge, D.E.C. Phosphorus: Chemistry, Biochemistry and Technology, 6th ed.; CRC Press: New York, NY, USA, 2013. [Google Scholar]
- Kawaguchi, S.I.; Ogawa, A. Future Trends in Organophosphorus Chemistry. In Organophosphorus Chemistry: From Molecules to Applications, 1st ed.; Iaroshenko, V., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2019; pp. 545–555. [Google Scholar]
- Eto, M. Organophosphorus Pesticides, 1st ed.; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Tajti, Á.; Keglevich, G. The importance of organophosphorus compounds as biologically active agents. In Organophosphorus Chemistry: Novel Developments; Keglevich, G., Ed.; Walter de Gruyter GmbH: Berlin, Germany; Boston, MA, USA, 2018. [Google Scholar]
- Xiang, C.B.; Bian, Y.J.; Mao, X.R.; Huang, Z.Z. Coupling reactions of heteroarenes with phosphites under silver catalysis. J. Org. Chem. 2012, 77, 7706–7710. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Ren, Y.; Lang, R.; Hu, X.; Xia, C.; Li, F. Palladium-catalyzed direct phosphonation of azoles with dialkylphosphites. Chem. Commun. 2012, 48, 5181–5183. [Google Scholar] [CrossRef]
- Trostyanskaya, I.G.; Beletskaya, I.P. Copper (II)-catalyzed regio-and stereoselective addition of H/P(O)R2 to alkynes. Tetrahedron 2014, 70, 2556–2562. [Google Scholar] [CrossRef]
- Muhammad, M.H.; Chen, X.L.; Yu, B.; Qu, L.B.; Zhao, Y.F. Applications of H-phosphonates for C element bond formation. Pure Appl. Chem. 2019, 91, 33–41. [Google Scholar] [CrossRef]
- Khemchyan, L.L.; Ivanova, J.V.; Zalesskiy, S.S.; Ananikov, V.P.; Beletskaya, I.P.; Starikova, Z.A. Unprecedented Control of Selectivity in Nickel-Catalyzed Hydrophosphorylation of Alkynes: Efficient Route to Mono-and Bisphosphonates. Adv. Synth. Catal. 2014, 356, 771–780. [Google Scholar] [CrossRef]
- Ananikov, V.P.; Ivanova, J.V.; Khemchyan, L.L.; Beletskaya, I.P. Unusual Control of Reaction Selectivity through a Subtle Change in the Ligand: Proof of Concept and Application in Pd-Catalyzed C–P Bond Formation. Eur. J. Org. Chem. 2012, 2012, 3830–3840. [Google Scholar] [CrossRef]
- Ananikov, V.P.; Beletskaya, I.P. Alkyne Insertion into the M-P and M-H Bonds (M= Pd, Ni, Pt, and Rh): A Theoretical Mechanistic Study of the C–P and C–H Bond-Formation Steps. Chem. Asian J. 2011, 6, 1423–1430. [Google Scholar] [CrossRef]
- Yurko, E.O.; Gryaznova, T.V.; Kholin, K.V.; Khrizanforova, V.V.; Budnikova, Y.H. External oxidant-free cross-coupling: Electrochemically induced aromatic C–H phosphonation of azoles with dialkyl-H-phosphonates under silver catalysis. Dalton Trans. 2018, 47, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Gansäuer, A.; Hildebrandt, S.; Vogelsang, E.; Flowers, R.A., II. Tuning the redox properties of the titanocene (III)/(IV)-couple for atom-economical catalysis in single electron steps. Dalton Trans. 2016, 45, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Lang, K.; Lu, H.; Wojtas, L.; Zhang, X.P. Asymmetric radical bicyclization of allylazidoformates via cobalt(II)-based metalloradical catalysis. J. Am. Chem. Soc. 2017, 139, 9164–9167. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, P.F.; Tiekink, M.J.; Breukelaar, W.B.; Broere, D.L.J.; van Leest, N.P.; van der Vlugt, J.I.; Reek, J.N.H.; de Bruin, B. Cobalt-Porphyrin-CatalysedIntramolecular Ring-Closing C–H Amination of Aliphatic Azides: A Nitrene-Radical Approach to Saturated Heterocycles. Chem. Eur. J. 2017, 23, 7945–7952. [Google Scholar] [CrossRef] [PubMed]
- Grotenhuis, C.; van den Heuvel, N.; van der Vlugt, J.I.; de Bruin, B. Catalytic Dibenzocyclooctene Synthesis via Cobalt(III)-Carbene Radical and ortho-Quinodimethane Intermediates. Angew. Chem. Int. Ed. 2018, 57, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Richrath, R.; Gansäuer, A. Merging Catalysis in Single Electron Steps with Photoredox Catalysis–Efficient and Sustainable Radical Chemistry. ACS Catal. 2019, 9, 3208–3212. [Google Scholar] [CrossRef]
- Gansäuer, A.; Fleckhaus, A.; Lafont, M.A.; Okkel, A.; Kotsis, K.; Anoop, A.; Neese, F. Catalysis via Homolytic Substitutions with C−O and Ti−O Bonds: Oxidative Additions and Reductive Eliminations in Single Electron Steps. J. Am. Chem. Soc. 2009, 131, 16989–16999. [Google Scholar] [CrossRef]
- Budnikova, Y.H.; Gryaznova, T.V.; Grinenko, V.V.; Dudkina, Y.B.; Khrizanforov, M.N. Eco-efficient electrocatalytic C–P bond formation. Pure Appl. Chem. 2017, 89, 311–330. [Google Scholar] [CrossRef]
- Costentin, C.M.; Savéant, J. Multielectron, multistep molecular catalysis of electrochemical reactions: Benchmarking of homogeneous catalysts. ChemElectroChem 2014, 1, 1226–1236. [Google Scholar] [CrossRef]
- Savéant, J.M. Molecular catalysis of electrochemical reactions. Mechanistic aspects. Chem. Rev. 2008, 108, 2348–2378. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.D.; Newell, R.H.; McNevin, M.J.; Muckerman, J.T.; Rakowski DuBois, M.; DuBois, D.L. Hydrogen oxidation and production using nickel-based molecular catalysts with positioned proton relays. J. Am. Chem. Soc. 2006, 128, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, T.; Spannring, P.; Riccardi, L.; Gansäuer, A. Mechanism-Based Condition Screening for Sustainable Catalysis in Single-Electron Steps by Cyclic Voltammetry. Angew. Chem. Int. Ed. 2018, 57, 5006–5010. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, T.; Hilche, T.; Klare, S.; Gansäuer, A. Condition Screening for Sustainable Catalysis in Single-Electron Steps by Cyclic Voltammetry: Additives and Solvents. ChemSusChem 2019, 12, 1–7. [Google Scholar] [CrossRef]
- Budnikova, Y.H.; Dudkina, Y.B. Progress of electrochemical C(sp2)-H phosphonation. Phosphorus Sulfur Silicon Relat. Elem. 2019. [Google Scholar] [CrossRef]
- Khrizanforov, M.N.; Strekalova, S.O.; Gryaznova, T.V.; Khrizanforova, V.V.; Budnikova, Y.H. New method of metal-induced oxidative phosphorylation of benzene. Russ. Chem. Bull. 2015, 64, 1926–1932. [Google Scholar] [CrossRef]
- Strekalova, S.O.; Khrizanforov, M.N.; Shamsieva, A.V.; Grinenko, V.V.; Gryaznova, T.V.; Musina, E.I.; Karasik, A.A.; Budnikova, Y.H. Direct phosphorylation of pyridine in the presence of Ni(BF4)2bpy and CoCl2bpy metal complexes. Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1545–1546. [Google Scholar] [CrossRef]
- Grinenko, V.V.; Khrizanforov, M.N.; Strekalova, S.O.; Khrizanforova, V.V.; Kholin, K.V.; Gryaznova, T.V.; Budnikova, Y.H. Electrooxidative phosphorylation of coumarins by bimetallic catalytic systems Ni(II)/Mn(II) or Co(II)/Mn(II). Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1660–1661. [Google Scholar] [CrossRef]
- Khrizanforov, M.N.; Strekalova, S.O.; Grinenko, V.V.; Khrizanforova, V.V.; Gryaznova, T.V.; Budnikova, Y.H. Various ways of CP bonds formation via selective electrochemical phosphorylation of aromatic CH bonds. Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1491–1493. [Google Scholar] [CrossRef]
- Gryaznova, T.V.; Khrizanforov, M.N.; Strekalova, S.O.; Budnikova, Y.H.; Sinyshin, O.G. Electrochemical oxidative phosphonation of azoles. Phosphorus Sulfur Silicon Relat. Elem. 2016, 191, 1658–1659. [Google Scholar] [CrossRef]
- Strekalova, S.O.; Khrizanforov, M.N.; Gryaznova, T.V.; Khrizanforova, V.V.; Budnikova, Y.H. Electrochemical phosphorylation of coumarins catalyzed by transition metal complexes (Ni—Mn, Co—Mn). Russ. Chem. Bull. 2016, 65, 1295–1298. [Google Scholar] [CrossRef]
- Khrizanforov, M.N.; Strekalova, S.O.; Kholin, K.V.; Khrizanforova, V.V.; Kadirov, M.K.; Gryaznova, T.V.; Budnikova, Y.H. Novel approach to metal-induced oxidative phosphorylation of aromatic compounds. Catal. Today 2017, 279, 133–141. [Google Scholar] [CrossRef]
- Strekalova, S.O.; Grinenko, V.V.; Gryaznova, T.V.; Kononov, A.I.; Dolengovski, E.L.; Budnikova, Y.H. Electrochemical phosphorylation of arenes catalyzed by cobalt under oxidative and reductive conditions. Phosphorus Sulfur Silicon Relat. Elem. 2018. [Google Scholar] [CrossRef]
- Khrizanforov, M.; Strekalova, S.; Khrizanforova, V.; Dobrynin, A.; Kholin, K.; Gryaznova, T.; Grinenko, V.; Gubaidullin, A.; Kadirov, M.K.; Budnikova, Y. Cobalt-Catalyzed Green Cross-Dehydrogenative C(sp2)-H/PH Coupling Reactions. Top. Catal. 2018, 61, 1949–1956. [Google Scholar] [CrossRef]
- Khrizanforov, M.N.; Strekalova, S.O.; Grinenko, V.V.; Kononov, A.I.; Dolengovski, E.L.; Budnikova, Y.H. CP bond formation via selective electrocatalytic CH phosphorylation. Phosphorus Sulfur Silicon Relat. Elem. 2019. [Google Scholar] [CrossRef]
- Khrizanforova, V.V.; Kholin, K.V.; Khrizanforov, M.N.; Kadirov, M.K.; Budnikova, Y.H. Electrooxidative CH/PH functionalization as a novel way to benzo[b]phosphole oxides mediated by catalytic amounts of silver acetate. New J. Chem. 2018, 42, 930–935. [Google Scholar] [CrossRef]
- Shimizu, G.K.H.; Vaidhyanathan, R.; Taylor, J.M. Phosphonate and sulfonate metal organic frameworks. Chem. Soc. Rev. 2009, 38, 1430–1449. [Google Scholar] [CrossRef]
- Mao, J.-G. Structures and luminescent properties of lanthanide phosphonates. Coord. Chem. Rev. 2007, 251, 1493–1520. [Google Scholar] [CrossRef]
- Zheng, Y.-Z.; Evangelisti, M.; Tuna, F.; Winpenny, R.E.P. Co–Ln mixed-metal phosphonate grids and cages as molecular magnetic refrigerants. J. Am. Chem. Soc. 2012, 134, 1057–1065. [Google Scholar] [CrossRef]
- Taylor, J.M.; Mah, R.K.; Moudrakovski, I.L.; Ratcliffe, C.I.; Vaidhyanathan, R.; Shimizu, G.K.H. Facile proton conduction via ordered water molecules in a phosphonate metal− organic framework. J. Am. Chem. Soc. 2010, 132, 14055–14057. [Google Scholar] [CrossRef]
- Khrizanforov, M.; Strekalova, S.; Kholin, K.; Khrizanforova, V.; Grinenko, V.; Gryaznova, T.; Budnikova, Y. One-stage synthesis of FcP(O)(OC2H5)2 from ferrocene and α-hydroxyethylphosphonate. RSC Adv. 2016, 6, 42701–42707. [Google Scholar] [CrossRef]
- Gao, C.Y.; Wang, F.; Tian, H.R.; Li, L.J.; Zhang, J.; Sun, Z.M. Particular handedness excess through symmetry-breaking crystallization of a 3D cobalt phosphonate. Inorg. Chem. 2015, 55, 537–539. [Google Scholar] [CrossRef]
- Jutand, A. Contribution of electrochemistry to organometallic catalysis. Chem. Rev. 2008, 108, 2300–2347. [Google Scholar] [CrossRef]
- Khrizanforova, V.V.; Musina, E.I.; Khrizanforov, M.N.; Gerasimova, T.P.; Katsyuba, S.A.; Spiridonova, Y.S.; Islamov, D.R.; Kataeva, O.N.; Karasik, A.A.; Sinyashin, O.G.; et al. Unexpected ligand effect on the catalytic reaction rate acceleration for hydrogen production using biomimetic nickel electrocatalysts with 1, 5-diaza-3, 7-diphosphacyclooctanes. J. Organomet. Chem. 2015, 789, 14–21. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Wang, Z.C.; Zhan, S.; Ye, J.S. A water-soluble iron electrocatalyst for water oxidation with high TOF. Appl. Catal. A 2015, 490, 128–132. [Google Scholar] [CrossRef]
- Kargin, Y.M.; Budnikova, Y.G. Electrochemistry of organophosphorus compounds. Russ. J. Gen. Chem. 2001, 71, 1393–1421. [Google Scholar] [CrossRef]
- Budnikova, Y.G.; Tazeev, D.I.; Kafiyatullina, A.G.; Yakhvarov, D.G.; Morozov, V.I.; Gusarova, N.K.; Trofimov, B.A.; Sinyashin, O.G. Activation of white phosphorus in the coordination sphere of nickel complexes with σ-donor ligands. Russ. Chem. Bull. 2005, 54, 942–947. [Google Scholar] [CrossRef]
- Romero, I.; Rodríguez, M.; Llobet, A.; Corbella, M.; Fernández, G.; Collomb, M.N. EPR and magnetic properties of [Mn(μ-Cl)2(bpy)]n: An unusual ferromagnetic interaction in a Mn(II) chloro-bridged polymer. Inorg. Chim. Acta 2005, 358, 4459–4465. [Google Scholar] [CrossRef]
Sample Availability: Samples of the all compounds are available from the authors. |

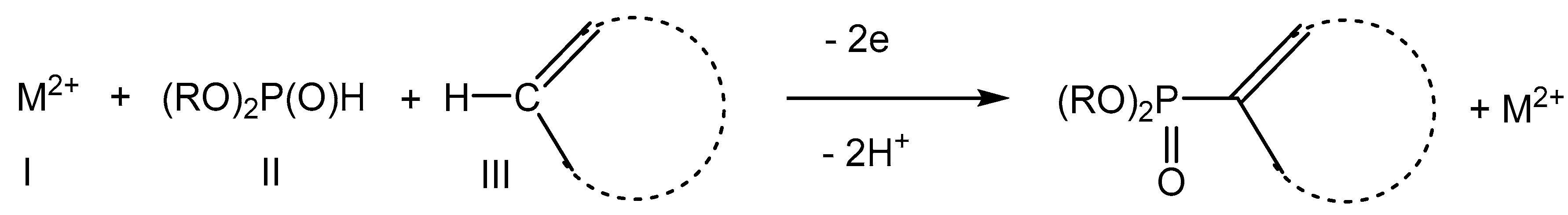

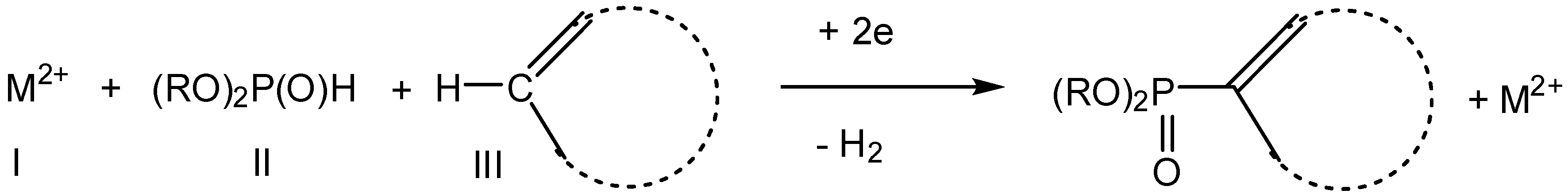

{kind=link}
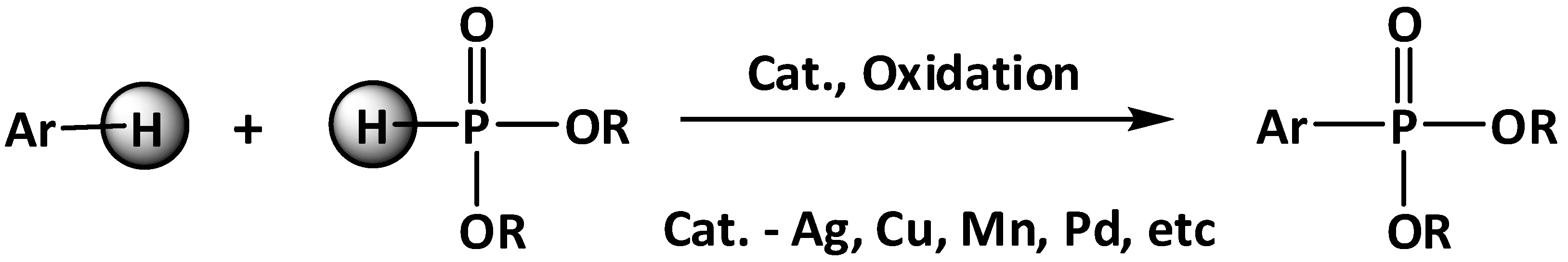{kind=link}
{kind=link}
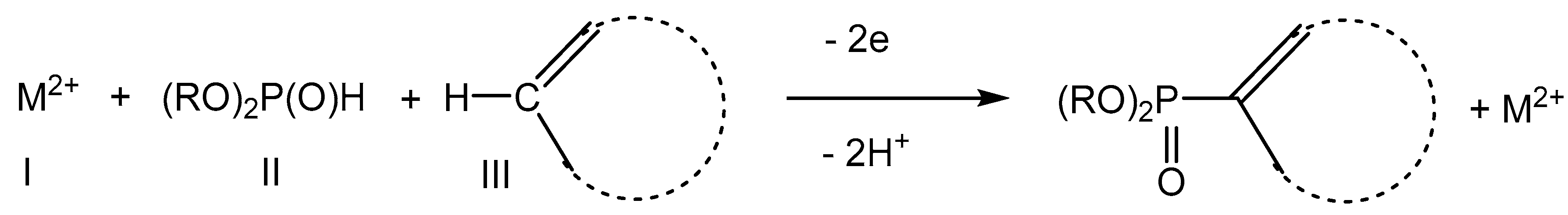{kind=link}
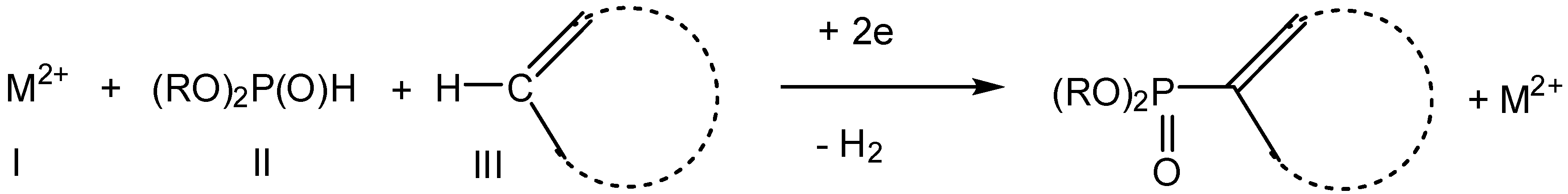{kind=link}
{kind=link}
| Catalytic System | Ratio [Complex:DEP] | ic/ip (v = 0.1V/s) | TOF, s−1 |
|---|---|---|---|
| CoCl2bpy: HP(O)(OEt)2 | 1:144 | - | - |
| Ni(BF4)2bpy: HP(O)(OEt)2 | 1:72 | 1.3 | 160 |
| MnCl2bpy: HP(O)(OEt)2 | 1:196 | 3.6 | 355 |
| MnCl2bpy/Ni(BF4)2bpy: HP(O)(OEt)2 | 1:180 | 6.1 | 690 |
| MnCl2bpy/CoCl2bpy: HP(O)(OEt)2 | 1:180 | - | - |
| CoCl2bpy/Ni(BF4)2bpy: HP(O)(OEt)2 | 1:24 | 2.7 | 721 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Strekalova, S.; Khrizanforov, M.; Budnikova, Y. Evaluation of Transition Metal Catalysts in Electrochemically Induced Aromatic Phosphonation. Molecules 2019, 24, 1823. https://doi.org/10.3390/molecules24091823
Strekalova S, Khrizanforov M, Budnikova Y. Evaluation of Transition Metal Catalysts in Electrochemically Induced Aromatic Phosphonation. Molecules. 2019; 24(9):1823. https://doi.org/10.3390/molecules24091823
Chicago/Turabian StyleStrekalova, Sofia, Mikhail Khrizanforov, and Yulia Budnikova. 2019. "Evaluation of Transition Metal Catalysts in Electrochemically Induced Aromatic Phosphonation" Molecules 24, no. 9: 1823. https://doi.org/10.3390/molecules24091823